

Abstract
Cyanohydroxybutene (CHB), a glycosinolate breakdown product, causes pancreatic injury when given to animals in large amounts. To determine the course of CHB-induced pancreatopathy, rats were given a single subcutaneous dose of CHB and the pancreas weighed and examined by light and electron microscopy and immunohistochemistry at intervals from 2 h to 28 days. The pancreatic lesion was unusual in that there was marked early oedema with limited inflammatory cell infiltration, rapid synchronous onset of acinar cell apoptosis and early advanced atrophy engendering only a limited regenerative response. Acinar cell apoptosis was atypical in that cell fragmentation was limited and phagocytosis delayed, resulting in extensive secondary necrosis. As ducts were unaffected by CHB, the crowded ducts making up the epithelial component of atrophic lobules could be clearly shown to derive from their condensation and proliferation, not the redifferentiation of pre-existing acinar cells, widely held to produce this lesion. Although the basis of CHB selectivity and toxicity for pancreatic acinar cells remains unknown, the potential therapeutic benefit of such an agent in patients with pancreatitis or pancreatic tumours warrants further investigation.
Keywords: pancreas, atrophy, apoptosis, regeneration, cyanohydroxybutene, tubular complexes
It has been known for some decades that seeds and plants containing glucosinolates and their breakdown products are toxic to stock animals in large quantities (Bell & Williams 1953; Nordfeldt et al. 1954). One of the breakdown products, cyanohydroxybutene (CHB), also known as crambene (Staack et al. 1998), has a LD50 of 200 mg/kg when administered subcutaneously in rats, causing liver necrosis and thyroid hyperplasia (Nishie & Daxenbichler 1980). CHB derived from Crambe abyssinica(the S-enantiomer), whether given orally or intravenously, is pancreatotoxic in rodents, inducing apoptosis of pancreatic acinar cells (Wallig et al. 1988,1992; Wallig & Jeffery 1990; Bhatia et al. 1998). When given as a single intravenous dose of 70 mg/kg to mice, apoptotic acinar cells reach 30–40 per high power field (HPF, magnification ×250) 12–24 h after injection but, at 48 h, substantial viable acinar tissue remains. Oral doses induce variable, incomplete pancreatic atrophy with regeneration at 96 h, but no long-term studies have been performed (Wallig et al. 1988; Wallig & Jeffery 1990). Synthetic racemic CHB is also pancreatotoxic inducing oedema and acinar cell vacuolation and depletion of zymogen granules in rats within hours of administration (Maher et al. 1991).
Atrophic pancreatic lobules comprising crowded ductlike structures within a collagenous stroma occur as interim or endstage lesions in human acute and chronic pancreatitis, cystic fibrosis and animal models of pancreatitis and pancreatic adenocarcinoma (reviewed in Iovanna 1996 and Bockman 1997). The ductlike structures are frequently referred to as ‘tubular complexes’ defined as ‘cylindrical tubes, sometimes connected, with a wide empty lumen lined by a monolayer epithelium of flattened duct-like cells in which mitoses are frequently seen’ (Iovanna 1996). These lesions are generally believed to develop through acinar lumen dilation, decrease in acinar cell height and subsequent loss of acinar cell secretory granules and endoplasmic reticulum to form cells morphologically resembling duct cells; such acinar cell ‘dedifferentiation’ or ‘redifferentiation’ is supported by immunohistochemical and in vitro studies (reviewed in Iovanna 1996; Bockman 1997). Others, however, studying some of these models, believe the lesions result from apoptotic deletion of acinar cells, condensation of ducts and proliferation of duct cells (Pour 1988; Walker et al. 1992; Wada et al. 1997). The resolution of this controversy, analogous to that involving similar processes in salivary glands (Takahashi et al. 1998; Walker & Gobe 1987), is of fundamental importance in determining the cellular mechanisms involved in pancreatitis, pancreatic atrophy and regeneration.
We were interested in the effects of CHB on the pancreas for two reasons. First, CHB or a similarly acting compound might be of benefit in the management of pancreatic cancer. Second, by deleting acinar cells, CHB might further our studies of the cellular origins of exocrine glandular regeneration. When our preliminary work demonstrated a single subcutaneous injection of synthetic CHB (150 mg/kg) in rats induced severe pancreatic atrophy within 4 days, we examined the time-course morphological changes after such a dose. It was found the CHB induced rapid and massive acinar cell apoptosis that progressed to ‘secondary necrosis’, a rare event in vivo. This outcome provided clear insights into the origin of ductlike structures in atrophic pancreatic lobules. An unexpected finding was the failure of acinar cell regeneration in this model.
Materials and methods
Synthetic CHB made according to the method of Das & Torssell (1983) was obtained from Research Directions, Brisbane, Australia.
Male Wistar rats weighing 200–250 g were caged in pairs and given food and water ad libitum with a 12-h light-dark cycle. Twelve groups of 10 rats were divided randomly into 6 test animals and 4 control animals. At time 0, test animals were given 150 mg/kg of CHB mixed in 0.5 ml sterile normal saline and controls were given 0.5 ml sterile normal saline subcutaneously.
For light microscopy, 4 experimental and 4 control animals were killed at 2, 4, 6, 12, 24, 48, 72, 96 h and 7, 10, 18, and 28 days using 60 mg intraperitoneal pentobarbitone. Animals were weighed and the pancreas removed, weighed and processed using routine methods. Additional pairs of experimental animals were killed at 18 and 60 h for electron microscopy and morphological study. Weights were recorded as mean ± standard error of the mean (SEM). Differences between means were analysed using Student's t-test.
