

Abstract
The extra cellular matrix (ECM) of the normal artery wall is a collection of fibrous proteins and associated glycoproteins embedded in a hydrated ground substance of glycosaminoglycans and proteoglycans. These distinct molecules are organized into a highly ordered network that are closely associated with the vascular cells that produce them. In addition to providing the architectural framework for the artery wall that imparts mechanical support and viscoelasticity, the ECM can regulate the behaviour of vascular cells, including their ability to migrate, proliferate and survive injury. The composition of the ECM is different within intimal lesions of atherosclerosis, which are composed of monocytes and lymphocytes from the circulation and smooth muscle cells (SMC) that migrate from the media to the intima (Ross 1993, 1999), and these differences may contribute to the altered phenotype of vascular cells within lesions. This review will briefly outline the ECM changes observed in atherosclerosis and restenosis and the potential relationship of these changes to altered vascular cell functions.
Keywords: smooth muscle, endothelial cell, monocyte, macrophage, phenotype
Vascular extracellular matrix in normal vessels
Normal blood vessel walls are composed of endothelial cells, SMC (smooth muscle cells), and ECM (the extracellular matrix) that vary within the different layers of the vessel wall (reviewed in Wight 1996). They are arranged in concentric layers: the intima, composed of the lining endothelial cells with minimal subendothelial ECM enriched in proteoglycans and hyaluronan (HA); the media, separated from the intima by a dense elastic membrane (internal elastic lamina), and composed of smooth muscle cell layers embedded in an ECM comprising elastic elements, collagen and proteoglycans (Table 1); and the adventitia, separated from the media by the external elastic lamina, and composed of fibrillar collagen, fibroblasts, and vasa vasora that nourish the vascular wall. The ECM of each of these layers imparts different properties to the vessel (Figure 1), with compressible inner layers allowing expansion of the vessel during systole and elastic recoil during diastole.
Table 1.
Comparison of the extracellular matrix in the normal media and in lesions of atherosclerosis*

*Expression levels have been summarized from the available data: -, none detectable; ±, variably detectable; +, detectable; + +, detectable at moderate levels; and + + +, detectable at high levels.
Figure 1.

A transmission electron micrograph of a developing monkey aorta. Within normal vessels, smooth muscle layers are embedded in a matrix composed of elastic elements, collagen and proteoglycans. Two endothelial cells can be seen at the lumen (although their nuclei are not apparent in this thin section). Beneath the endothelial cells are newly forming elastic fibres (E) that are separated from the endothelium by basement membrane and collagen fibrils. A layer of smooth muscle lies beneath the forming elastic fibres, which is separated from another layer of smooth muscle by well-formed elastic laminae (EL). Reprinted with permission from Ross R. (1988) In Heart Disease:A Textbook of Cardiovascular Medicine, 3rd edition. (Ed. E. Braunwald) Philadelphia: W.B. Saunders Co., 1135–1152.
Collagens are composed of a triple helix of three polypeptide α chains, each having a gly-x-y repeating sequence (Prockop & Kivirikko 1995). In vessels, types I and III are predominant and assemble into cross-banded fibrils that provide tensile strength to the vessel wall. Collagens type IV, VI and VIII are nonfibrillar collagens. Within basement membranes beneath endothelial cells and surrounding media SMC, collagen types IV and VIII form 3-dimensional networks (Yurchenco & Schittny 1990; Shuttleworth 1997) that serve as an anchoring substrate and help form a permeability barrier. Self-association and disulphide bonding of type VI collagen result in high molecular weight aggregates that are localized between fibrils of collagens type I and III (Katsuda et al. 1992). Collagen XVIII, the precursor of the endothelial cell inhibitor endostatin, is also expressed in normal media and associated with elastic fibres in the multiple elastic membranes of the aorta and large arteries (Miosge et al. 1999).
While collagens provide tensile strength, elastin assembled into elastic fibres provides elastic recoil needed to accommodate the pulsatile nature of blood flow as well as haemodynamic and pressure changes (Rosenbloom et al. 1993). Fibrillin, a 350-kDa glycoprotein which associates with itself or with other components of the ECM, forms a microfibrillar network that serves as scaffolding for deposition of elastin and assembly of elastic fibres (Reinhardt et al. 1995). Expression of emilin, an extracellular matrix glycoprotein, also precedes elastin deposition and is thought to be involved in elastogenesis (Bressan et al. 1993). Elastic fibres are synthesized by SMC and are arranged in concentric lamellae that separate the different layers of the vessel and form boundaries between layers of SMC. Mice made hemizygous for the elastin gene are normal in terms of arterial compliance, but to be compliant they increase the number of rings of elastic lamellae and SMC 2.5-fold (Li et al. 1998a).
Proteoglycans and HA are hydrophilic molecules that represent the third general component of the ECM. Proteoglycans consist of a core protein linked to one or more polysaccharides that have diverse roles in regulating connective tissue structure and permeability (Iozzo & Murdoch 1996; Rosenberg et al. 1997). HA is a huge molecule that consists of many repeats of a simple disaccharide stretched end-to-end, which often serves as a backbone for large proteoglycan complexes (Fraser et al. 1997). HA binds a large amount of water forming a viscous hydrate gel, which allows the ECM to resist compression forces. In addition to interacting with other matrix constituents, proteoglycans and HA interact with vascular cells.
The adhesive glycoproteins fibronectin and laminin form connections between other ECM and cells via specific integrin receptors. Fibronectin is a multifunctional adhesive protein present in the plasma and also synthesized by vascular cells. Fibronectin is a large (approximately 450 kDa) disulphide-linked, glycoprotein dimer that binds collagen, fibrin and proteoglycans via specific domains as well as vascular cells through specific integrins (Ruoslahti 1988). Laminin, an even larger (approximately 820 kDa) trimeric glycoprotein, is the most abundant glycoprotein in endothelial and SMC basement membranes (Timpl & Brown 1994). Laminin binds cells through specific integrins and interacts with other ECM, such as collagen type IV and heparin sulphate.
Another group of glycoproteins, termed matricellular proteins (Bornstein 1995), are a class of secreted proteins that interact with other ECM constituents, multiple specific cell surface receptors, as well as growth factors, to modulate cell–matrix interactions (Sage & Bornstein 1991). This group includes osteopontin, SPARC (also known as osteonectin), tenascin and thrombospondin. As indicated in Table 1, their expression in normal vessels is limited.
The extracellular matrix surrounding vascular cells is altered after injury and in developing lesions of atherosclerosis
After balloon injury of a normal or diseased vessel and in atherosclerosis, the physiologic healing response results in the formation of a neointima. The neointima after balloon injury of a normal vessel is composed primarily of SMC that migrate from the media into the intima (Clowes et al. 1986). In contrast, in atherosclerosis, the inflammatory response initiates neointimal accumulation of monocytes and lymphocytes followed by SMC migration and proliferation (Ross 1993, 1999). In both cases, analysis of experimental models of balloon injury and atherosclerosis, as well as of more advanced human lesions, has demonstrated alterations in the ECM within the neointima (Table 1). With the formation of intimal lesions, the phenotype of the SMC changes from a ‘contractile’ state, in which the SMC are filled with myofilaments and contain a relatively poorly developed Golgi apparatus and rough endoplasmic reticulum, to a ‘synthetic’ phenotype (Figure 2) characterized by an abundance of rough endoplasmic reticulum and Golgi bodies with few and sometimes no evident myofilaments (Thyberg et al. 1990).
