

Abstract
In renovascular hypertensive rats, low doses of angiotensin converting enzyme (ACE) inhibitors have been found to prevent myocardial hypertrophy independent of blood pressure level. This finding would suggest humoral rather than mechanical control of myocyte growth. The aim of this study was to examine the effect of nonantihypertensive doses of ACE inhibitor on myocardial hypertrophy and necrosis in hypertensive rats. Renovascular hypertension (RHT) was induced in four-week-old Wistar rats. Twenty-eight animals were treated for four weeks with three doses of ramipril (0.01, 0.1 or 1.0 mg/kg/day, which are unable to lower blood pressure. Fourteen animals were not treated (RHT group). A sham operated, age/sex-matched group was used as control (n = 10). Myocardial histology was analysed in 3 μm thick sections of the ventricle stained with either haematoxylin-eosin, reticulin silver stain or Masson's trichrome. There was a significant correlation between systolic blood pressure and left ventricular to body weight ratio in both sets of animals: untreated plus controls and ramipril-treated rats. ACE inhibition prevented myocyte and perivascular necrosis and fibrosis in a dose-dependent manner. We conclude that myocardial hypertrophy in rats with renovascular hypertension is directly related to arterial pressure, and that this relationship is not affected by nonantihypertensive doses of ACE inhibitor. Myocardial necrosis/fibrosis and coronary artery damage induced by angiotensin II are prevented by ACE inhibitor in a dose-dependent manner, despite the presence of arterial hypertension.
Keywords: pressure overload, coronary artery damage, myocardial fibrosis, ramipril
Myocardial hypertrophy in arterial hypertension is commonly thought to be the consequence of an increased pressure load to the heart. However, recent observations using antihypertensive therapy suggest that prevention and regression of cardiac hypertrophy is disassociated from the blood pressure reduction (Linz et al. 1989; Schölkens et al. 1991; Baker et al. 1994; Chevalier et al. 1994). For example, treatment with an angiotensin-converting enzyme (ACE) inhibitor prevented the increase in left ventricular mass in rats, independent of its effect on afterload (Baker et al. 1994; Chevalier et al. 1994). Even a low dose of ACE inhibitor, which was unable to normalize blood pressure in rats with renovascular hypertension, resulted in regression of cardiac mass(Linz et al. 1989; Schölkens et al. 1991). Moreover, rats receiving a chronic infusion of angiotensin II (AII) were found to have an increased left ventricular mass, even though the pressor activity of AII was blocked or a nonantihypertensive dose of AII was used (Dostal & Baker 1992; Susic et al. 1996). Altogether, these results suggested a direct trophic or growth effect of AII on the myocyte.
Contrariwise, Battle et al. (1993) detected a significant correlation between the heart weight and the arterial blood pressure in rats with unilateral renal ischaemia, independent of ACE inhibitor treatment. In addition, Rhaleb et al. (1994) observed that only an antihypertensive dose of ramipril could abolish left ventricular hypertrophy in rats with aortic coarctation. These data suggested that in these experimental conditions the mechanical load to the heart plays a major role in myocyte growth.
In addition to this controversy, several studies over the last decade have supported the hypothesis that myocardial dysfunction accompanying myocardial hypertrophy is due to an abnormal collagen accumulation rather than an increased myocardial mass (Narayan et al. 1989; Brilla et al. 1991; Cicogna et al. 1994). However, it is not clear whether this fibrosis is the result of a reactive or reparative process.
Several investigators have shown that both cardiomyocyte necrosis and coronary vascular injury are associated with increased serum levels of AII(Tan et al. 1991; Rodrigues et al. 1992; Campbell et al. 1995; Kabour et al. 1995). Campbell et al. (1995) suggested that, while the necrotic response occurred within the first two weeks of unilateral renal ischaemia in rats, there was a progressive increase in perivascular and interstitial collagen over an eight-week follow-up period. This is in disagreement with our previous findings of sustained injury to the myocardium throughout a similar period, indicating that the AII-induced myocardial fibrosis is closely related to myocardial necrosis (Okoshi et al. 1997).