For quantification of apoptosis, apoptotic cells and bodies, identified using the morphological criteria of Kerr et al. (1995), were counted in 10 high-power fields (× 400), selected at random, in a histological slide from each animal at 2, 4, 6 and 12 h with the proviso that mostly acinar tissue filled the field. A group of tightly clustered apoptotic bodies, presumably derived from a single cell, was recorded as a single count. An estimate of the total number of acinar cells per HPF in each slide was made for calculation of an apoptotic index (apoptotic count as a percentage of total acinar cells present). Counts/HPF and apoptotic indices were recorded as means ± SEM for each group. Differences between means were analysed using Student's t-test. Terminal d-UTP nick-end labelling (TUNEL) was not used because it is our experience and the experience of others that it is not always specific for apoptosis, and ultimately, apoptosis must be confirmed morphologically (Ansari et al. 1993; Grasl-Kraupp et al. 1995).
Immunohistochemistry for cytokeratin and amylase was performed to identify cells in sections as duct (Schüssler et al. 1992; Bouwens et al. 1995) or acinar (Bendayan 1984), respectively. For cytokeratin, deparaffinized sections were pretreated with 0.1% trypsin, then 0.3% hydrogen peroxide in methanol followed by mouse monoclonal AE1/AE3 anticytokeratin (DAKO Corp., Carpinteria, California, USA) which is a pan-cytokeratin checked for specificity with known positive control tissue (normal rat pancreas) at a dilution of 1/40. Secondary antibody was rat antimouse biotinylated IgG (Jackson ImmunoResearch, West Grove, Pennsylvania, USA) used at a dilution of 1/400. Antibody-binding was demonstrated using the peroxidase-labelled streptavidin biotin complex method (DAKO strept ABComplex/HRP) and reactions were developed with 3,3′-diaminobenzidine tetrahydrochloride solution (Zymed, San Francisco, California). For amylase, deparaffinized sections were boiled in Target Retrieval Solution (DAKO) then placed in 0.3% hydrogen peroxide in methanol. Primary antibody was antirabbit immunoglobulin (DAKO) used at a dilution of 1/500 and secondary antibody was antirabbit goat biotinylated IgG (Jackson ImmunoResearch) used at a dilution of 1/400. Antigen-binding was demonstrated using the peroxidase-streptavidin method developed with Vector VIP peroxidase substrate (Vector Laboratories, Burlingame, California). All sections were lightly counterstained with haematoxylin.
For electron microscopy, two rats from each test group were deeply anaesthetized with intraperitoneal pentabarbitone sodium. A catheter was retrogradely inserted into the abdominal aorta and the vasculature flushed in sequence with heparinized normal saline then 1% paraformaldehyde and 1.2% glutaraldehyde in cacodylate buffer and finally 4% paraformaldehyde and 5% glutaraldehyde in cacodylate buffer (Karnovsky 1965). Pancreas was removed immediately, diced and immersed in the final perfusate for two hours, then stored in cacodylate buffer. The tissue was postfixed in 1% osmium tetroxide, stained en bloc in 5% aqueous uranyl acetate, dehydrated through a series of graded alcohols, cleared in propylene oxide, and embedded in an epon-araldite mixture. Semithin sections (1–8 μm) were cut on an LKB Ultratome V and stained with toluidine blue for viewing. Ultrathin sections from selected areas were picked up on uncoated copper grids, stained with lead citrate and examined with a JEOL-1200 EX11 electron microscope.
All animal experimentation followed guidelines prescribed by the National Health and Medical Research Council of Australia with University of Queensland Animal Experimental Ethics Committee Approval No. Path/501/94/QRIRF.
Results
General observations
Control rats showed normal weight gain reaching 180% at 28-days (Figure 1). Experimental rats showed a fall in body weight over the first week and thereafter body weight remained unchanged, indicating failure of normal growth (Figure 1). At 18 and 28 days CHB-treated rats had muscle wasting and abdominal distension.
Figure 1.
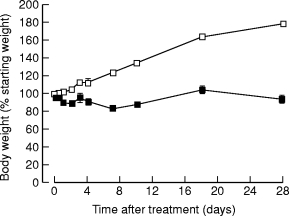
Body weights of animals as a percentage of starting weight after a single subcutaneous injection of saline (□) or CHB (▪). (n = 4, results expressed as means ± SEM.)
At autopsy, control rats had normal viscera and a pancreatic weight that was constant as a proportion of body weight (Figure 2). Experimental animals showed pancreatic oedema from 2 h, actual pancreatic weight reaching 4.41 ± 0.71 g at 6 h (compared with 0.78 ± 0.14 g in controls, P < 0.001), then falling. Atrophy of the pancreas was apparent at 7 days and persisted, actual pancreatic weight falling to 0.44 ± 0.04 g at this time compared with 1.80 ± 0.08 g in controls (P < 0.001). Changes in pancreatic weight as a percentage of body weight are shown in Figure 2. Abdominal distension at later stages was found to be due to dilated bowel containing poorly digested food.
Figure 2.
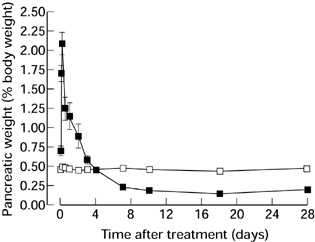
Pancreatic weight as a percentage of body weight in animals after a single subcutaneous injection of saline (□) or CHB (▪). (n = 4, results expressed as means ± SEM.)
Light microscopy
Control animals showed histologically normal pancreas (Jamieson 1988) (Figure 3A). From 2 h test animals showed mild dilation of acinar lumens and acinar cell vacuolation and depletion of zymogen granules.
Figure 3.
