

Abstract
The interrelationship between acinar cell apoptosis and tubular complex formation was examined in caerulein-induced pancreatitis using histology, immunohistochemistry, electron microscopy and DNA gel electrophoresis. Rats were given 8 hourly subcutaneous injections of caerulein, 24μg/kg, for up to 2 days. Morphologically and biochemically typical apoptosis affected 4.6 and 8.9% of acinar cells at 1 and 2 days, respectively, resulting in removal of most acinar cells by 2 days. Consequently, pancreatic ducts, the lining cells expressing bcl-2 and therefore resistant to apoptosis, became much more closely approximated to form the basis of tubular complexes; small numbers of immunohistochemically discrete acinar cells in their lining were either pre-apoptotic resistant to it or newly formed. Proliferation of duct-like lining cells was associated with apoptosis, an increase in islet cells and acinar cell regeneration. There was evidence of duct to acinar cell differentiation but the main increase in acinar cell numbers appeared to derive from proliferation of newly formed acinar cells.
Keywords: caerulein, pancreatitis, apoptosis, tubular complexes, regeneration
Administration of supramaximal doses of caerulein by continuous intravenous infusion or intermittent intraperitoneal or subcutaneous injection causes acute pancreatitis in rats (Lampel & Kern 1977; Adler et al. 1979; Jurkowska et al. 1992; Gorelick et al. 1993). Caerulein-induced pancreatitis is characterized by early interstitial oedema, inflammatory cell infiltration and acinar cell vacuolation, formation of tubular complexes and subsequent acinar cell regeneration. Tubular complexes are defined as cylindrical tubes lined by a monolayer epithelium of more or less flattened duct-like cells in which mitoses are seen (Iovanna 1996). The lining cells are interpreted to derive from metaplasia or redifferentiation of acinar cells to cells with a duct cell-like phenotype (Gorelick et al. 1993; Iovanna 1996; Bockman 1997).
Several recent studies have documented apoptosis of pancreatic acinar cells after administration of caerulein in rats (Kaiser et al. 1995; Sandoval et al. 1996; Fujimoto et al. 1997; Gukovskaya et al. 1997; Hahm et al. 1998; Kimura et al. 1998a,b). They have looked mainly at early time points and variations of the original model including prior neutrophil depletion (Sandoval et al. 1996; Fujimoto et al. 1997), adrenalectomy (Kimura et al. 1998a) or low dose lipopolysaccharide administration (Kimura et al. 1998b). Thus, there is yet no overview of the role of acinar cell apoptosis in the sequence of morphological changes occurring in caerulein-induced pancreatitis. Moreover, while acinar cell apoptosis and tubular complex formation are now recognized to be common concomitant events in pancreatic pathology (Iovanna 1996; Bockman 1997), hypotheses of the origin of tubular complexes, conceived prior to documentation of extensive acinar cell apoptosis in pancreatic models, have not been modified.
Here we produce caerulein-induced pancreatitis in a way known to cause pancreatic DNA depletion (indicating cell loss) and tubular complex formation (Jurkowska et al. 1992). We demonstrate extensive apoptotic acinar cell loss. This necessitates review of original hypotheses of the origins of tubular complexes. There are also implications for the source of acinar cell regeneration that occurs subsequently.
Materials and methods
Experimental procedures
Animal experiments were performed under National Health and Medical Research Committee guidelines and approved by the University of Queensland Animal Experimentation Ethics Committee prior to project commencement (PATH%573%92%d). Animals were monitored for clinical signs of pain or distress. As treated animals showed normal behaviour patterns, no analgaesia was given.