Therefore, the purpose of this study was to further investigate the influence of AII on the heart in order to address these ongoing controversies. Accordingly, the specific objectives of this study were: (i) to determine the association between myocardial hypertrophy and pressure overload in the experimental model of renovascular hypertension, (ii) to test whether this association is influenced by ACE inhibitor treatment, and (iii) to analyse how nonantihypertensive doses of ACE inhibitor affect the remodeling process in the myocardium.
Methods
Animals and groups
Forty-two, four-week-old, male Wistar rats weighing 120–140 g were used in the study. Their care and use conformed with all NIH guidelines and the protocol was approved by the University's Animal Use Committee. The animals were anaesthetized with thiopental sodium and a 1.5-cm incision was made posteriorly, below and parallel to the inferior edge of the rib cage. The left kidney was exposed and the renal artery was isolated and carefully separated from the vein. A solid silver clip with a 0.25-mm opening was placed on the artery to induce unilateral renal ischaemia and renovascular hypertension (RHT). Ten sex-and age-matched rats were sham-operated and used as controls. All animals were housed in a temperature-controlled room (24°C) with a 12-h light/dark cycle. Food and water were supplied ad libitum.
The renovascular hypertensive rats were randomly divided into four groups as follows: RHT (untreated, n = 14), RAM0.01 (n = 7), RAM0.1 (n = 9) and RAM1 (n = 12), treated with either ramipril 0.01, 0.1 or 1 mg/kg/day respectively, given orally by gavage one day after the surgery and throughout the study period of 4 weeks. In our laboratory, pilot experiments have shown that these doses did not prevent hypertension. The lowest dose that would fully prevent arterial hypertension in rats with unilateral renal ischaemia was found to be 10 mg/kg/day. The rats that were either untreated or treated with nonantihypertensive doses of ramipril, systolic blood pressure became significantly greater than normal rats after approximately one week of unilateral ischaemia.
At the end of the four-week period, the rats were anaesthetized and the carotid blood pressure was recorded. Typically, this was performed 3–6 h after the last dose of ramipril was given. After thoracotomy, the heart was excised and the left (including the septum) and right ventricles were separated and weighed.
Morphological study
Coronal sections of the left and right ventricle were fixed in 10% buffered formalin. Serial 3-μm paraffin-embedded cross-sections were taken for histological assessment and stained with either haematoxylin-eosin to distinguish areas of necrosis and cellular infiltrate, silver for reticulin fibers, or Masson's trichrome to assess the degree of interstitial remodeling. Masson's trichrome highlights the connective tissue, which in the heart consists mostly of collagen.
Two pathologists, who were blinded as to the source of the tissue sections, independently examined the slides and rated the necrosis and fibrosis, if present, as mild, moderate or severe depending on the following features: mild — small foci of necrosis or fibrosis affecting few myocytes; moderate — one or more foci of necrosis/fibrosis affecting an area smaller than 0.05 mm2; severe — one or more foci of necrosis/fibrosis affecting an area greater than 0.05 mm2. The areas of necrosis or fibrosis were measured using a binocular microscope attached to a video camera. This system was connected to a personal computer equipped with image analysis software (OPTIMAS 4.1, Image Corporation, USA). Vascular damage was also defined as mild, moderate or severe depending on the following criteria: mild — perivascular oedema and small inflammatory cellular infiltration without necrosis; moderate — perivascular oedema and inflammatory cell infiltration with necrosis of the myocytes adjacent to the vessels or increased perivascular fibrosis; severe — perivascular necrosis/fibrosis with necrosis of arterial smooth muscle cells and surrounding myocytes. In general, the pathologists' assessments were similar. When there was marked disagreement, the slides in question were jointly re-analysed in order to arrive at a consensus.
Cellular cross-sectional area (CSA,μm2) of at least 50 myocytes randomly chosen per heart was measured. The sections stained with silver were projected at a magnification of x400 using the same system described above for measuring myocardial necrosis or fibrosis areas.