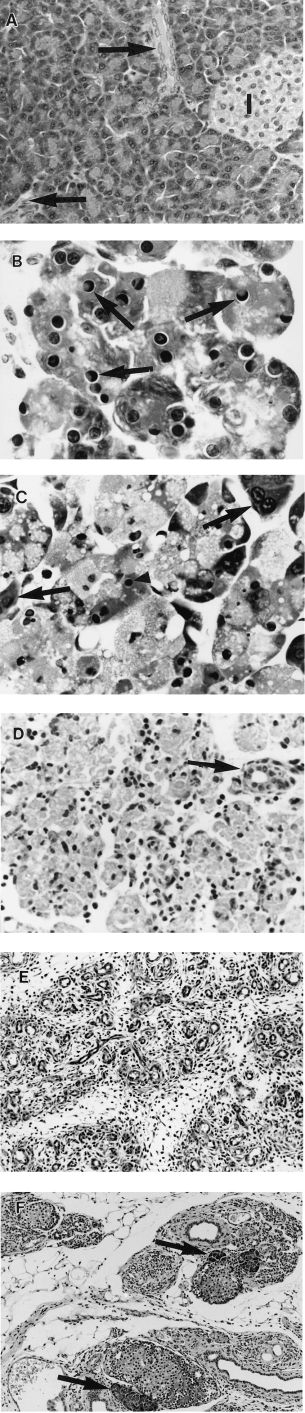
Pancreatic morphology after a single subcutaneous injection of saline or CHB. All H & E. (A) 48 h after saline. There is wide separation of ducts (arrows) and islets (I) by closely packed acinar cells (×360). (B) 12 h after CHB. Note numerous apoptotic acinar cells with characteristic nuclear morphology (arrows) (×1280). (C) 18 h after CHB. Most acinar cells contain pyknotic nuclear remnants (arrowheads) and show cytoplasmic swelling and vacuolation; a few appear normal (arrows) (×960). (D) 48 h after CHB. Advanced secondary necrosis affecting all acinar cells in field. Intact duct is indicated by arrow (×380). (E) 96 h after CHB. No acinar cells remain. Atrophic lobules comprise crowded ducts in a connective tissue stroma (×210). (F) 28 days after CHB. Sparse regenerative acini are seen adjacent to islets (arrows). Note few ducts in a collagenous stroma and prominent fatty infiltration (×210).
Apoptotic acinar cells, evident at 6 h, showed sharply defined crescents of clumped chromatin against the nuclear envelope but infrequent fragmentation. Their number reached 178 ± 10/HPF at 12 h (compared with 0.85 ± 0.13/HPF in controls, P < 0.001) or 23.6 ± 7.43% of acinar cells (compared with 0.001% in controls) (Figure 3B). By 18 h most acinar cells had chromatin changes of apoptosis but swollen vacuolated cytoplasm indicative of ‘secondary necrosis’ (Figure 3C) which subsequently progressed (Figure 3D). By 96 h no acinar cells remained (Figure 3E). A few regenerative acini appeared by 18 days, particularly adjacent to islets of Langerhans, but thereafter they did not increase appreciably in number (Figure 3F).
Intercalated ducts were mildly dilated at 4 h, duct cell mitoses were prominent at 48 h, and at 96 h, lobules comprised groups of ducts within a connective tissue stroma (Figure 3E). Small numbers of apoptotic bodies continued to be seen within duct lumens and epithelium. By 7 days ducts had larger lumens and flattened lining epithelial cells. Thereafter the number of ducts decreased with few remaining at 18 and 28 days (Figure 3F).
Interlobular oedema was present from 2 h and intralobular oedema from 4 h; both persisted for 72 h. The interstitial spaces were acellular before small numbers of mononuclear phagocytes appeared about vessels at 4 h and within acini at 24 h. They reached moderate numbers at 48 h, peaked at 72 h, then declined markedly by 7 days. Sparse neutrophils were present from 12 h and mitotic mononuclear phagocytes at 48 h.
Enlarged mitotically active fibroblasts were seen at 48 h. By 96 h fibroblasts and collagen enveloped lobules and at 7 days fibroblasts were less prominent and collagen was found both in and around lobules. At 28 days the pancreas comprised largely fat, collagen and islets (Figure 3F). Islets were not studied in detail.
Immunohistochemistry
In controls ducts were positive for cytokeratin and acinar cells negative (Figure 4A). At 48 h in test animals, when few viable acinar cells remained, cytokeratin marked dispersed intact ducts and duct cells (Figure 4B). At 96 h ducts of atrophic lobules, the only remaining epithelium, were positive for cytokeratin (Figure 4C). Amylase was demonstrated in apoptotic cells at 18 h, confirming their acinar cell origin. There were no or rare amylase-containing acinar cells at 72 and 96 h (Figure 4D). The periphery of islets also showed amylase staining at 96 h (Figure 4D).
Figure 4.
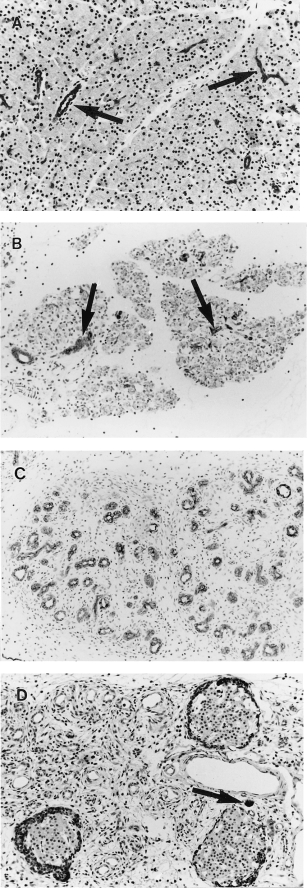
Pancreatic immunohistochemistry after a single subcutaneous injection of saline or CHB. All with haematoxylin counterstain. (A) 24 h after saline. Widely spaced keratin-positive ducts (arrows) are separated by closely packed keratin negative acinar cells (×170) (B) 48 h after CHB. Widely spaced keratin-positive ducts (arrows) separated by keratin-negative nonviable acinar cells (×190). (C) 96 h after CHB. Lobules comprise crowded keratin-positive ducts separated by loose connective tissue. No acinar cells are seen (×190). (D) 96 h after CHB. Ducts are negative for amylase. Note isolated amylase positive acinar cell (arrow) and periinsular amylase positivity (×170).