Figure 2.
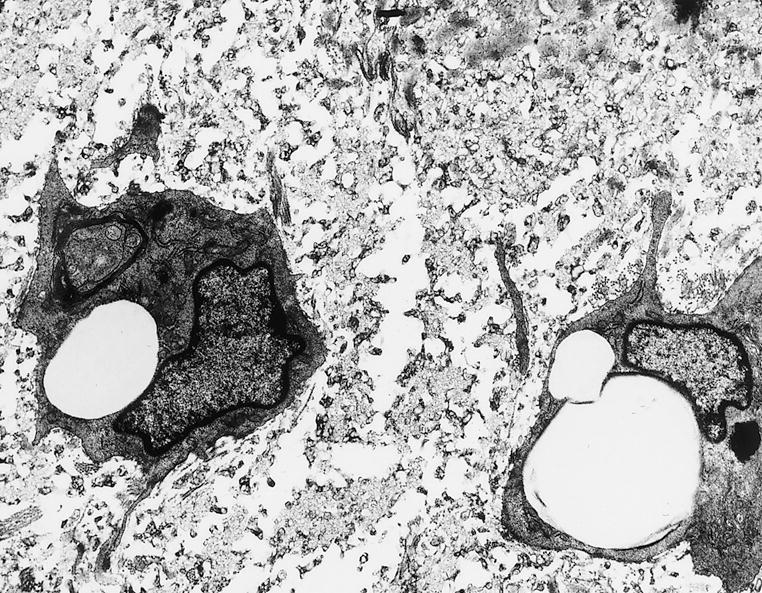
Smooth muscle cells within atherosclerotic lesions have abundant rough endoplasmic reticula. A transmission electron micrograph of a deep layer of a fatty streak in the thoracic aorta of a nonhuman primate six months after initiation of a high cholesterol diet. The extracellular space between the smooth muscle cells in the deep layer of the fatty streak contains collagen, elastin and numerous extracellular liposomes. The smooth muscle cells have abundant rough endoplasmic reticula and few myofilaments. Reprinted with permission from Masuda J. & Ross R. (1990) Arteriosclerosis10, 164–177.
Lipid- and macrophage-rich areas of lesions contain less collagen (Voss & Rauterberg 1986). In contrast, fibrous plaques contain areas rich in types I and III collagen (Voss & Rauterberg 1986). Type V and VI collagen are observed diffusely distributed in the intimal space associated with cross-banded collagen fibres and beaded filaments respectively (Katsuda et al. 1992), while type IV collagen increases in multilayered basement membrane beneath endothelial cells and around SMC in lesions (Shekhonin et al. 1985; Katsuda et al. 1992). Type VIII collagen is expressed in response to balloon injury(Sibinga et al. 1997), and was identified in a differential screen of the rat carotid artery four days after injury in which the migratory response was enhanced (Bendeck et al. 1996). Type VIII collagen has also been demonstrated to accumulate in the intimal space in response to cholesterol diet in rabbits (Plenz et al. 1999a), in both SMC (Plenz et al. 1999b) and macrophages (Weitkamp et al. 1999).
Disruption or loss of elastic fibres is frequently observed in atherosclerosis (Fulop et al. 1998), while the content and distribution of proteoglycans and HA also change (Table 1). In lesions from hypercholesterolemic nonhuman primates at different stages of lesion development, strong immunostaining for decorin, biglycan, versican and HA is observed in both intermediate and advanced lesions, with decorin being more predominant in macrophage-rich areas and versican most prevalent in areas rich in SMC (Evanko et al. 1998). Selective deposits of versican (Wight et al. 1997) and biglycan and decorin (Riessen et al. 1994) are also observed in the ECM of restenotic human vessels. The cell surface proteoglycans syndecan-1 and syndecan-4 are also increased after vascular injury (Cizmeci-Smith et al. 1997).
In situ fibronectin assembly by neointimal SMC has been demonstrated in tissue sections from balloon-injured rat carotid artery 12 days after injury (Pickering et al. 2000). Electron microscopic analysis also demonstrates newly deposited fibronectin assembled into a fibrillar network associated with the surface of synthetic SMC during early atherosclerotic and restenotic lesion development (Kakolyris et al. 1995; Thyberg et al. 1997).
Expression of the matricellular proteins is significantly increased in developing lesions (Table 1). Osteopontin (Giachelli et al. 1993), SPARC (Raines et al. 1992), tenascin (Hedin et al. 1991; Wallner et al. 1999), and thrombospondin (Wight et al. 1985; Reed et al. 1995; Riessen et al. 1998) are all increased after arterial injury in the atherosclerotic lesions. In addition to being localized to lesions of atherosclerosis and balloon-injured vessels, antibody blockade of osteopontin (Liaw et al. 1997) and thrombospondin (Chen et al. 1999) reduces neointimal thickening after carotid denudation and thrombospondin-enhanced re-endothelialization.
In addition to changes in the ECM composition after injury and in atherosclerotic lesions, there is evidence of matrix degration by matrix metalloproteinases (MMPs) (Galis et al. 1994; Halpert et al. 1996; Nikkari et al. 1996; Sukhova et al. 1999) and cathepsins (Sukhova et al. 1998). MMP expression is particularly elevated in the shoulders and cores of lesions and may contribute to plaque destabilization. Cleaved type I collagen is localized in atheromatous lesions with MMP-1- and MMP-13-positive macrophages (Sukhova et al. 1999). Macrophage-derived proteinases have the capacity to degrade every component of the extracellular matrix, even in the presence of high-affinity proteinase inhibitors (Jones & Werb 1980; Owen & Campbell 1999). The proteinases are required for the response after injury, as evidenced by: 1) MMP blockade inhibits SMC migration in the rat carotid (Bendeck et al. 1996); 2) gene transfer of tissue inhibitor of metalloproteinase (TIMP)-2 into SMC after rat carotid balloon injury inhibits SMC migration and accumulation (Forough et al. 1996; Cheng et al. 1998); 3) the accumulation of SMC after electric injury of the mouse femoral artery is impaired in animals deficient in TIMP-1 (Lijnen et al. 1999). Conversely, overexpression of MMP-9 enhances SMC migration and alters remodelling in the injured rat carotid artery (Mason et al. 1999). In many cases, the proteolytic events are probably concentrated near the cell surface where they will be effective even in the presence of high concentrations of inhibitors of apoptosis (Werb 1997). Thus, new ECM deposition, as well as localized ECM degradation, is observed in neointimal lesions.