Statistical analysis
Pressure, body weight, ventricular weights and the ratio of left ventricular weight to body weight were expressed as mean ± SD and compared by one way analysis of variance and post hoc Tukey test. The cross-sectional area data were analysed by nonparametric Kruskall-Wallis test. Systolic blood pressure and left ventricular/body weight ratio values were fitted to linear regression for all groups treated with ramipril and for RHT plus control groups. The linear regression lines were compared by analysis of variance for comparison of regression model for several samples (Ostle & Mensing 1975). The analyses of the presence of myocardial or perivascular damage in all groups were done by using the simultaneous confidence intervals for contrasts among binomial populations (Goodman 1964). The microsections were separated into two categories: none/mild damage and moderate/severe damage. Statistical significance was taken to be P < 0.05.
Results
Systolic blood pressure recorded before sacrifice, heart and body weight data are summarised in Table 1. Systolic blood pressure was significantly increased in the untreated RHT group (187 ± 26 mmHg) and in ramipril treated groups (RAM0.01, 191 ± 25 mmHg; RAM0.1, 167 ± 24 mmHg; RAM1, 165 ± 18 mmHg) compared to control group (134 ± 13 mmHg, P < 0.05). Body weight and right ventricular weight did not differ among the five groups. The ratios of left ventricular to body weight in RHT and the three RAM groups were comparable to each other and significantly greater than those of controls, showing myocardial hypertrophy. These results indicated that left ventricular hypertrophy was not prevented by nonantihypertensive doses of ACE inhibitor. There was a significant correlation between systolic blood pressure and left ventricular to body weight ratio for the two sets of data (Figure 1). The linear regression slopes were similar in hypertensive ramipril-treated and hypertensive untreated plus control rats (0.009 and 0.010 mg/g/mmHg, respectively, P > 0.05). These results suggested that left ventricular hypertrophy was directly related to arterial pressure with no influence of ACE inhibition. For each mmHg rise in systolic blood pressure, the increase in normalized LV weight was the same for controls plus untreated renovascular hypertensive rats and for all ACE inhibitor-treated rats. The myocyte cross-sectional area data (Figure 2) further confirmed the hypertrophy in ramipril treated rats.
Table 1.
Group values for systolic blood pressure, recorded before sacrifice, body and ventricular weights
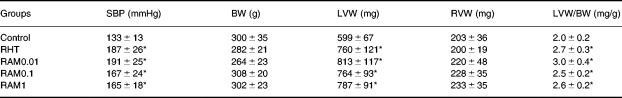
RHT, rats with renovascular hypertension; RAM0.01, RAM0.1 and RAM, RHT treated with either 0.01, 0.1, or 1 mg/kg/day of ramipril, respectively; SBP, systolic blood pressure; BW, body weight; LVW and RVW, left and right ventricular weight, respectively.
*P < 0.05 vs. Control (anova).
Figure 1.
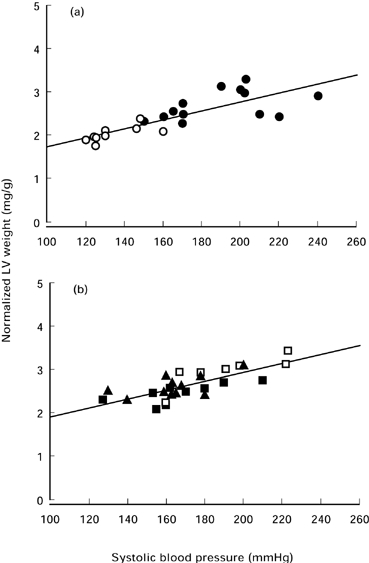
The relationship between normalized left ventricular weight to body weight and systolic blood pressure. (a) ○ Controls; • Untreated renovascular hypertensive rats. y = 0.72 + 0.10 x; r = 0.82; P < 0.001 (b) Three groups of hypertensive rats treated with nonantihypertensive doses of ramipril. □ RAM0.01; ▪ RAM0.1; ▴ RAM1. y = 1.05 + 0.009 x; r = 0.71; P < 0.001 The slopes of both linear regressions were statistically similar (anova for comparison of regression model for several samples).