Electron microscopy
Controls showed normal pancreatic ultrastructure (Ekholm et al. 1962a, b). In test animals acinar cell apoptosis was slightly increased at 6 h and markedly increased at 12 h, when large numbers of adjacent cells were often affected (Figure 5A). Apoptotic cells showed sharply defined crescents of chromatin abutting the nuclear envelope, prominent nucleolar remnants, whorling of endoplasmic reticulum and structural preservation of organelles (Figure 5A) but cellular fragmentation to form apoptotic bodies was uncommon. At 18 h, apoptotic cells, identified by their nuclear characteristics, remained in situ, but showed dilatation of endoplasmic reticulum and nuclear envelopes, swelling and rupture of mitochondria and rupture of plasma membranes (Figure 5B), so-called ‘secondary necrosis’ (Wyllie et al. 1980). This process progressed such that, by 48 h, acinar cells were reduced to degraded cellular material associated with small numbers of intraepithelial macrophages containing degraded cellular material in phagosomes or residual bodies (Figure 5C). By 96 h, acinar cell debris had been removed (Figure 5D). Ducts and duct cells survived, showing increased mitotic activity, particularly at 60 and 72 h (Figure 5C,D). Small numbers of ductal intraepithelial apoptotic bodies and surrounding collapsed basement membrane were identified (Figure 5D).
Figure 5.
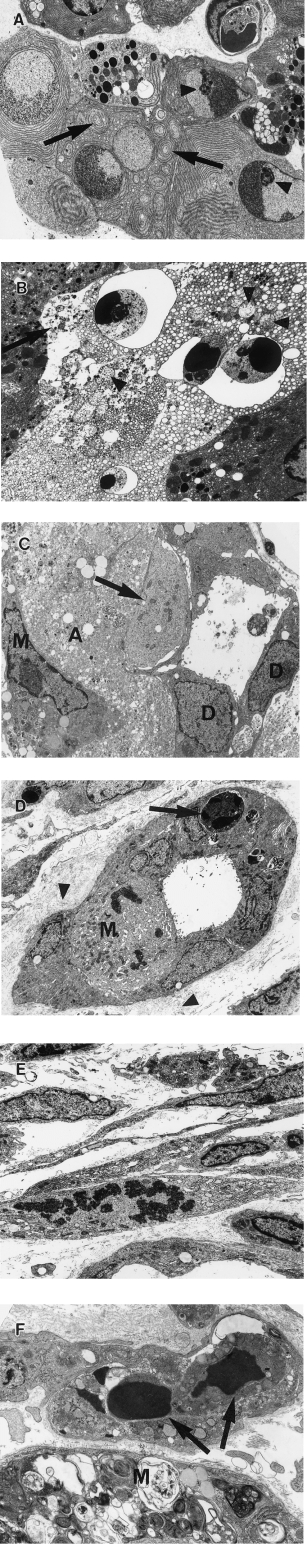
Pancreatic ultrastructure after a single subcutaneous injection of CHB. (A) 12 h after CHB. Adjacent apoptotic acinar cells show well-demarcated crescentic clumped chromatin, large nucleolar remnants (arrowhead) and whorling of endoplasmic reticulin (arrows) (×3200). (B) 18 h after CHB. Apoptotic acinar cells show nuclear fragments with crescentic clumped chromatin but dilation of endoplasmic reticulin, swollen mitochondria (arrowheads) and plasma membrane rupture (arrow). Contrast with adjacent viable acinar cells (×3200). (C) 48 h after CHB. Note viable duct epithelial cells (D), residual acinar cell cytoplasmic debris (A) and intra-acinar macrophage (M) laden with residual bodies. The pale cell in the duct epithelium (arrow) is also likely to represent an intraepithelial macrophage (×3700). (D) 96 h after CHB. Duct with typical indented nuclei and sparse organelles of lining cells. Note mitotic lining cell (M), intraepithelial apoptotic body (arrow), and adjacent collapsed redundant basement membrane (arrowhead) (×2650). (E) 96 h after CHB. Activated and mitotic interstitial fibroblasts (×3500). (F) 48 h after CHB. Capillary with intraluminal apoptotic bodies of presumed endothelial cell origin (arrows). Note adjacent intra-acinar macrophage (M) with residual body-laden cytoplasm (×7500).
From 48 to 96 h, prominent, activated and mitotic fibroblasts were seen (Figure 5E). At first collagen was sparse but increased in amount towards 7 days. At 48 h, mitoses in interstitial macrophages were confirmed and endothelial cell apoptosis was present in interstitial capillaries (Figure 5F); this continued over succeeding days. By 18 days, isolated regenerative acini comprised acinar cells closely resembling acinar cells in control glands.
Discussion
Pancreatic atrophy induced by a single dose of subcutaneous CHB resembles that seen by us after pancreatic duct ligation (Walker 1987; Walker et al. 1992) and the administration of ethionine and a protein-depleted diet (Walker et al. 1993). In all, there was marked acinar cell apoptosis preceding lobular regression to groups of crowded ducts within a condensed stroma. The relatively synchronous onset of apoptosis in the majority of acinar cells within 12 h of CHB administration, however, contrasts with the slow onset of apoptosis and gradual increase peaking about the third day that occurs in the other models (Walker 1987; Walker et al. 1993). Previously recorded peak rates of pancreatic acinar cell apoptosis are 6.6% 3 days after common bile duct ligation (Kaiser et al. 1995), 9.5% after 6 weeks administration of a copper-deficient diet (Rao et al. 1993), 13.4/HPF (×400) 3 days after administration of high-dose ethionine and a protein-depleted diet (Walker et al. 1993), and 30–40/HPF (×250) 12–24 h after CHB (Bhatia et al. 1998). Twelve hours after CHB administration in this study, 23.6% of acinar cells or 178/HPF(×400) showed changes of apoptosis. This high count reflects not only the relatively synchronous initiation of apoptosis in acinar cells but also accumulation of affected cells through failure of phagocytic removal; the latter usually causes disappearance of apoptotic cells within 1–2 h of formation (Barres et al. 1992; Coles et al. 1993). Synchronous onset of apoptosis is frequently seen in vitro (Harmon et al. 1991) but has only rarely been recorded in vivo(Ogasawara et al. 1993). Marked depletion of pancreatic glutathione within 1–2 h of CHB administration (Wallig & Jeffery 1990; Wallig et al. 1992) suggests CHB causes oxidative injury that initiates apoptosis (Clutton 1997) in acinar cells. As the effect of CHB appears to be dose-related (Wallig et al. 1992), a sufficiently high dose, as given here, might be expected to initiate apoptosis in most cells within a relatively short timeframe.