Randomly bred male Sprague-Dawley rats weighing 170–250 g were fed a standard pellet diet and allowed free access to water. Pancreatitis was induced, using the method of Jurkowska et al. (1992), by subcutaneous injection of caerulein (synthetic caerulein, C-9026, Sigma Chemical Co., St Louis, Mo., U.S.A.), 24μg/kg, every 8 h for 2 days. The caerulein was dissolved in gelatin (G-2625, Sigma Chemical Co.) in the ratio 75% gelatin to 25% stock solution to prolong its absorption. The dose was determined by trial as the least dose producing a relatively uniform but maximum pancreatic lesion. Saline control animals were subjected to the same procedures and handling as experimental animals but with saline substituted for caerulein. Groups of 6 rats were assessed 1, 2, 4 and 7 days after the first injection.
Rats used to assess pancreatic weight were weighed prior to dissection. The entire pancreas was dissected free, trimmed of fat and lymph nodes and weighed. The mean pancreatic weight and mean pancreatic weight as a percentage of body weight (pancreatic weight index) ±standard error of the mean (SEM) were calculated for each group. Differences between experimental and saline control groups were assessed using Student's t-test.
Pancreas for histological study was fixed in 4% formalin in phosphate buffered saline at pH 7.2, dehydrated in graded alcohols, cleared in xylol and embedded in paraffin wax. Three and 5μm sections were stained with haematoxylin and eosin (H & E).
Levels of apoptosis were calculated from counts of apoptotic and total acinar cells in 10 random high power microscopic fields (×400) in a 5μm H & E section from each animal. The number of acinar cells counted varied between animals and with time after caerulein administration. In 2 day experimental animals, immunoperoxidase stains for amylase were used to confirm intact acinar cells, which were otherwise difficult to identify. The following criteria were used for apoptotic counts: a single condensed cell or large single apoptotic body with typically marginated nuclear chromatin was given a single count; multiple apoptotic bodies clustered together were given a single count; any doubtful cells were disregarded (Gobé & Axelsen 1987). Rates of apoptosis (percentage of total acinar cells) were expressed as the mean ±SEM for each group of animals. Differences between experimental and saline control groups were assessed using Student's t-test. Terminal d-UTP nick-end labelling (TUNEL) was not used to identify apoptosis as it is our experience and the experience of others that it is not specific for apoptosis and ultimately apoptosis must be confirmed using morphological criteria (Ansari et al. 1993; Grasl-Kraupp et al. 1995; Gukovskaya et al. 1996).
Immunohistochemistry
Immunohistochemistry for cytokeratin, bcl-2 and amylase was performed to assist in the identification of epithelial cells in sections as duct or acinar in type (Bendayan 1984; Schüssler et al. 1992; Bouwens & De Blay 1996; Wada et al. 1997). For cytokeratin and amylase, sections were pretreated with prediluted Proteinase K (DAKO Corp., Carpinteria, California, USA). In all cases, endogenous peroxidase was inhibited by treating sections with 1% hydrogen peroxide and 0.1% sodium azide in Tris buffered saline (TBS). Non-specific antibody binding was inhibited by incubation with 4% nonfat skim milk powder in TBS followed by 10% nonimmune goat serum. Primary antibodies were mouse monoclonal AE1/AE3 anticytokeratin (DAKO) recognizing cytokeratins (CK) 1–8, 10, 14–16 and 19 (Sun et al. 1985), bcl-2 oncogene, clone 124 (DAKO) at dilutions of 1/40 or rabbit antihuman α–amylase (DAKO) at a dilution of 1/500. Secondary antibodies were rat antimouse biotinylated IgG (Jackson Immunoresearch, West Grove, Pennsylvania) at a dilution of 1%300 for cytokeratin and bcl-2 and goat antirabbit biotinylated IgG (Jackson Immunoresearch) at a dilution of 1/400 for amylase. Cytokeratin and bcl-2 localization were demonstrated using the peroxidase-labelled streptavidin biotin complex method (DAKO strept AB Complex/HRP) and reactions were developed with 3,3′-diaminobenzidine tetrahydrochloride solution (DAB) (Zymed, San Francisco, California). Amylase localization was demonstrated using the peroxidase-streptavidin method developed with vector VIP peroxidase substrate (Vector Laboratories, Burlingame, California). Double staining for cytokeratin and amylase was performed with the above reagents with blocking before each primary antibody. All sections were lightly counterstained with haematoxylin. Monoclonal antibodies to human CK7 and 19 react specifically with rat pancreatic duct epithelial cells (Bouwens et al. 1995). The antibody to bcl-2 used recognizes rat bcl-2 protein in Western blots (Wada et al. 1997). Specificity of the antibodies used for identification of duct and acinar cells was checked by inclusion of known positive control tissue (normal rat pancreas) in each run. Negative controls comprised test tissues without the addition of primary antibody.