Figure 2.
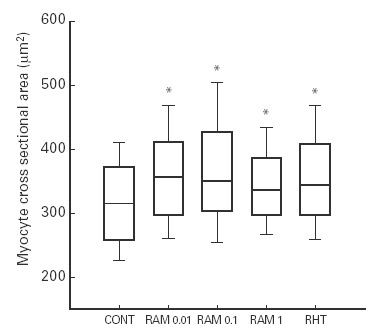
Myocyte cross-sectional area in controls (CONT) and in renovascular hypertensive rats with no treatment (RHT) or treated with nonantihypertensive doses of ramipril (RAM0.01, RAM0.1 and RAM1). The lower boundary of the boxes indicates the 25th percentile, the line within the boxes marks the median, and the upper boundary of the boxes indicates the 75th percentile. Error bars above and below the boxes indicate the 90th and 10th percentiles. *P < 0.05 vs. Control.
Histologically the RHT and RAM0.01 groups were undistinguishable from each other (Table 2 and Figure 3 a–d). The myocardial and vascular damages were moderate/severe in both groups. There were several foci of myocyte necrosis either involving only a group of few cells or an area greater than 0.05 mm2. Some hearts presented changes corresponding to subendocardial myocardium ischaemia. Many coronary arteries presented hyalinization and fibrinoid necrosis of smooth muscle cells in the medial layer. There was an adventitial lymphomononuclear inflammatory exudate and oedema, involving the adjacent myocytes. The silver staining demonstrated collapse of the reticular framework at the sites of myocyte loss. There was increased perivascular and interstitial fibrosis and scarring.
Table 2.
Prevalence (%) of moderate/severe myocyte and perivascular damage in all groups

Groups are the same as in Table 1.
*P < 0.05 vs. Control (Goodman test).
Figure 3.
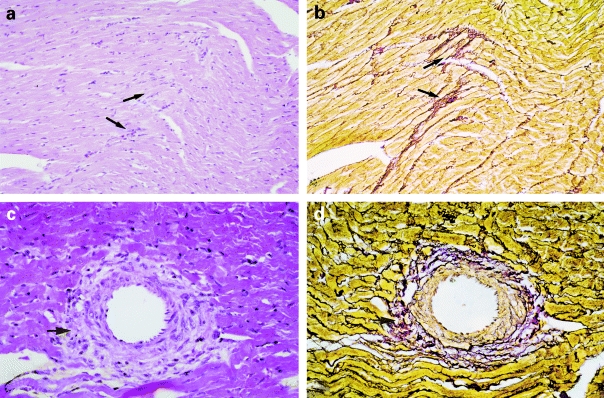
Photomicrographs of the myocardium. In panels a, b, c and d tissue from renovascular hypertensive (RHT) rat illustrates myocyte necrosis and increased interstitial collagen (a,b magnification ×50) and perivascular inflammatory infiltrate and necrosis (c,d magnification ×80). There was a close association between interstitial/perivascular inflammatory process seen in H & E (a,c arrows) and the stromal collapse as demonstrated by silver staining (b,d arrows).
The RAM0.1 group presented only mild damage of the myocardium with very small areas of necrosis (Table 2 and Figure 4a). However, the vascular damage was mainly moderate with perivascular oedema and inflammatory exudate involving adjacent myocytes.
Figure 4.
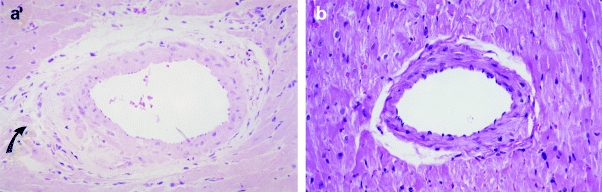
Photomicrographs of the myocardium (×80). In panels a and b, tissue from hypertrophied myocardium of RHT treated with either 0.1 or 1 mg/kg of ramipril, respectively, illustrate a moderate perivascular inflammatory infiltrate (arrow) and absence of damage. H & E staining.