Rapid removal of apoptotic acinar cells after duct ligation and after administration of ethionine and a protein-depleted diet is facilitated by the insidious onset of apoptosis, allowing time for ingress and multiplication of mononuclear phagocytes (Walker 1987). In contrast, the rapid and synchronous onset of apoptosis after CHB administration overwhelms the capacity of viable epithelial cells and tissue macrophages to rapidly remove the apoptotic cells. As a consequence, most apoptotic cells remain in situ undergoing progressive swelling, rupture of organelle and plasma membranes and degradative change referred to as ‘secondary necrosis’ (Wyllie et al. 1980). This is typically seen where apoptotic bodies are formed in vitro or shed into lumina from epithelial surfaces where phagocytosis does not occur. A comparable in vivo lesion is seen in liver of susceptible mice after intraperitoneal administration of anti-Fas antibody (Ogasawara et al. 1993).
Another feature in this study was the limited fragmentation of apoptotic acinar cells compared with that seen, for example, after duct ligation (Walker 1987). Microfilament disrupting agents such as cytochalasin B inhibit cell fragmentation during apoptosis but allow nuclear condensation and fragmentation (Cotter et al. 1992). In the first hours after CHB administration and at lower doses, however, apoptosis proceeds to cell fragmentation with intraepithelial macrophages at 12 h engorged with phagocytosed apoptotic bodies (Kelly, unpublished observations), making it unlikely that CHB prevents microfilament reorganization. There is some evidence that surrounding viable cells or phagocytes are required for apoptotic cell budding and fragmentation (Ellis et al. 1991; Driscoll 1992; Lang et al. 1994; Little & Flores 1996). When all surrounding cells are themselves apoptotic or full of apoptotic bodies, this process may be impaired. It is of interest that an illustration of the massive synchronous hepatocyte apoptosis in mouse liver previously mentioned also shows limited cell fragmentation (Ogasawara et al. 1993) although fragmentation of apoptotic hepatocytes is usual in other models (Searle et al. 1987).
During the period of acinar cell death and removal, duct cell viability was demonstrated by light and electron microscopy as well as immunohistochemistry, only extraordinarily rare amylase-positive acinar cells surviving. Thus, in this model, ducts and duct cells were the only possible source of the crowded ducts in atrophic lobules seen at 4 days. These latter structures, having the immunohistochemical profile of duct cells, must therefore result from the condensation of preexisting ducts after dead acinar cells are removed, the proliferation of surviving duct cells or a combination of both. The origin of such ducts, also called ‘ductules’ and ‘tubular complexes’, in various settings remains controversial. Concurrent acinar cell loss, duct cell proliferation and acinar cell regeneration in some models clearly make interpretation of electron microscopic and immunohistochemical preparations difficult and ‘redifferentiation’ in some of these circumstances certainly cannot be excluded (Gorelick et al. 1993). What we have demonstrated is that reduction of the exocrine pancreas to ductlike structures can occur in the almost complete absence of acinar cells, making ‘redifferentiation’ of acinar cells unnecessary for their formation. It is interesting that the reduction in whole pancreatic weight after CHB to 24% of control weight at 1 week is similar to that after ethionine and a protein-depletion diet (28% at 10 days) (Walker et al. 1993) and pancreas distal to a ligature (37% at 1 week) (Walker et al. 1992) where a similar origin for these ducts has been proposed. Ongoing apoptosis of duct epithelial cells is presumably responsible for the progressive reduction in duct numbers seen after 96 h, as it is after duct ligation (Walker et al. 1992).
Despite early cell death and oedema, inflammatory cell infiltration, as previously reported (Wallig et al. 1988; Wallig & Jeffery 1990), is delayed, reaching moderate density only at 48 h, the number of neutrophils remaining small throughout. In contrast, cerulein administration excites a vigorous inflammatory response (Fujimoto et al. 1997). It may be that after CHB, acinar cell contents initially remain largely membrane-bound limiting activation of inflammatory mediators such as complement. The marked oedema might result from duct rupture (Walker 1987) or vascular injury, demonstrated after caerulein administration, potential mediators including bradykinin B2 and oxygen free radicals (Steer et al. 1991; Kelly et al. 1993; Shibuya et al. 1996). Enlargement and proliferation of fibroblasts, collagen deposition, apoptosis of capillary endothelial cells, and stromal fatty infiltration are remarkably similar to those occurring in pancreas after duct obstruction (Pound & Walker 1981; Walker et al. 1992).