Cellular response to the extracellular matrix is regulated by specific matrix receptors
The principal matrix receptors on vascular cells include: integrins, members of the diverse family of transmembrane heterodimers that lack any inherent kinase activity (Shattil & Ginsberg 1997); CD44, a broadly expressed cell surface protein that is a receptor for hyaluronate (Aruffo et al. 1990); receptor for HA-mediated motility (RHAMM), which has been implicated in the regulation of SMC migration (Cheung et al. 1999); and the discoidin domain receptors, previously, orphan tyrosine kinases that have been identified as collagen receptors (Schlessinger 1997). The integrin profiles of endothelial cells and SMC in the normal vascular wall have been characterized (Conforti et al. 1992; Skinner et al. 1994), and are shown in Table 2. Both the adhesive and signalling properties of the integrins are critical for their biological activities (Shattil & Ginsberg 1997). Changes in integrin and other adhesion receptors during atherogenesis have not been well defined.
Table 2.
Integrins expressed in normal vessels*

*Expression levels have been summarized from the available data: -, none detectable; ±, variably detectable; +, detectable at low levels; and + +, detectable at higher levels.
The effects of different extracellular matrices on vascular cell functions
When normal medial SMC are placed in culture, within a few days they lose their contractility and myofilaments and develop an extensive rough endoplasmic reticulum and a large Golgi complex (Hultgardh-Nilsson et al. 1997), similar to the features of SMC in developing lesions (Thyberg et al. 1990). Interestingly, laminin, a constituent of normal media, has been shown to inhibit the shift of cultured SMCs from the ‘contractile’ phenotype, while fibronectin promotes the shift to the ‘synthetic’ phenotype (Hedin et al. 1988). Culture of SMC on polymerized collagen type I inhibits SMC proliferation and mimics many of the properties of medial SMC (Koyama et al. 1996; Raines et al. 2000), and on rigid gels of type IV collagen may even more closely reproduce the phenotype of medial SMC (Hirose et al. 1999). Four other matrix-associated glycoproteins (osteopontin, SPARC, thrombospondin and tenascin) present in lesions of atherosclerosis (Table 1), have been shown to exert common ‘antiadhesive’ functions involved in cell migration and proliferation (Sage & Bornstein 1991) and osteopontin and vitronectin promote SMC adhesion and migration (Liaw et al. 1995; Slepian et al. 1998). Thus, different ECM environments of the SMC can modulate SMC phenotype and responsiveness.
Each of the types of ECM listed in Table 1 within the local environment surrounding vascular cells creates distinct environments for these cells. Collagens, specifically polymerized collagens type I and IV surrounding SMC, promote a more quiescent and ‘contractile’ SMC phenotype similar to normal medial SMC in vivo (Koyama et al. 1996; Hirose et al. 1999; Raines et al., in press). However, evaluation of calcification of cultures of bovine SMC has shown that films of monomeric type I collagen enhance mineralized nodule formation, calcium incorporation, von Kossa staining, and alkaline phosphatase activity, while films of type IV collagen inhibit mineralization parameters (Watson et al. 1998). Addition of soluble type VIII collagen, which accumulates in developing lesions, stimulates SMC migration in vitro, and attachment to type VIII collagen increases production of MMP-2 and MMP-9 in rat SMC isolated from carotid neointima after balloon injury, but fails to alter MMP production in rat SMC isolated from normal media (Hou et al. 2000). Similarly, mutation of one allele of elastin, previously thought to play a purely structural role, is sufficient to induce subendothelial proliferation of SMC (Li et al. 1998b).
Collagens also modulate monocytes/macrophages that infiltrate the artery wall early in lesion initiation and are present throughout lesion development (Ross 1993, 1999). Monomeric films of collagen type I enhance acquisition of resident macrophage traits, such as expression of CD71, cell spreading, uptake of modified lipoproteins, and release of MMP-9 (Wesley et al. 1998). Integrin engagement of ECM, such as with fibronectin, has also been shown to be a primary signal transduction pathway regulating monocyte immediate-early gene induction, including a number of inflammatory mediators (Yurochko et al. 1992).
Heparan sulphate species produced by SMC have long been recognized as potent inhibitors of SMC proliferation (Fritze et al. 1985). In addition to direct effects, a number of growth factors bind to heparin sulphate-rich ECM (Taipale & Keski-Oja 1997), and thus can serve as a local site of storage. Heparin has also been critical for oligomerization of fibroblast growth factor (FGF) and subsequent stimulation of endothelial cells and SMC (Spivak-Kroizman et al. 1994). Recently, heparanase degradation of syndecan-1 ectodomain, soluble heparan sulphate proteoglycan shed from vascular cell surfaces after injury, produces heparin sulphate fragments, which activate FGF-stimulated proliferation (Kato et al. 1998). Glypican-1, the only member of the family of glycosylphosphatidylinositol-anchored cell surface heparan sulphate proteoglycans expressed in vascular cells, binds vascular endothelial growth factor and acts as an extracellular chaperone that enhances its activity (Gengrinovitch et al. 1999). In addition, the proteoglycans biglycan and versican and HA have been involved in migration and proliferation of vascular cells (Wight et al. 1992). Biglycan expression is upregulated in migrating endothelial cells, and is localized to the tips and edges of lamellopodia on migrating cells (Kinsella et al. 1997) and in lesions of atherosclerosis (Evanko et al. 1998). All migrating and proliferating human SMC display abundant HA- and versican-rich coats whose appearance is coordinated with cell detachment and mitotic cell rounding (Evanko et al. 1999). Reduction in this coat is sufficient to enhance adhesion and decrease SMC migration and proliferation. In animal models of balloon injury of vessels, administration of HA has inhibited neointimal macrophage influx (Ferns et al. 1995) and SMC accumulation (Savani & Turley 1995).
Fibronectin fibril assembly, another feature of atherosclerotic lesions (Thyberg et al. 1997; Pickering et al. 2000) is necessary for SMC growth (Mercurius & Morla 1998; our unpublished observations), but is also important as an adhesion substrate for the survival of endothelial cells (Meredith et al. 1993; Chen et al. 1997). Multiple splice variants of fibronectin have been identified, and differential regulation of the multiple splice variants may further modulate vascular cell adhesion, migration and proliferation (Schwarzbauer 1991). A film of fibronectin also promotes calcification of SMC in vitro (Watson et al. 1998).
The matricellular proteins − SPARC, tenascin and thrombospondin − promote antiadhesive and antiproliferative responses in endothelial cells and SMC (Sage & Bornstein 1991), while osteopontin promotes adhesion and migration of SMC (Liaw et al. 1995), but also inhibits calcification (Wada et al. 1999) and apoptosis (Scatena et al. 1998). The modular domain structure of matricellular proteins allows them to independently bind cells and matrix components and a multiplicity of binding partners simultaneously. However, their ability to bind a large number of integrins and other receptors has made it difficult to assess the relative contribution of specific binding interactions. Loss of either of these binding partners may have significant impact on the ECM and vascular cell behaviour. In embryonic mov 13 mice that lack type I collagen, SPARC is not observed in the ECM despite normal levels of synthesis by mov 13 cells (Iruela-Arispe et al. 1996). The ability of matricellular proteins to bind growth factors, as shown for platelet-derived growth factor binding to SPARC (Raines et al. 1992), provides an alternative mechanism for the matricellular proteins to modulate vascular cell responses.