The treatment with the dose of 1.0 mg/kg/day of ramipril (RAM1 group) completely prevented myocardial and vascular damage (Table 2 and Figure 4b). The histology of this group was statistically similar to the control hearts (Table 2).
Discussion
Treatment with three different nonantihypertensive doses of ACE inhibitor allowed the investigation of the hypertrophic response over a wide range of arterial pressures. The finding that LV weight correlates with systolic blood pressure, with and without ACE inhibition, indicates that myocardial hypertrophy in young rats with renovascular hypertension is strongly affected by load conditions. Myocardial hypertrophy is known to be mild or absent in arterial hypertension induced by chronic inhibition of nitric oxide synthase in rats (Matsubara et al. 1998) and therefore is thought to be associated with the absence of an abnormal elevation of AII in this model (Arnal et al. 1993). This would explain the myocardial hypertrophy in rats treated with nonantihypertensive doses of ramipril. With unilateral renal ischaemia, it is reasonable to assume that elevations in arterial pressure are directly related to increased levels of circulating AII. Thus, the low doses of ACE inhibitor did not fully prevent the AII increase and the resulting hypertension, hypertrophy and myocardial damage.
Our results differ from previous observations in adults rats. Schölkens et al. (1991) were able to prevent myocardial hypertrophy in renovascular hypertensive rats with a nonantihypertensive dose of ramipril (0.01 mg/kg/day). While Rhaleb et al. (1994) using the same dose of ramipril were not able to prevent myocardial hypertrophy in adult hypertensive rats with aortic coarctation, both hypertension and hypertrophy were prevented with a dose of 1 mg/kg/day. It is important to note that our rats underwent unilateral renal ischaemia at four weeks of age. It is possible that immature rats may have a more intensive hypertrophic response to a given stimulus than do adult rats. For exemple, a significant correlation between right ventricular weight and pulmonary arterial pressure was obtained only in young rats exposed to intermittent high-altitude hypoxaemia (Kolar et al. 1989). Also, ACE inhibitor appears to exert its influence to a greater degree during periods of rapid growth. When the effects of enalapril on normotensive rats, treated from either 4 or 10 weeks of age onwards, were evaluated, a more pronounced effect on the cardiovascular connective tissue of the younger animals was noted (Keeley et al. 1992).
Tan et al. (1991) also reported myocardial necrosis in hypertensive rats which was prevented by ACE inhibition. They also analysed hypertensive rats with no renal ischaemia and did not find myocardial damage. They concluded that myocyte injury was caused by angiotensin II and was unrelated to hypertension. Others (Rodrigues et al. 1992) using rats with renovascular hypertension found widespread necrotizing changes of the intramural coronary arteries surrounded by areas of myocyte necrosis. Other studies performed with a chronic infusion of AII in rats demonstrated that the cytotoxic effects of AII are receptor mediated (Kabour et al. 1995) and related to the local release of neural catecholamines within the heart (Henegar et al. 1995).
Interestingly, several investigators (Kabour et al. 1994; Campbell et al. 1995) have found that the myocardial damage induced by chronic infusion of AII is primarily an acute process occurring in the first 2–3 days of elevated plasma AII levels. These results suggest a cardioprotective receptor downregulation (Kabour et al. 1994). However, our findings in rats with unilateral renal ischaemia for 4–8 weeks suggest a progressive injury to the myocardium (Okoshi et al. 1997).
The perivascular and interstitial inflammatory process observed in the hypertensive rats with and without the lowest dose of ramipril was partially prevented with a 10-fold greater dose and fully prevented with the highest dose. These results as well as those by Kabour et al. (1995) indicate a dose-dependent protective effect of ACE inhibition. Finally, since none of the doses in this study prevented arterial hypertension, the vascular injury induced by AII is not due to hypertension.
The close association between the interstitial/perivascular inflammatory process and stromal collapse as demonstrated by silver staining indicates that myocardial fibrosis related to renovascular hypertension is primarily a reparative process secondary to myocyte necrosis. The full prevention of myocyte necrosis in the RAM0.1 group, which had moderate vascular damage, indicates that the myocyte injury is produced by a direct effect of AII and/or AII-released catecholamines and is not directly related to myocardial ischaemia as suggested by Rodrigues et al. (1992).