The limited acinar cell regeneration detected morphologically after CHB administration was reflected in the residual low pancreatic weight and evidence of pancreatic insufficiency in treated animals; the latter included failure of normal growth, muscle wasting and poorly digested food in the small intestine. In contrast, pancreatic regeneration after caerulein and ethinione administration is rapid and complete once the causative agent is removed (Fitzgerald 1960; Elsässer et al. 1986). The difference may be more complete loss of acinar cells after high dose CHB, limiting regeneration from this source (Adler et al. 1979). Lower dose CHB, leaving residual acinar cells, allows regeneration to occur (Wallig & Jeffery 1990). Pancreatic stem cells capable of acinar and islet cell replenishment have variously been identified as centroacinar cells (Elsässer et al. 1986; Pour 1988; Walker et al. 1992) or lining cells of intercalated (Zajicek et al. 1990; Gu et al. 1994) or common pancreatic ducts (Bonner-Weir et al. 1993). Loss of or damage to these cells by CHB is an alternative explanation for the failure of acinar cell replenishment in this model. Immunohistochemical staining for amylase was prominent in the periphery of islets at 96 h but the staining cells otherwise appeared identical to islet cells, a finding reported during islet cell replenishment in transgenic mice (Gu et al. 1994). The few regenerative acini seen after CHB most commonly surrounded islets, consistent with the observed trophic effects of islets on adjacent exocrine tissue (Tsubouchi et al. 1987). Further work on pancreatic regeneration in a number of settings is underway.
In summary, we have shown that, after administration of a single high dose of CHB, massive acinar cell apoptosis precedes secondary necrosis, ductlike structures in atrophic lobules are derived from ducts, and pancreatic regeneration is negligible. As selective induction of pancreatic acinar cell apoptosis has potential therapeutic benefit to patients with pancreatitis (Saluja et al. 1996; Bhatia et al. 1998) and pancreatic tumours with acinar differentiation, further studies of CHB and similarly acting compounds are warranted.
References
- Adler G, Hupp T, Kern HF. Course and spontaneous regression of acute pancreatitis in the rat. Virchows Arch. [A] Path. Anat. Histol. 1979;382:31–47. doi: 10.1007/BF01102739. [DOI] [PubMed] [Google Scholar]
- Ansari B, Coates PJ, Greenstein BD, Hall PA. In situ end-labelling detects DNA strand breaks in apoptosis and other physiological and pathological states. J. Pathol. 1993;170:1–8. doi: 10.1002/path.1711700102. [DOI] [PubMed] [Google Scholar]
- Barres BA, Hart IK, Coles HSR, Barne JF, Voyvodic JT, Richardson WD, Raff MC. Cell death and control of cell survival in the oligodendrocyte lineage. Cell. 1992;70:31–46. doi: 10.1016/0092-8674(92)90531-g. [DOI] [PubMed] [Google Scholar]
- Bell JM, Williams K. Growth-depressing factors in rapeseed oilmeal. Can. J. Agr. Sci. 1953;33:201–209. [Google Scholar]
- Bendayan M. Concentration of amylase along its secretory pathway in the pancreatic acinar cell as revealed by high resolution immunocytochemistry. Histochem. J. 1984;16:85–108. doi: 10.1007/BF01003438. [DOI] [PubMed] [Google Scholar]
- Bhatia M, Wallig MA, Hofbauer B, Lee H-S, Frossard J-L, Steer ML, Saluja AK. Induction of apoptosis in pancreatic acinar cells reduces the severity of acute pancreatitis. Biochem. Biophys. Res. Comm. 1998;246:476–483. doi: 10.1006/bbrc.1998.8519. [DOI] [PubMed] [Google Scholar]
- Bockman D. Morphology of the exocrine pancreas related to pancreatitis. Microsc. Res. Technical. 1997;37:509–519. doi: 10.1002/(SICI)1097-0029(19970601)37:5/6<509::AID-JEMT13>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Bonner-Weir S, Baxter LA, Schuppin GT, Smith FE. A second pathway for regeneration of adult exocrine and endocrine pancreas. Diabetes. 1993;42:1715–1720. doi: 10.2337/diab.42.12.1715. [DOI] [PubMed] [Google Scholar]
- Bouwens L, Braet F, Heimberg H. Identification of rat pancreatic duct cells by their expression of cytokeratins 7, 19, and 20 in vivo and after isolation and culture. J. Histochem. Cytochem. 1995;43:245–253. doi: 10.1177/43.3.7532655. [DOI] [PubMed] [Google Scholar]
- Clutton S. The importance of oxidative stress in apoptosis. Br. Med. Bull. 1997;53:662–668. doi: 10.1093/oxfordjournals.bmb.a011637. [DOI] [PubMed] [Google Scholar]
- Coles HSR, Burne JF, Raff MC. Large-scale normal cell death in the developing rat kidney and its reduction by epidermal growth factor. Development. 1993;118:777–784. doi: 10.1242/dev.118.3.777. [DOI] [PubMed] [Google Scholar]
- Cotter TG, Lennon SV, Glynn JM, Green DR. Microfilament-disrupting agents prevent the formation of apoptotic bodies in tumor cells undergoing apoptosis. Cancer Res. 1992;52:997–1005. [PubMed] [Google Scholar]
- Das NB, Torssell KBG. Silyl nitronates, nitrile oxides, and derived 2-isoxazolines in organic synthesis. Functionalization of butadiene, a novel route to furans and 2-isoxazolines as an alternative to aldol-type condensations. Tetrahedron. 1983;39:2247–2243. [Google Scholar]