Electron microscopy
For electron microscopy, animals were anaesthetized and the pancreas perfused in sequence with firstly, 1% heparinized saline, secondly, 1% paraformaldehyde and 1.25% glutaraldehyde in cacodylate buffer and finally 4% paraformaldehyde and 5% glutaraldehyde in cacodylate buffer via a catheter inserted into the abdominal aorta. The pancreas was immediately removed, trimmed of fat and lymph nodes, diced, and immersed in final perfusate for two hours. The diced tissue was then stored in cacodylate buffer. The tissue was postfixed in 1% osmium tetroxide, stained en bloc in 5% aqueous uranyl acetate, dehydrated through a series of graded alcohols, cleared in propylene oxide, and embedded in an epon/araldite mixture. Semithin sections (1μm) were cut on an Ultratome V microtome, and stained with toluidine blue for viewing. Ultrathin sections from selected areas were stained with lead citrate and examined with a JEOL-1200 EX11 electron microscope.
DNA Analysis
For pancreatic DNA analysis, 100 mg of frozen tissue was homogenized in TES lysis buffer (10 mmol/l Tris HCl pH 7.8, 1 mmol/l ethylenediaminetetraacetic acid (EDTA) pH 8.0, 100 mmol/l NaCl) and 1%(wt/vol) sodium dodecyl sulphate and the extracts incubated with proteinase K 100μg/ml (Sigma Chemical Co.) overnight at 37 °C. RNA was degraded by incubation with 5μg/ml ribonuclease A (Sigma Chemical Co.) at 37 °C for 45 mins. DNA was extracted by addition of equal volumes of phenol and chloroform, then ethanol-precipitated in the presence of 0.3 mol/l sodium acetate (1 vol. sodium acetate/2 vol. ethanol). Pellets of DNA were air dried then dissolved in TE buffer (10 mmol/l Tris HCl pH 7.8, 1 mmol/l EDTA pH 7.8). The sample was treated with EDTA (10 mmol/l) plus ribonuclease A (350μg/ml) at 37 °C for 15 mins then heated at 56 °C for 10 mins before analysis by electrophoresis on a 1.8/ agarose gel containing ethidium bromide. A preparation of pGem (Promega Corporation, Rozelle, Australia) was run concurrently as a DNA fragment size marker. The gel was photographed under UV light.
Results
Pancreatic weights
After caerulein, the mean pancreatic weight index at day 2 was identical to that of the saline controls (Table 1). By day 4, it had decreased by 42/ and by 7 days by 36/ relative to saline controls (Table 1).
Table 1.
Mean pancreatic weight and pancreatic weight index ±SEM (n = 6) for saline and caerulein groups 2, 4 and 7 days after commencement of injections
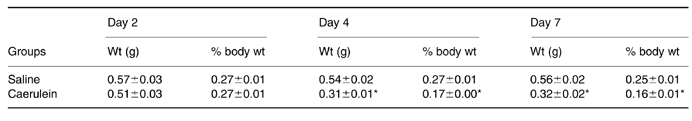
Caerule in group differs significantly from saline group, P < 0.001.