In summary, this study demonstrates that renovascular hypertensive rats develop myocardial hypertrophy that is directly related to systolic blood pressure. The linear regression between blood pressure and ventricular hypertrophy is not influenced by nonantihypertensive doses of ACE inhibitor. On the other hand, perivascular and myocyte necrosis/fibrosis are preventable by such doses of ACE inhibitor.
Acknowledgments
We gratefully acknowledge the technical assistance of Vitor M. Souza and José Carlos Georgete.
This work was supported by Conselho Nacional de Pesquisa, Grant CNPq 301028/95–8.
References
- 1.Arnal JFEL, Amranl AI, Chatellier G, Menard J, Michel JB. Cardiac weight in hypertension induced by nitric oxide synthase blockade. Hypertension. 1993;22:380–387. doi: 10.1161/01.hyp.22.3.380. [DOI] [PubMed] [Google Scholar]
- 2.Baker KM, Chernin MI, Wixson SK, Aceto JF. Renin-angiotensin system involvement in pressure-overload cardiac hypertrophy in rats. Amer. J. Physiol. 1994;259:H324–H332. doi: 10.1152/ajpheart.1990.259.2.H324. [DOI] [PubMed] [Google Scholar]
- 3.Battle T, Schnell C, Bunkenburg B, Heudes D, Wood JM, Ménard J. Continuous versus intermittent angiotensin converting enzyme inhibition in renal hypertensive rats. Hypertension. 1993;22:188–196. doi: 10.1161/01.hyp.22.2.188. [DOI] [PubMed] [Google Scholar]
- 4.Brilla CG, Janicki JS, Weber KT. Impaired diastolic function and coronary reserve in genetic hypertension. Role of interstitial fibrosis and medial thickening of intramyocardial coronary arteries. Circ. Res. 1991;69:107–115. doi: 10.1161/01.res.69.1.107. [DOI] [PubMed] [Google Scholar]
- 5.Campbell SE, Janicki JS, Weber KT. Temporal differences in fibroblast proliferation and phenotype expression in response to chronic administration of angiotensin II or aldosterone. Journal of Mol. Cell. Cardiol. 1995;27:1545–1560. doi: 10.1016/s0022-2828(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 6.Chevalier B, Callenselamrani F, Heymes C, Swynghedauw B. Molecular basis of regression of cardiac hypertrophy. Amer. J. Cardiol. 1994;73:C10–C17. doi: 10.1016/0002-9149(94)90618-1. [DOI] [PubMed] [Google Scholar]
- 7.Cicogna AC, Matsubara BB, Okoshi MP, Matsubara LS, Janicki JS. Hypertrophy versus collagen concentration as a factor of increased myocardial passive stiffness (abstract) FASEB Jounal. 1994;8:A794. [Google Scholar]
- 8.Dostal DE, Baker KM. Angiotensin II stimulation of left ventricular hypertrophy in adult rat heart. Am. J. Hypertension. 1992;5:276–280. doi: 10.1093/ajh/5.5.276. [DOI] [PubMed] [Google Scholar]
- 9.Goodman LA. Simultaneous confidence intervals for contrasts among multinomial populations. Annals of Mathematical Statistics. 1964;35:716–725. [Google Scholar]
- 10.Henegar JR, Brower GL, Kabour A, Janicki JS. Cathecholamines response to chronic ANG II infusion and its role in myocyte and coronary vascular damage. Am. J. Physiol. 1995;269:H1564–H1569. doi: 10.1152/ajpheart.1995.269.5.H1564. [DOI] [PubMed] [Google Scholar]
- 11.Kabour A, Henegar JR, Devineni VR, Janicki JS. Prevention of angiotensin II induced myocyte necrosis and coronary vascular damage by lisinopril and losartan in the rat. Cardiovasc. Res. 1995;29:543–548. [PubMed] [Google Scholar]