- Driscoll M. Molecular genetics of cell death in the nematode Caenorhabditis elegans. J. Neurobiol. 1992;23:1327–1351. doi: 10.1002/neu.480230919. [DOI] [PubMed] [Google Scholar]
- Ekholm R, Zelander T, Edlund Y. The ultrastructural organization of the pancreas: I. Acinar cells. J. Ultrastruct. Res. 1962a;7:61–72. doi: 10.1016/s0022-5320(62)80029-x. [DOI] [PubMed] [Google Scholar]
- Ekholm R, Zelander T, Edlund Y. The ultrastructural organization of the pancreas: II. Centro-acinar cells, intercalary and intralobular ducts. J. Ultrastruct. Res. 1962b;7:73–83. doi: 10.1016/s0022-5320(62)80029-x. [DOI] [PubMed] [Google Scholar]
- Ellis RE, Jacobson DM, Horvitz HR. Genes required for the engulfment of cell corpses during programmed cell death in Caenorhabditis elegans. Genetics. 1991;129:79–94. doi: 10.1093/genetics/129.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsässer HP, Adler G, Kern HF. Time course and cellular source of pancreatic regeneration following acute pancreatitis in the rat. Pancreas. 1986;1:421–429. doi: 10.1097/00006676-198609000-00006. [DOI] [PubMed] [Google Scholar]
- Fitzgerald PJ. The problem of the precursor cell of regenerating pancreatic acinar epithelium. Lab. Invest. 1960;9:67–85. [PubMed] [Google Scholar]
- Fujimoto K, Hosotani R, Doi R, Wada M, Lee J-U, Koshiba T, Miyamoto Y, Imamura M. Role of neutrophils in cerulein-induced pancreatitis in rats: Possible involvement of apoptosis. Digestion. 1997;58:421–430. doi: 10.1159/000201478. [DOI] [PubMed] [Google Scholar]
- Gorelick FS, Adler G, Kern HF. Cerulein-induced pancreatitis. In: Go VLW, et al., editors. The Pancreas: Biology, Pathobiology and Disease. 2. New York: Raven Press Ltd; 1993. pp. 501–526. [Google Scholar]
- Grasl-Kraupp B, Ruttkay-Nedeky B, Koudelka H, Bukowska K, Bursch W, Schulte-Hermann R. In situ detection of fragmented DNA (TUNEL) assay fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology. 1995;21:1465–1468. doi: 10.1002/hep.1840210534. [DOI] [PubMed] [Google Scholar]
- Gu D, Lee M-S, Krahl T, Sarvetnick N. Transitional cells in the regenerating pancreas. Development. 1994;120:1873–1881. doi: 10.1242/dev.120.7.1873. [DOI] [PubMed] [Google Scholar]
- Harmon BV, Takano YS, Winterford CM, Gobe GC. The role of apoptosis in the response of cells and tumours to mild hyperthermia. Int. J. Radiat. Biol. 1991;59:489–501. doi: 10.1080/09553009114550441. [DOI] [PubMed] [Google Scholar]
- Iovanna JL. Redifferentiation and apoptosis of pancreatic cells during acute pancreatitis. Int. J. Pancreatol. 1996;20:77–84. doi: 10.1007/BF02825505. [DOI] [PubMed] [Google Scholar]
- Jamieson JD. The exocrine pancreas and salivary glands. In: Weiss L, editor. Cell and Tissue Biology: ATextbook of Histology. Baltimore: Urban and Scharzenberg; 1988. pp. 716–749. [Google Scholar]
- Kaiser AM, Saluja AK, Sengupta A, Saluja M, Steer ML. Relationship between severity, necrosis, and apoptosis in five models of experimental acute pancreatitis. Am. J. Physiol. 1995;269:C1295–C1304. doi: 10.1152/ajpcell.1995.269.5.C1295. [DOI] [PubMed] [Google Scholar]
- Karnovsky MJ. A formaldehyde-glutaraldehyde fixative of high osmolality for use in electron microscopy. J. Cell. Biol. 1965;27:137A–138A. [Google Scholar]
- Kelly DM, McEntee GP, Delaney C, McGeeney KF, Fitzpatrick JM. Temporal relationship of acinar and microvascular changes in caerulein-induced pancreatitis. Br. J. Surg. 1993;80:1174–1176. doi: 10.1002/bjs.1800800936. [DOI] [PubMed] [Google Scholar]
- Kerr JFR, Gobe GC, Winterford CM, Harmon BV. Anatomical methods in cell death. Meth. Cell. Biol. 1995;46:1–27. doi: 10.1016/s0091-679x(08)61921-4. [DOI] [PubMed] [Google Scholar]
- Lang R, Lustig M, Francois F, Sellinger M, Plesken H. Apoptosis during macrophage-dependent ocular tissue remodelling. Development. 1994;120:3395–3403. doi: 10.1242/dev.120.12.3395. [DOI] [PubMed] [Google Scholar]
- Little GH, Flores A. Programmed cell death in the anuran tadpole tail requires expression of a cell surface glycoprotein. Comp. Biochem. Physiol. 1996;113B:289–293. doi: 10.1016/0305-0491(95)02026-8. [DOI] [PubMed] [Google Scholar]
- Maher M, Chernenko G, Barrowman JA. The acute pancreatotoxic effects of the plant nitrile 1-cyano-2-hydroxy-3-butene. Pancreas. 1991;6:168–174. doi: 10.1097/00006676-199103000-00007. [DOI] [PubMed] [Google Scholar]
- Nishie K, Daxenbichler ME. Toxicology of glucosinolates, related compounds (nitriles, R-goitrin, isothiocyanates) and vitamin U found in Cruciferae. Fd. Cosmet. Toxicol. 1980;18:159–172. doi: 10.1016/0015-6264(80)90070-x. [DOI] [PubMed] [Google Scholar]
- Nordfeldt S, Gellerstedt N, Falkmer S. Studies on rapeseed meal and its goitrogenic effect on pigs. Acta Pathol. Microbiol. Scand. 1954;35:217–236. doi: 10.1111/j.1699-0463.1954.tb00864.x. [DOI] [PubMed] [Google Scholar]