Fragments of the extracellular matrix have distinct activities from the native molecules
ECM fragments generated as a result of local release of MMPs and cathepsins have different effects on vascular cells than their native counterparts, including the generation of novel regulators of angiogenesis (Sage 1997). A 20-kDa C-terminal fragment of collagen XVIII, endostatin, specifically inhibits endothelial proliferation and blocks angiogenesis (O'Reilly et al. 1997). Administration of endostatin to apolipoprotein E -/- mice (20–36 weeks), which develop lesions of atherosclerosis, reduces intimal neovascularization and decreased plaque area at the aortic origin (Moulton et al. 1999). The noncollagenous domains of α2(IV), α3(IV), and α6(IV) chains of collagen type IV regulate endothelial cell adhesion and migration and potently inhibit angiogenesis (Petitclerc et al. 2000). In cultured human SMC, addition of fragments of type I collagen induces cell rounding through calpain-mediated cleavage of focal adhesion proteins (Carragher et al. 1999). This effect is dominant irrespective of the ECM, to which the SMC are adherent, and may provide a mechanism for de-adhesion of cells from ECM. Elastin peptides promote monocyte migration (Senior et al. 1980), and induce a dose-dependent and endothelium-dependent vasorelation mediated by the elastin/laminin receptor and by endothelial nitric oxide production (Faury et al. 1998). On human monocytes, elastin peptides also mobilize calcium and stimulate a respiratory burst and enzyme secretion (Fulop et al. 1986). In SMC, elastin peptides stimulate production of MMPs and decrease secretion of their inhibitors, TIMPs (Tummalapalli & Tyagi 1999).
The extracellular domain of the cell surface proteoglycan syndecan-1 is shed after injury, and heparanase treatment of the shed ectodomain generates heparin sulphate fragments that markedly enhance FGF activity (Kato et al. 1998). HA in its native form exists as a high molecular weight polymer, but during inflammation lower molecular weight fragments accumulate. In macrophages, the HA fragments induce chemokine gene expression (McKee et al. 1996), including MMP regulation (Horton et al. 1999), and stimulate nitric oxide synthase through a nuclear factor-κB-dependent mechanism (McKee et al. 1997).
Several examples exist for cleavage-induced exposure of cryptic binding sites in vascular glycoproteins. Cleavage of fibronectin results in the release of a 120-kDa cell-binding fragment that is chemotactic for human monocytes (Clark et al. 1988). The same fibronectin fragment stimulates tumour necrosis factor secretion by human monocytes (Beezhold & Personius 1992). More recently, it has been shown that this fragment also modulates integrin α5 expression, which has been proposed to promote monocyte accumulation (Trial et al. 1999). Thrombin cleavage of osteopontin promotes endothelial cell attachment and spreading (Senger et al. 1994). This cleavage exposes a cryptic adhesive sequence recognized by α9 β1, an activity not found in native osteopontin (SMith et al. 1996). Plasminolysis of SPARC produces a copper-binding peptide that stimulates angiogenesis (Sage 1997).
Perspective
Vascular ECM is critical for maintenance of vascular integrity and imparts tensile strength, viscoelasticity, elastic recoil and compressibility through the distinct properties of the different constituents. As reviewed here, the ECM composition is altered during the formation of intimal lesions, which changes the physical properties of the ECM, including the generation of fragments with distinct activities. However, in addition to changing the physical nature of the ECM, the composition of the ECM can regulate vascular cell responses, including survival, migration and proliferation, all of which can significantly contribute to remodelling of the vascular wall. An outgrowth of our increasing knowledge of the molecular interactions of ECM receptors with different ECM constituents is the development of peptide mimetics that can interfere specifically with some of these processes. Further understanding of these cell–matrix interactions promises to provide novel therapeutic targets for the prevention of unfavourable remodelling of the artery wall in atherosclerosis and restenosis.
Acknowledgments
This manuscript is dedicated to the memory of the late Russell Ross who would have enjoyed writing a review in honor of his dear friend and colleague, Neville Woolf. Russ's first studies as a young investigator focused on cellular interactions with the extracellular matrix in wound repair. Within the last five years, our laboratory has returned to extracellular matrix to examine how it may alter cellular responses in atherogenesis. Although he left us much too soon, a wonderful legacy remains in the people he trained, the colleagues he inspired, and our memories of a truly caring and supportive mentor, colleague and friend and an enthusiastic scientist who was passionate in his pursuit of understanding the cellular and molecular mechanisms involved in the formation of lesions of atherosclerosis.
The editorial assistance of Barbara Droker is gratefully acknowledged.