Light microscopy
Pancreas from saline-treated control rats appeared normal with widely spaced ducts separated by closely packed acinar cells (Figure 1 a). Twenty-four hours after the first caerulein dose, the pancreas showed oedematous separation of lobules and acini with a moderate interstitial infiltrate of neutrophils and mononuclear phagocytes; enlarged interstitial fibroblasts were also conspicuous. Acini showed cellular vacuolization, partial degranulation and dilated lumens; extensive loss of acinar cells had already occurred. By day 2, pancreatic lobules mainly comprised tubular complexes in a loose connective tissue stroma with few remaining acinar cells (Figure 1b). Lobules and tubular complexes within lobules were separated by an expanded interstitial space and inflammatory cell infiltrate. At 4 days, the epithelial component was remarkably similar to that at 2 days but lobules and tubular complexes within them were more closely approximated and now separated by fibrosis and a persistent moderate mononuclear inflammatory cell infiltrate. By day 7, the lobular architecture had returned to a near normal appearance though mild fibrosis and a mild, patchy inflammatory cell infiltrate persisted (Figure 1c).
Figure 1.
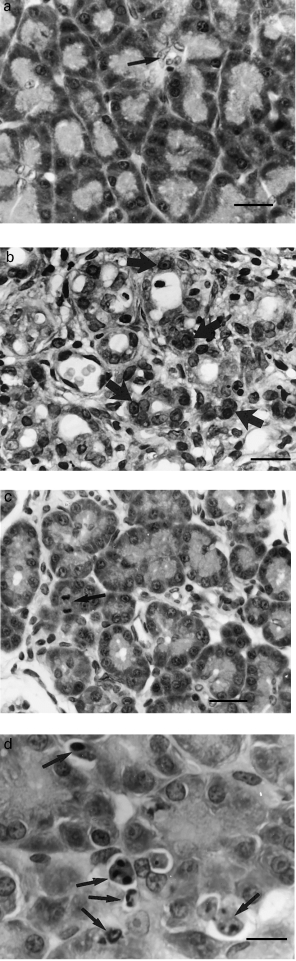
(a) Pancreas of control animal treated with saline/gelatin for 2 days. Arrow, intercalated duct. (b) Pancreas 2 days after commencement of caerulein injections. Tubular complexes are lined by flattened duct-like cells and acinar cells (arrows) are difficult to identify. (c) Pancreas 7 days after commencement of caerulein injections. Acinar cells are once again abundant and readily identifiable. Note mitotic acinar cell (arrow). (d) Apoptotic acinar cells and bodies (arrows) 1 day after commencement of caerulein injections. (H & E; a, b, c, bar = 20 μm; d, bar = 10 μm)
At 1, 2 and 4 days after the first dose of caerulein, abundant cell death by apoptosis was seen by light microscopy. Apoptotic cells at 24 h could clearly be identified as acinar cell in origin by their size, staining and location. They showed sharply demarcated condensed chromatin abutting the nuclear envelope, cell shrinkage, and cell and nuclear fragmentation to form apoptotic bodies. The latter were seen as rounded eosinophilic or basophilic structures often containing pyknotic nuclear chromatin and apparently lying within cleared spaces (Figure 1d). Apoptotic bodies were seen within the epithelium, in acinar lumens or free within the interstitium. Apoptotic cells and bodies at 2 days were smaller and less distinctive. Their acinar cell origin was confirmed by their immunohistochemical positivity for amylase. At 4 days, small apoptotic bodies were seen within tubular complexes, their number appearing to be inversely related to the number of differentiated acinar cells present. Their cell of origin was not apparent morphologically or immunohistochemically. As a consequence a cell specific apoptotic index could not be calculated for 4 days.
The acinar cell apoptotic index at 1 day was 4.65 ±0.04% (compared with 0.03 ± 0.01% in saline controls, P < 0.001) and at 2 days 8.9 ± 1.55% (compared with 0.01 ± 0.01% in saline controls, P < 0.001). At 7 days, an acinar cell apoptotic index could again be accurately calculated. At this time, the apoptotic index had almost returned to control levels (0.27 ± 0.03% compared with 0% in saline controls, P < 0.001).