- 12.Kabour A, Henegar JR, Janicki JS. Angiotensin II (AII)-induced myocyte necrosis: role of the AII receptor. J. Cardiovasc. Pharmacol. 1994;23:547–553. doi: 10.1097/00005344-199404000-00005. [DOI] [PubMed] [Google Scholar]
- 13.Keeley FW, Elmoselli A, Leenem FHH. Enalapril suppresses normal accumulation of elastin and collagen in cardiovascular tissues of growing rats. Amer. J. Cardiol. 1992;262:H1013–H1021. doi: 10.1152/ajpheart.1992.262.4.H1013. [DOI] [PubMed] [Google Scholar]
- 14.Kolar F, Ostadal B, Prochazka J, Pelouch V, Widimsky J. Comparison of cardiopulmonary response to intermittent high-altitude hypoxia in young and adult rats. Respiration. 1989;56:57–62. doi: 10.1159/000195778. [DOI] [PubMed] [Google Scholar]
- 15.Linz W, Schölkens BA, Ganten D. Converting enzyme inhibition specifically prevents the development and induces regression of cardiac hypertrophy in rats. Clin. Exp. Hypertension. 1989;11:1325–1350. doi: 10.3109/10641968909038172. [DOI] [PubMed] [Google Scholar]
- 16.Matsubara BB, Matsubara LS, Zornoff LAM, Franco M, Janicki JS. Left ventricular adaptation to chronic pressure overload induced by inhibition of nitric oxide synthase in rats. Bas. Res. Cardiol. 1998;93:173–181. doi: 10.1007/s003950050084. [DOI] [PubMed] [Google Scholar]
- 17.Narayan S, Janicki JS, Shroff SG, Pick R, Weber KT. Myocardial collagen and mechanics after preventing hypertrophy in hypertensive rats. Am. J. Hypertension. 1989;2:675–682. doi: 10.1093/ajh/2.9.675. [DOI] [PubMed] [Google Scholar]
- 18.Okoshi MP, Matsubara LS, Franco M, Cicogna AC, Matsubara BB. Myocyte necrosis is the basis for fibrosis in renovascular hypertensive rats. Braz. J. Med. Biol. Res. 1997;30:1135–1144. doi: 10.1590/s0100-879x1997000900013. [DOI] [PubMed] [Google Scholar]
- 19.Ostle P, Mensing RW. [3rd edn] Iowa City: Iowa State University Press; 1975. Statistics in Research. [Google Scholar]
- 20.Rhaleb N-E, Yang X-P, Scili AG, Carretero OA. Role of kinins and nitric oxide in the antihypertensive effect of ramipril. Hypertension. 1994;23(2):865–868. doi: 10.1161/01.hyp.23.6.865. [DOI] [PubMed] [Google Scholar]
- 21.Rodrigues MAM, Bregagnollo EA, Montenegro MR, Tucci PJF. Coronary vascular and myocardial lesions due to experimental constriction of the abdominal aorta. Int. J. Cardiol. 1992;35:253–257. doi: 10.1016/0167-5273(92)90184-5. [DOI] [PubMed] [Google Scholar]
- 22.Schölkens BA, Linz W, Martorana PA. Experimental cardiovascular benefits of angiotensin-converting enzyme inhibitors: beyond blood pressure reduction. J. Cardiovasc. Pharmacol. 1991;18(Suppl. 2):526–530. [PubMed] [Google Scholar]
- 23.Susic D, Nuñez E, Frohlich ED, Prakash O. Angiotensin II increases left ventricular mass without affecting myosin isoform mRNAs. Hypertension. 1996;28:265–268. doi: 10.1161/01.hyp.28.2.265. [DOI] [PubMed] [Google Scholar]
- 24.Tan LB, Jalil JE, Pick R, Janicki JS, Weber KT. Cardiac myocyte necrosis induced by angiotensin II. Circ. Res. 1991;69:1185–1195. doi: 10.1161/01.res.69.5.1185. [DOI] [PubMed] [Google Scholar]