References
- 1.Aruffo A, Stamenkovic I, Melnick M. CD44 is the principal cell surface receptor for hyaluronate. Cell. 1990;61:1303–1313. doi: 10.1016/0092-8674(90)90694-a. [DOI] [PubMed] [Google Scholar]
- 2.Beezhold DH, Personius C. Fibronectin fragments stimulate tumor necrosis factor secretion by human monocytes. J. Leukoc. Biol. 1992;51:59–64. doi: 10.1002/jlb.51.1.59. [DOI] [PubMed] [Google Scholar]
- 3.Bendeck MP, Irvin C, Reidy MA. Inhibition of matrix metalloproteinase activity inhibits smooth muscle cell migration but not neointimal thickening after arterial injury. Circ. Res. 1996;78:38–43. doi: 10.1161/01.res.78.1.38. [DOI] [PubMed] [Google Scholar]
- 4.Bornstein P. Diversity of function is inherent in matricellular proteins: an appraisal of thrombospondin 1. J. Cell Biol. 1995;130:503–506. doi: 10.1083/jcb.130.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bressan GM, Daga-Gordini D, Colombatti A, et al. Emilin, a component of elastic fibers preferentially located at the elastin–microfibrils interface. J. Cell Biol. 1993;121:201–212. doi: 10.1083/jcb.121.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carragher NO, Levkau B, Ross R, Raines EW. Degraded collagen fragments promote rapid disassembly of smooth muscle focal adhesions that correlates with cleavage of pp125 (FAK), paxillin, and talin. J. Cell Biol. 1999;147:619–630. doi: 10.1083/jcb.147.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen CS, Mrksich M, Huang S, et al. Geometric control of cell life and death. Science. 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- 8.Chen D, Asahara T, Krasinski K, et al. Antibody blockade of thrombospondin accelerates reendothelialization and reduces neointima formation in balloon-injured rat carotid artery. Circulation. 1999;100:849–854. doi: 10.1161/01.cir.100.8.849. [DOI] [PubMed] [Google Scholar]
- 9.Cheng L, Mantile G, Pauly R, et al. Adenovirus-mediated gene transfer of the human tissue inhibitor of metalloproteinase-2 blocks vascular smooth muscle cell invasiveness in vitro and modulates neointimal development in vivo. Circulation. 1998;98:2195–2201. doi: 10.1161/01.cir.98.20.2195. [DOI] [PubMed] [Google Scholar]
- 10.Cheung WF, Cruz TF, Turley EA. Receptor for hyaluronan-mediated motility (RHAMM), a hyaladherin that regulates cell responses to growth factors. Biochem. Soc. Trans. 1999;27:135–142. doi: 10.1042/bst0270135. [DOI] [PubMed] [Google Scholar]
- 11.Cizmeci-Smith G, Langan E, Youkey J, et al. Syndecan-4 is a primary-response gene induced by basic fibroblast growth factor and arterial injury in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1997;17:172–180. doi: 10.1161/01.atv.17.1.172. [DOI] [PubMed] [Google Scholar]
- 12.Clark RA, Wikner NE, Doherty DE, Norris DA. Cryptic chemotactic activity of fibronectin for human monocytes resides in the 120-kDa fibroblastic cell-binding fragment. J. Biol. Chem. 1988;263:12115–12123. [PubMed] [Google Scholar]
- 13.Clowes AW, Clowes MM, Reidy MA. Kinetics of cellular proliferation after arterial injury. III. Endothelial and smooth muscle growth in chronically denuded vessels. Lab. Invest. 1986;54:295–303. [PubMed] [Google Scholar]
- 14.Conforti G, Dominguez-Jimenez C, Zanetti A, et al. Human endothelial cells express integrin receptors on the luminal aspect of their membrane. Blood. 1992;80:437–446. [PubMed] [Google Scholar]
- 15.Evanko SP, Raines EW, Ross R, et al. Proteoglycan distribution in lesions of atherosclerosis depends on lesion severity, structural characteristics, and the proximity of platelet-derived growth factor and transforming growth factor-beta. Am. J. Pathol. 1998;152:533–546. [PMC free article] [PubMed] [Google Scholar]
- 16.Evanko SP, Angello JC, Wight TN. Formation of hyaluronan- and versican-rich pericellular matrix is required for proliferation and migration of vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1999;19:1004–1013. doi: 10.1161/01.atv.19.4.1004. [DOI] [PubMed] [Google Scholar]
- 17.Faury G, Garnier S, Weiss AS, et al. Action of tropoelastin and synthetic elastin sequences on vascular tone and on free Ca2+ level in human vascular endothelial cells. Circ. Res. 1998;82:328–336. doi: 10.1161/01.res.82.3.328. [DOI] [PubMed] [Google Scholar]
- 18.Ferns GA, Konneh M, Rutherford C, et al. Hyaluronan (HYAL-BV 5200) inhibits neo-intimal macrophage influx after balloon-catheter induced injury in the cholesterol-fed rabbit. Atherosclerosis. 1995;114:157–164. doi: 10.1016/0021-9150(94)05479-3. [DOI] [PubMed] [Google Scholar]
- 19.Forough R, Koyama N, Hasenstab D, et al. Overexpression of tissue inhibitor of matrix metalloproteinase-1 inhibits vascular smooth muscle cell functions in vitro and in vivo. Circ. Res. 1996;79:812–820. doi: 10.1161/01.res.79.4.812. [DOI] [PubMed] [Google Scholar]
- 20.Fraser JR, Laurent TC, Laurent UB. Hyaluronan: its nature, distribution, functions and turnover. J. Intern. Med. 1997;242:27–33. doi: 10.1046/j.1365-2796.1997.00170.x. [DOI] [PubMed] [Google Scholar]
- 21.Fritze LM, Reilly CF, Rosenberg RD. An antiproliferative heparan sulfate species produced by postconfluent smooth muscle cells. J. Cell Biol. 1985;100:1041–1049. doi: 10.1083/jcb.100.4.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fulop T, Jacob MP, Varga Z, et al. Effect of elastin peptides on human monocytes: Ca2+ mobilization, stimulation of respiratory burst and enzyme secretion. Biochem. Biophys. Res. Commun. 1986;141:92–98. doi: 10.1016/s0006-291x(86)80339-4. [DOI] [PubMed] [Google Scholar]
- 23.Fulop T, Jacob MP, Khalil A, et al. Biological effects of elastin peptides. Pathol. Biol. (Paris) 1998;46:497–506. [PubMed] [Google Scholar]
- 24.Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J. Clin. Invest. 1994;94:2493–2503. doi: 10.1172/JCI117619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gengrinovitch S, Berman B, David G, et al. Glypican-1 is a VEGF165 binding proteoglycan that acts as an extracellular chaperone for VEGF165. J. Biol. Chem. 1999;274:10816–10822. doi: 10.1074/jbc.274.16.10816. [DOI] [PubMed] [Google Scholar]