Mitoses were rarely seen in sections from saline control animals, but 2 days after commencement of caerulein injections, considerable numbers of duct-like cells lining tubular complexes were in mitosis and, on day 4, mitoses were seen in these and acinar cells, the number of the latter varying greatly between animals. On day 7, acinar cell mitoses alone were conspicuous (Figure 1c). The mean numbers of duct and acinar cell mitoses per 10 high power fields in treated animals at 2, 4 and 7 days (n = 6; results expressed as the ratio duct cell mitoses:acinar cell mitoses) were 4.5:0.0, 0.5:6.2 and 0.0:12.5, respectively. More detailed studies of the regenerative phase are currently underway.
Immunohistochemistry
In saline-treated control rats, duct epithelial cells were positive for cytokeratin and bcl-2 (Figure 2a), acinar cells for amylase, and blood vessel endothelium and muscle cells for bcl-2; islets, inflammatory and connective tissue cells other than those in blood vessels were negative with all antibodies tested. As intervening acinar cells were depleted by apoptosis, bcl-2 and cytokeratin positive ducts became more closely approximated (Figure 2b,c); few bcl-2 and cytokeratin negative epithelial cells remained at 2 and 4 days (Figure 2c). Although the epithelial tissue component appeared similar on days 2 and 4, by day 4 ducts (tubular complexes) were more closely packed (Figure 2c). Pancreatic regeneration commencing about 4 days resulted in renewed separation of ducts as acinar cells increased in number; at 7 days, the appearance approached that in saline controls (Figure 2d).
Figure 2.
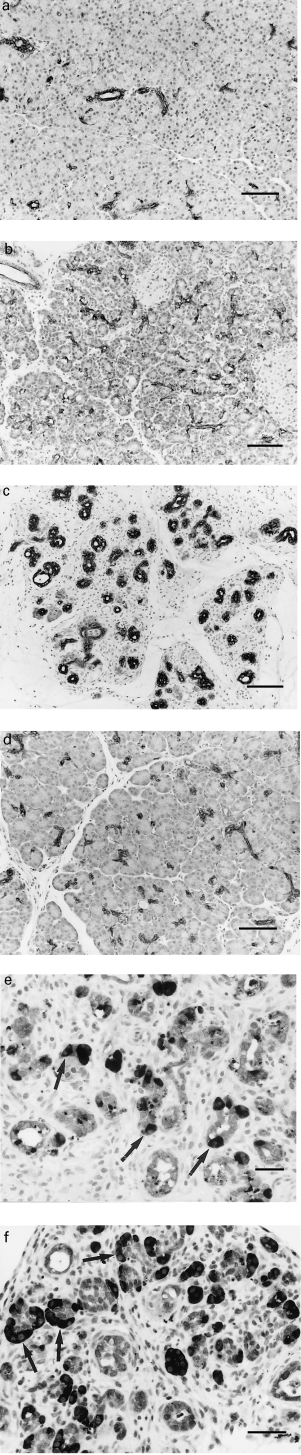
Immunohistochemistry. (a) Pancreas of untreated animal. Ducts are widely separated by closely packed keratin-negative acini. (Cytokeratin). (b) Pancreas 1 day after commencement of caerulein injections. Relatively more tubular complexes. (bcl-2). (c) Pancreas 4 days after commencement of caerulein injections. Tubular complexes with few keratin negative epithelial cells present. (Cytokeratin). (d) Pancreas 7 days after commencement of caerulein injections. Nearly normal. (bcl-2). (e) Pancreas 2 days after commencement of caerulein injections. Note numerous small darkly staining amylase positive apoptotic bodies (dark specks) within epithelium and mainly single residual acinar cells (arrows). (Double staining for amylase (dark) and cytokeratin (light)). (f) Pancreas 4 days after commencement of caerulein injections. Note absence of the amylase positive apoptotic bodies seen in (e) and clustered amylase positive acinar cells, some seen as demilunes surrounding lightly staining complexes lined by duct-like cells (arrows). (Double staining for amylase (dark) and cytokeratin (light)). (a, b, c, d, bar = 50 μm; e, f bar = 25 μm; counterstain haematoxylin).