- Ogasawara J, Watanabe-Fukunaga R, Adachi M, Matsuzawa A, Kasugai T, Kitamura Y, Itoh N, Suda T, Nagata S. Lethal effect of the anti-Fas antibody in mice. Nature. 1993;364:806–809. doi: 10.1038/364806a0. [DOI] [PubMed] [Google Scholar]
- Pound AW, Walker NI. Involution of the pancreas after ligation of the pancreatic ducts. I: a histological study. Br. J. Exp. Path. 1981;62:547–558. [PMC free article] [PubMed] [Google Scholar]
- Pour PM. Mechanism of pseudoductular (tubular) formation during pancreatic carcinogenesis in the hamster model. Am. J. Pathol. 1988;130:335–344. [PMC free article] [PubMed] [Google Scholar]
- Rao MS, Yeldandi AV, Subbarao V, Reddy JK. Role of apoptosis in copper deficiency-induced pancreatic involution in the rat. Am. J. Pathol. 1993;142:1952–57. [PMC free article] [PubMed] [Google Scholar]
- Saluja A, Hofbauer B, Yamaguchi Y, Yamanaka K, Steer M. Induction of apoptosis reduces the severity of caerulein-induced pancreatitis in mice. Biochem. Biophys. Res. Commun. 1996;220:875–878. doi: 10.1006/bbrc.1996.0498. [DOI] [PubMed] [Google Scholar]
- Schüssler MH, Skoudy A, Ramaekers F, Real FX. Intermediate filaments as differentiation markers of normal pancreas and pancreas cancer. Am. J. Pathol. 1992;140:559–568. [PMC free article] [PubMed] [Google Scholar]
- Searle J, Harmon B, Bishop C, Kerr J. The Significance of cell death by apoptosis in hepatobiliary disease. J. Gastroenterol. Hepatol. 1987;2:77–96. [Google Scholar]
- Shibuya K, Sunamura M, Yamauchi J-I, Takeda K, Kobari M, Matsuno S. Analysis of the derangement of the pancreatic microcirculation in a rat caerulein pancreatitis model using an intravital microscope system. Tohoku J. Exp. Med. 1996;180:173–186. doi: 10.1620/tjem.180.173. [DOI] [PubMed] [Google Scholar]
- Staack R, Kingston S, Wallig MA, Jeffery EH. A comparison of the individual and collective effects of four glucosinolate breakdown products from brussels sprouts on induction of detoxification enzymes. Toxicol. Appl. Pharmacol. 1998;149:17–23. doi: 10.1006/taap.1997.8340. [DOI] [PubMed] [Google Scholar]
- Steer ML, Rutledge PL, Powers RE, Saluja M, Saluja AK. The role of oxygen-derived free radicals in two models of experimental acute pancreatitis: Effects of catalase, superoxide dismutase, dimethylsulfoxide, and allopuranol. Klin. Wochenschr. 1991;69:1012–1017. doi: 10.1007/BF01645149. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Schoch E, Walker NI. Origin of acinar cell regeneration after atrophy of the rat parotid induced by duct obstruction. Int. J. Exp. Path. 1998;79:293–301. doi: 10.1046/j.1365-2613.1998.710405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubouchi S, Kano E, Suzuki H. Demonstration of expanding cell populations in mouse pancreatic acini and islets. The Anat. Record. 1987;218:111–115. doi: 10.1002/ar.1092180203. [DOI] [PubMed] [Google Scholar]
- Wada M, Doi R, Hosotani R, Lee J-U, Fujimoto K, Koshiba T, Miyamoto Y, Fukoka S-I. Expression of Bcl-2 and PCNA in duct cells after pancreatic duct ligation in rats. Pancreas. 1997;15:176–182. doi: 10.1097/00006676-199708000-00010. [DOI] [PubMed] [Google Scholar]
- Walker NI. Ultrastructure of the rat pancreas after experimental duct ligation. I. The role of apoptosis and intraepithelial macrophages in acinar cell depletion. Am. J. Pathol. 1987;126:439–451. [PMC free article] [PubMed] [Google Scholar]
- Walker NI, Gobe GC. Cell death and cell proliferation during atrophy of the rat parotid gland induced by duct obstruction. J. Pathol. 1987;153:333–344. doi: 10.1002/path.1711530407. [DOI] [PubMed] [Google Scholar]
- Walker NI, Winterford CM, Kerr JFR. Ultrastructure of the rat pancreas after duct ligation. II: Duct and stromal cell proliferation, differentiation, and deletion. Pancreas. 1992;7:420–434. doi: 10.1097/00006676-199207000-00002. [DOI] [PubMed] [Google Scholar]
- Walker NI, Winterford CM, Williamson RM, Kerr JFR. Ethionine-induced atrophy of rat pancreas involves apoptosis of acinar cells. Pancreas. 1993;8:443–449. doi: 10.1097/00006676-199307000-00007. [DOI] [PubMed] [Google Scholar]
- Wallig MA, Gould DH, Fettman MJ. Selective pancreatotoxicity in the rat induced by the naturally occurring plant nitrile 1-cyano-2-hydroxy-3-butene. Fd. Chem. Toxic. 1988;26:137–147. doi: 10.1016/0278-6915(88)90110-x. [DOI] [PubMed] [Google Scholar]
- Wallig MA, Jeffery EH. Enhancement of pancreatic and hepatic glutathione levels in rats during cyanohydroxybutene intoxication. Fundam. Appl. Toxicol. 1990;14:144–159. doi: 10.1016/0272-0590(90)90240-k. [DOI] [PubMed] [Google Scholar]
- Wallig MA, Kore AM, Crawshaw J, Jeffery EH. Separation of the toxic and glutathione-enhancing effects of the naturally occurring nitrile cyanohydroxybutene. Fundam. Appl. Toxicol. 1992;19:598–606. doi: 10.1016/0272-0590(92)90099-4. [DOI] [PubMed] [Google Scholar]
- Wyllie AH, Kerr JFR, Currie AR. Cell death. The significance of apoptosis. Int. Rev. Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- Zajicek G, Arber N, Scwartz-Arad D, Ariel I. Streaming pancreas: Islet cell kinetics. Diabetes Res. 1990;13:121–125. [PubMed] [Google Scholar]