- 26.Giachelli CM, Bae N, Almeida M, et al. Osteopontin is elevated during neointima formation in rat arteries and is a novel component of human atherosclerotic plaques. J. Clin. Invest. 1993;92:1686–1696. doi: 10.1172/JCI116755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Halpert I, Sires UI, Roby JD, et al. Matrilysin is expressed by lipid-laden macrophages at sites of potential rupture in atherosclerotic lesions and localizes to areas of versican deposition, a proteoglycan substrate for the enzyme. Proc. Natl. Acad. Sci. USA. 1996;93:9748–9753. doi: 10.1073/pnas.93.18.9748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hedin U, Bottger BA, Forsberg E, et al. Diverse effects of fibronectin and laminin on phenotypic properties of cultured arterial smooth muscle cells. J. Cell Biol. 1988;107:307–319. doi: 10.1083/jcb.107.1.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hedin U, Holm J, Hansson GK. Induction of tenascin in rat arterial injury. Relationship to altered smooth muscle cell phenotype. Am. J. Pathol. 1991;139:649–656. [PMC free article] [PubMed] [Google Scholar]
- 30.Hirose M, Kosugi H, Nakazato K, Hayashi T. Restoration to a quiescent and contractile phenotype from a proliferative phenotype of myofibroblast-like human aortic smooth muscle cells by culture on type IV collagen gels. J. Biochem. (Tokyo) 1999;125:991–1000. doi: 10.1093/oxfordjournals.jbchem.a022407. [DOI] [PubMed] [Google Scholar]
- 31.Horton MR, Shapiro S, Bao C, et al. Induction and regulation of macrophage metalloelastase by hyaluronan fragments in mouse macrophages. J. Immunol. 1999;162:4171–4176. [PubMed] [Google Scholar]
- 32.Hou G, Mulholland D, Gronska MA, Bendeck MP. Type VIII collagen stimulates smooth muscle cell migration and matrix metalloproteinase synthesis after arterial injury. Am. J. Pathol. 2000;156:467–476. doi: 10.1016/S0002-9440(10)64751-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hultgardh-Nilsson A, Lovdahl C, Blomgren K, et al. Expression of phenotype- and proliferation-related genes in rat aortic smooth muscle cells in primary culture. Cardiovasc. Res. 1997;34:418–430. doi: 10.1016/s0008-6363(97)00030-8. [DOI] [PubMed] [Google Scholar]
- 34.Iozzo RV, Murdoch AD. Proteoglycans of the extracellular environment: clues from the gene and protein side offer novel perspectives in molecular diversity and function. Faseb J. 1996;10:598–614. [PubMed] [Google Scholar]
- 35.Iruela-Arispe ML, Vernon RB, Wu H, et al. Type I collagen-deficient Mov-13 mice do not retain SPARC in the extracellular matrix: implications for fibroblast function. Dev. Dyn. 1996;207:171–183. doi: 10.1002/(SICI)1097-0177(199610)207:2<171::AID-AJA5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 36.Jones PA, Werb Z. Degradation of connective tissue matrices by macrophages. II. Influence of matrix composition on proteolysis of glycoproteins, elastin, and collagen by macrophages in culture. J. Exp. Med. 1980;152:1527–1536. doi: 10.1084/jem.152.6.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kakolyris S, Karakitsos P, Tzardi M, Agapitos E. Immunohistochemical detection of fibronectin in early and advanced atherosclerosis. In Vivo. 1995;9:35–40. [PubMed] [Google Scholar]
- 38.Kato M, Wang H, Kainulainen V, et al. Physiological degradation converts the soluble syndecan-1 ectodomain from an inhibitor to a potent activator of FGF-2. Nat. Med. 1998;4:691–697. doi: 10.1038/nm0698-691. [DOI] [PubMed] [Google Scholar]
- 39.Katsuda S, Okada Y, Minamoto T, et al. Collagens in human atherosclerosis. Immunohistochemical analysis using collagen type-specific antibodies. Arterioscler. Thromb. 1992;12:494–502. doi: 10.1161/01.atv.12.4.494. [DOI] [PubMed] [Google Scholar]
- 40.Kinsella MG, Tsoi CK, Jarvelainen HT, Wight TN. Selective expression and processing of biglycan during migration of bovine aortic endothelial cells. The role of endogenous basic fibroblast growth factor. J. Biol. Chem. 1997;272:318–325. doi: 10.1074/jbc.272.1.318. [DOI] [PubMed] [Google Scholar]
- 41.Koyama H, Raines EW, Bornfeldt KE, et al. Fibrillar collagen inhibits arterial smooth muscle proliferation through regulation of Cdk2 inhibitors. Cell. 1996;87:1069–1078. doi: 10.1016/s0092-8674(00)81801-2. [DOI] [PubMed] [Google Scholar]
- 42.Li DY, Faury G, Taylor DG, et al. Novel arterial pathology in mice and humans hemizygous for elastin. J. Clin. Invest. 1998a;102:1783–1787. doi: 10.1172/JCI4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li DY, Brooke B, Davis EC, et al. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998b;393:276–280. doi: 10.1038/30522. [DOI] [PubMed] [Google Scholar]
- 44.Liaw L, Lombardi DM, Almeida MM, et al. Neutralizing antibodies directed against osteopontin inhibit rat carotid neointimal thickening after endothelial denudation. Arterioscler. Thromb. Vasc. Biol. 1997;17:188–193. doi: 10.1161/01.atv.17.1.188. [DOI] [PubMed] [Google Scholar]
- 45.Liaw L, Skinner MP, Raines EW, et al. The adhesive and migratory effects of osteopontin are mediated via distinct cell surface integrins. Role of αvβ3 in smooth muscle cell migration to osteopontin in vitro. J. Clin. Invest. 1995;95:713–724. doi: 10.1172/JCI117718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lijnen HR, Soloway P, Collen D. Tissue inhibitor of matrix metalloproteinases-1 impairs arterial neointima formation after vascular injury in mice. Circ. Res. 1999;85:1186–1191. doi: 10.1161/01.res.85.12.1186. [DOI] [PubMed] [Google Scholar]
- 47.Mason DP, Kenagy RD, Hasenstab D, et al. Matrix metalloproteinase-9 overexpression enhances vascular smooth muscle cell migration and alters remodeling in the injured rat carotid artery. Circ. Res. 1999;85:1179–1185. doi: 10.1161/01.res.85.12.1179. [DOI] [PubMed] [Google Scholar]
- 48.McKee CM, Lowenstein CJ, Horton MR, et al. Hyaluronan fragments induce nitric-oxide synthase in murine macrophages through a nuclear factor kappaB-dependent mechanism. J. Biol. Chem. 1997;272:8013–8018. doi: 10.1074/jbc.272.12.8013. [DOI] [PubMed] [Google Scholar]
- 49.McKee CM, Penno MB, Cowman M, et al. Hyaluronan (HA) fragments induce chemokine gene expression in alveolar macrophages. The role of HA size and CD44. J. Clin. Invest. 1996;98:2403–2413. doi: 10.1172/JCI119054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mercurius KO, Morla AO. Inhibition of vascular smooth muscle cell growth by inhibition of fibronectin matrix assembly. Circ. Res. 1998;82:548–556. doi: 10.1161/01.res.82.5.548. [DOI] [PubMed] [Google Scholar]
- 51.Meredith JE, Fazeli B, Schwartz MA. The extracellular matrix as a cell survival factor. Mol. Biol. Cell. 1993;4:953–961. doi: 10.1091/mbc.4.9.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miosge N, Sasaki T, Timpl R. Angiogenesis inhibitor endostatin is a distinct component of elastic fibers in vessel walls. Faseb J. 1999;13:1743–1750. doi: 10.1096/fasebj.13.13.1743. [DOI] [PubMed] [Google Scholar]