Most apoptotic cells and bodies on days 1 and 2 stained for amylase (Figure 2e), but apoptotic bodies in tubular complexes on day 4, in most cases, were negative for amylase, bcl-2 and cytokeratin; a small proportion were positive for cytokeratin, and rare ones were positive for amylase. The relationship of residual acinar cells to duct-like cells in the tubular complexes on days 2 and 4 was best appreciated in sections double stained for cytokeratin and amylase. On day 2, residual amylase positive acinar cells were mostly seen as single cells or as small cell clusters in the lining of tubular complexes (Figure 2e). On day 4, amylase positive cells, many in mitosis, most often occurred as demilunes surrounding cytokeratin positive tubular complexes (Figures 2f and 3). Some of the amylase positive cells at day 4 also stained for cytokeratin (Figure 3).
Figure 3.
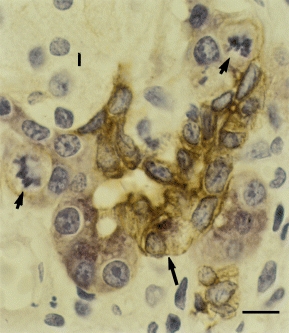
Immunohistochemistry. Pancreas 4 days after commencement of caerulein injections. Note clustered and mitotic (short arrows) acinar cells (pink). An occasional cell (long arrow) appears cytokeratin (brown) and amylase positive (pink). Islet cell (I) (Double staining for amylase and cytokeratin; bar = 7 μm).
Electron microscopy
One and 2 days after caerulein administration, many acinar cells showed ultrastructural changes of apoptosis. Nuclei showed sharply demarcated condensation of chromatin against the nuclear envelope, convolution of the nuclear outline, and budding to form several, usually membrane-bounded nuclear fragments retaining characteristically segregated condensed chromatin (Figure 4a). There was associated convolution of the cellular membrane, disruption of junctional complexes and concentric whorling of the rough endoplasmic reticulum (RER) with cellular cleavage to form apoptotic bodies often occurring between the whorls. Most bodies were taken up by intraepithelial macrophages that sometimes contained several with variable degrees of structural preservation (Figure 4b), indicative of a high apoptotic rate. Other bodies were taken up by adjacent acinar cells or shed into the acinar lumen where they underwent secondary degeneration or ‘necrosis’. At 4 days, apoptotic bodies were mostly intraepithelial, small or degraded; their cell of origin could not be elucidated from their structure. They were found between epithelial cells (Figure 4c) or within intraepithelial macrophages or duct-like cells. Some macrophages at this time contained large residual bodies, indicative of considerable phagocytic activity. Loss of acinar cells resulted in convolution and collapse of redundant acinar basement membrane at the margins of tubular complexes.
Figure 4.