- 53.Moulton KS, Heller E, Konerding MA, et al. Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation. 1999;99:1726–1732. doi: 10.1161/01.cir.99.13.1726. [DOI] [PubMed] [Google Scholar]
- 54.Nikkari ST, Geary RL, Hatsukami T, et al. Expression of collagen, interstitial collagenase, and tissue inhibitor of metalloproteinases-1 in restenosis after carotid endarterectomy. Am. J. Pathol. 1996;148:777–783. [PMC free article] [PubMed] [Google Scholar]
- 55.O'reilly MS, Boehm T, Shing Y, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 56.Owen CA, Campbell EJ. The cell biology of leukocyte-mediated proteolysis. J. Leukoc. Biol. 1999;65:137–150. doi: 10.1002/jlb.65.2.137. [DOI] [PubMed] [Google Scholar]
- 57.Petitclerc E, Boutaud A, Prestayko A, et al. New functions for non-collagenous domains of human collagen type IV. Novel integrin ligands inhibiting angiogenesis and tumor growth in vivo. J. Biol. Chem. 2000;275:8051–8061. doi: 10.1074/jbc.275.11.8051. [DOI] [PubMed] [Google Scholar]
- 58.Pickering JG, Chow LH, Li S, et al. α5β1 integrin expression and luminal edge fibronectin matrix assembly by smooth muscle cells after arterial injury. Am. J. Pathol. 2000;156:453–465. doi: 10.1016/s0002-9440(10)64750-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Plenz G, Dorszewski A, Volker W, et al. Cholesterol-induced changes of type VIII collagen expression and distribution in carotid arteries of rabbit. Arterioscler. Thromb. Vasc. Biol. 1999a;19:2395–2404. doi: 10.1161/01.atv.19.10.2395. [DOI] [PubMed] [Google Scholar]
- 60.Plenz G, Reichenberg S, Koenig C, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) modulates the expression of type VIII collagen mRNA in vascular smooth muscle cells and both are codistributed during atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1999b;19:1658–1668. doi: 10.1161/01.atv.19.7.1658. [DOI] [PubMed] [Google Scholar]
- 61.Prockop DJ, Kivirikko KI. Collagens: molecular biology, diseases, and potentials for therapy. Annu. Rev. Biochem. 1995;64:403–434. doi: 10.1146/annurev.bi.64.070195.002155. [DOI] [PubMed] [Google Scholar]
- 62.Raines EW, Koyama H, Carragher NO. The extracellular matrix dynamically regulates smooth muscle cell responsiveness to PDGF. Ann. NY Acad. Sci. 2000;902:39–52. doi: 10.1111/j.1749-6632.2000.tb06299.x. [DOI] [PubMed] [Google Scholar]
- 63.Raines EW, Lane TF, Iruela-Arispe ML, et al. The extracellular glycoprotein SPARC interacts with platelet-derived growth factor (PDGF) -AB and -BB and inhibits the binding of PDGF to its receptors. Proc. Natl. Acad. Sci. USA. 1992;89:1281–1285. doi: 10.1073/pnas.89.4.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reed MJ, Iruela-Arispe L, O'brien ER, et al. Expression of thrombospondins by endothelial cells. Injury is correlated with TSP-1. Am. J. Pathol. 1995;147:1068–1080. [PMC free article] [PubMed] [Google Scholar]
- 65.Reinhardt DP, Chalberg SC, Sakai LY. The structure and function of fibrillin. Ciba Found. Symposium. 1995;192:128–143. doi: 10.1002/9780470514771.ch7. [DOI] [PubMed] [Google Scholar]
- 66.Riessen R, Isner JM, Blessing E, et al. Regional differences in the distribution of the proteoglycans biglycan and decorin in the extracellular matrix of atherosclerotic and restenotic human coronary arteries. Am. J. Pathol. 1994;144:962–974. [PMC free article] [PubMed] [Google Scholar]
- 67.Riessen R, Kearney M, Lawler J, Isner JM. Immunolocalization of thrombospondin-1 in human atherosclerotic and restenotic arteries. Am. Heart J. 1998;135:357–364. doi: 10.1016/s0002-8703(98)70105-x. [DOI] [PubMed] [Google Scholar]
- 68.Rosenberg RD, Shworak NW, Liu J, et al. Heparan sulfate proteoglycans of the cardiovascular system. Specific structures emerge but how is synthesis regulated? J. Clin. Invest. 1997;100:S67–S75. [PubMed] [Google Scholar]
- 69.Rosenbloom J, Abrams WR, Mecham R. Extracellular matrix 4: the elastic fiber. Faseb J. 1993;7:1208–1218. [PubMed] [Google Scholar]
- 70.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 71.Ross R. Atherosclerosis — an inflammatory disease. N. Engl. J. Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 72.Ruoslahti E. Fibronectin and its receptors. Annu. Rev. Biochem. 1988;57:375–413. doi: 10.1146/annurev.bi.57.070188.002111. [DOI] [PubMed] [Google Scholar]
- 73.Sage EH. Pieces of eight: bioactive fragments of extracellular proteins as regulators of angiogenesis. Trends Cell Biol. 1997;7:182–186. doi: 10.1016/S0962-8924(97)01037-4. [DOI] [PubMed] [Google Scholar]
- 74.Sage EH, Bornstein P. Extracellular proteins that modulate cell–matrix interactions. SPARC, tenascin, and thrombospondin. J. Biol. Chem. 1991;266:14831–14834. [PubMed] [Google Scholar]
- 75.Savani RC, Turley EA. The role of hyaluronan and its receptors in restenosis after balloon angioplasty: development of a potential therapy. Int. J. Tissue React. 1995;17:141–151. [PubMed] [Google Scholar]
- 76.Scatena M, Almeida M, Chaisson ML, et al. NF-κB mediates αvβ3 integrin-induced endothelial cell survival. J. Cell Biol. 1998;141:1083–1093. doi: 10.1083/jcb.141.4.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schlessinger J. Direct binding and activation of receptor tyrosine kinases by collagen. Cell. 1997;91:869–872. doi: 10.1016/s0092-8674(00)80477-8. [DOI] [PubMed] [Google Scholar]
- 78.Schwarzbauer JE. Fibronectin: from gene to protein. Curr. Opin. Cell Biol. 1991;3:786–791. doi: 10.1016/0955-0674(91)90051-y. [DOI] [PubMed] [Google Scholar]
- 79.Senger DR, Perruzzi CA, Papadopoulos-Sergiou A, Van De Water L. Adhesive properties of osteopontin: regulation by a naturally occurring thrombin-cleavage in close proximity to the GRGDS cell-binding domain. Mol. Biol. Cell. 1994;5:565–574. doi: 10.1091/mbc.5.5.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Senior RM, Griffin GL, Mecham RP. Chemotactic activity of elastin-derived peptides. J. Clin. Invest. 1980;66:859–862. doi: 10.1172/JCI109926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shattil SJ, Ginsberg MH. Perspectives series: cell adhesion in vascular biology. Integrin signaling in vascular biology. J. Clin. Invest. 1997;100:1–5. doi: 10.1172/JCI119500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shekhonin BV, Domogatsky SP, Muzykantov VR, et al. Distribution of type I III, IV and V collagen in normal and atherosclerotic human arterial wall: immunomorphological characteristics. Coll. Relat. Res. 1985;5:355–368. doi: 10.1016/s0174-173x(85)80024-8. [DOI] [PubMed] [Google Scholar]
- 83.Shuttleworth CA. Type VIII collagen. Int. J. Biochem. Cell Biol. 1997;29:1145–1148. doi: 10.1016/s1357-2725(97)00033-2. [DOI] [PubMed] [Google Scholar]
- 84.Sibinga NE, Foster LC, Hsieh CM, et al. Collagen VIII is expressed by vascular smooth muscle cells in response to vascular injury. Circ. Res. 1997;80:532–541. doi: 10.1161/01.res.80.4.532. [DOI] [PubMed] [Google Scholar]
- 85.Skinner MP, Raines EW, Ross R. Dynamic expression of α1β1 and α2β1 integrin receptors by human vascular smooth muscle cells. α2β1 integrin is required for chemotaxis across type I collagen-coated membranes. Am. J. Pathol. 1994;145:1070–1081. [PMC free article] [PubMed] [Google Scholar]
- 86.Slepian MJ, Massia SP, Dehdashti B, et al. β3-integrins rather than β1-integrins dominate integrin–matrix interactions involved in postinjury smooth muscle cell migration. Circulation. 1998;97:1818–1827. doi: 10.1161/01.cir.97.18.1818. [DOI] [PubMed] [Google Scholar]
- 87.Smith LL, Cheung HK, Ling LE, et al. Osteopontin N-terminal domain contains a cryptic adhesive sequence recognized by α9β1 integrin. J. Biol. Chem. 1996;271:28485–28491. [PubMed] [Google Scholar]
- 88.Spivak-Kroizman T, Lemmon MA, Dikic I, et al. Heparin-induced oligomerization of FGF molecules is responsible for FGF receptor dimerization, activation, and cell proliferation. Cell. 1994;79:1015–1024. doi: 10.1016/0092-8674(94)90032-9. [DOI] [PubMed] [Google Scholar]
- 89.Sukhova GK, Schönbeck U, Rabkin E, et al. Evidence for increased collagenolysis by interstitial collagenases-1 and -3 in vulnerable human atheromatous plaques. Circulation. 1999;99:2503–2509. doi: 10.1161/01.cir.99.19.2503. [DOI] [PubMed] [Google Scholar]
- 90.Sukhova GK, Shi GP, Simon DI, et al. Expression of the elastolytic cathepsins S and K in human atheroma and regulation of their production in smooth muscle cells. J. Clin. Invest. 1998;102:576–583. doi: 10.1172/JCI181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Taipale J, Keski-Oja J. Growth factors in the extracellular matrix. Faseb J. 1997;11:51–59. doi: 10.1096/fasebj.11.1.9034166. [DOI] [PubMed] [Google Scholar]
- 92.Thyberg J, Blomgren K, Roy J, et al. Phenotypic modulation of smooth muscle cells after arterial injury is associated with changes in the distribution of laminin and fibronectin. J. Histochem. Cytochem. 1997;45:837–846. doi: 10.1177/002215549704500608. [DOI] [PubMed] [Google Scholar]
- 93.Thyberg J, Hedin U, Sjolund M, et al. Regulation of differentiated properties and proliferation of arterial smooth muscle cells. Arteriosclerosis. 1990;10:966–990. doi: 10.1161/01.atv.10.6.966. [DOI] [PubMed] [Google Scholar]
- 94.Timpl R, Brown JC. The laminins. Matrix Biol. 1994;14:275–281. doi: 10.1016/0945-053x(94)90192-9. [DOI] [PubMed] [Google Scholar]
- 95.Trial J, Baughn RE, Wygant JN, et al. Fibronectin fragments modulate monocyte VLA-5 expression and monocyte migration. J. Clin. Invest. 1999;104:419–430. doi: 10.1172/JCI4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tummalapalli CM, Tyagi SC. Responses of vascular smooth muscle cell to extracellular matrix degradation. J. Cell. Biochem. 1999;75:515–527. [PubMed] [Google Scholar]
- 97.Voss B, Rauterberg J. Localization of collagen types I III, IV and V, fibronectin and laminin in human arteries by the indirect immunofluorescence method. Pathol. Res. Pract. 1986;181:568–575. doi: 10.1016/S0344-0338(86)80151-0. [DOI] [PubMed] [Google Scholar]
- 98.Wada T, McKee MD, Steitz S, Giachelli CM. Calcification of vascular smooth muscle cell cultures: inhibition by osteopontin. Circ. Res. 1999;84:166–178. doi: 10.1161/01.res.84.2.166. [DOI] [PubMed] [Google Scholar]
- 99.Wallner K, Li C, Shah PK, et al. Tenascin-C is expressed in macrophage-rich human coronary atherosclerotic plaque. Circulation. 1999;99:1284–1289. doi: 10.1161/01.cir.99.10.1284. [DOI] [PubMed] [Google Scholar]
- 100.Watson KE, Parhami F, Shin V, Demer LL. Fibronectin and collagen I matrixes promote calcification of vascular cells in vitro, whereas collagen IV matrix is inhibitory. Arterioscler. Thromb. Vasc. Biol. 1998;18:1964–1971. doi: 10.1161/01.atv.18.12.1964. [DOI] [PubMed] [Google Scholar]
- 101.Weitkamp B, Cullen P, Plenz G, et al. Human macrophages synthesize type VIII collagen in vitro and in the atherosclerotic plaque. Faseb J. 1999;13:1445–1457. doi: 10.1096/fasebj.13.11.1445. [DOI] [PubMed] [Google Scholar]
- 102.Werb Z. ECM and cell surface proteolysis: regulating cellular ecology. Cell. 1997;91:439–442. doi: 10.1016/s0092-8674(00)80429-8. [DOI] [PubMed] [Google Scholar]
- 103.Wesley RB, Meng X, Godin D, Galis ZS. Extracellular matrix modulates macrophage functions characteristic to atheroma: collagen type I enhances acquisition of resident macrophage traits by human peripheral blood monocytes in vitro. Arterioscler. Thromb. Vasc. Biol. 1998;18:432–440. doi: 10.1161/01.atv.18.3.432. [DOI] [PubMed] [Google Scholar]
- 104.Wight TN. The vascular extracellular matrix. In: FUster V, Ross R, TOpol EJ, editors. Atherosclerosis and Coronary Artery Disease. Philadelphia: Lippincott-Raven Publishers; 1996. pp. 421–440. [Google Scholar]
- 105.Wight TN, Kinsella MG, Qwarnström EE. The role of proteoglycans in cell adhesion, migration and proliferation. Curr. Opin. Cell Biol. 1992;4:793–801. doi: 10.1016/0955-0674(92)90102-i. [DOI] [PubMed] [Google Scholar]
- 106.Wight TN, Lara S, Riessen R, et al. Selective deposits of versican in the extracellular matrix of restenotic lesions from human peripheral arteries. Am. J. Pathol. 1997;151:963–973. [PMC free article] [PubMed] [Google Scholar]
- 107.Wight TN, Raugi GJ, Mumby SM, Bornstein P. Light microscopic immunolocation of thrombospondin in human tissues. J. Histochem. Cytochem. 1985;33:295–302. doi: 10.1177/33.4.3884704. [DOI] [PubMed] [Google Scholar]
- 108.Yurchenco PD, Schittny JC. Molecular architecture of basement membranes. Faseb J. 1990;4:1577–1590. doi: 10.1096/fasebj.4.6.2180767. [DOI] [PubMed] [Google Scholar]
- 109.Yurochko AD, Liu DY, Eierman D, Haskill S. Integrins as a primary signal transduction molecule regulating monocyte immediate-early gene induction. Proc. Natl. Acad. Sci. USA. 1992;89:9034–9038. doi: 10.1073/pnas.89.19.9034. [DOI] [PMC free article] [PubMed] [Google Scholar]