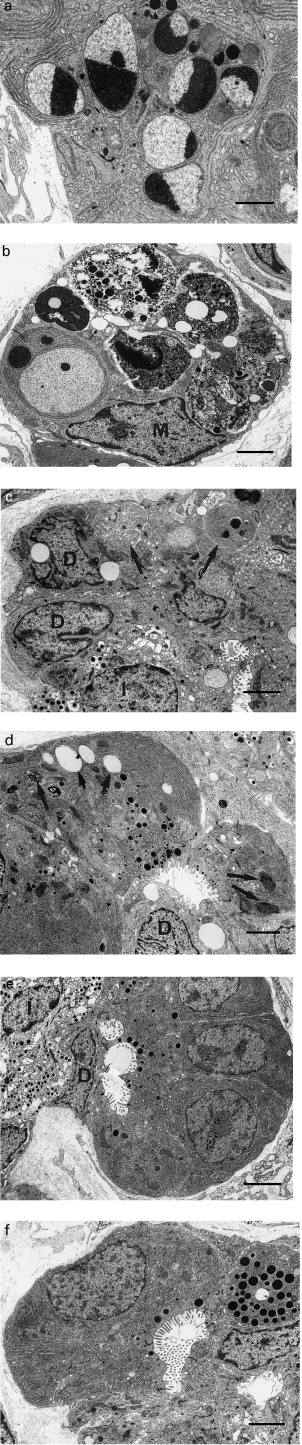
(a) Apoptotic acinar cell 2 days after commencement of caerulein injections. (b) Intraepithelial macrophage (M) containing multiple phagocytosed apoptotic bodies showing varying states of presentation 2 days after commencement of caerulein injections. (c) Small intercellular apoptotic bodies (arrows) in tubular complex of pancreas 4 days after commencement of caerulein injections. Note duct-like cells (D) and single islet cell (I) (d) Tubular complex 2 days after commencement of caerulein injections. Note duct-like (D) and altered acinar cells containing vacuoles (short arrows) and residual bodies (long arrows). (e) Tubular complex 4 days after commencement of caerulein injections. Note duct-like cell (D), islet cell (I), zymogen granules (arrows) and clustered presumably newly forming acinar cells with sparse zymogen granules and absence of vacuoles and residual bodies. (f) Tubular complex 4 days after commencement of caerulein injections. Note developing acinar differentiation in elongated cell with elongated nucleus, broad luminal border, relatively limited RER and sparse small luminal zymogen granules. a, bar = 1.25 μm; b, c, d, bar = 2 μm; e, f, bar = 2.5 μm.
Cells lining tubular complexes at 2 days mostly resembled normal duct epithelial cells. Residual acinar cells were separated by duct-like cells and had zymogen granules that were small and deformed, reduced RER, distorted cell shape, and reduced luminal microvilli (Figure 4d). Many contained vacuoles and small residual bodies. At 4 days, tubular complexes gave rise to single islet cells with increased frequency (Figure 4c,e) and clustered acinar cells corresponding to the amylase positive demilunes identified immunohistochemically (Figure 4e). The latter had sparse, small luminal zymogen granules and were usually devoid of vacuoles and residual bodies (Figure 4e). Occasional cells had features intermediate between duct and acinar cells (Figure 4f), suggesting differentiation of duct to acinar cells. At 2 and 4 days, mitoses were often identified in duct-like cells and at 4 days within acinar cells. By 7 days, acinar cells were again numerous and acinar cell mitosis remained prominent. By 4 days, interstitial collagen deposition was prominent.
DNA gel electrophoresis
An example of the electrophoretic pattern of DNA extracted from the pancreases of saline and caerulein groups is shown in Figure 5. One and two days after the commencement of caerulein injections, a ladder-like distribution pattern of low molecular weight DNA, characteristic of the production of mono-and oligonucleosomal-sized fragments in apoptosis, was seen. DNA extracted from saline controls (days 1 and 2) and animals 7 days after commencement of caerulein injections was of high molecular weight and showed no evidence of DNA fragmentation, correlating with the negligible apoptotic rates in these animals.
Figure 5.
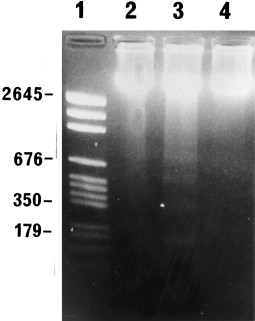
Agarose gel electrophoresis of DNA extracted from saline controls (2 days) (lane 2) and animals 2 days (lane 3) and 7 days (lane 4) after commencement of caerulein injections. Note typical ladder pattern of apoptosis in lane 3. DNA molecular weight markers, lane 1.
Discussion
As previously reported, administration of caerulein to rats caused pancreatic oedema, neutrophil and macrophage infiltration, acinar cell vacuolation and tubular complex formation preceding acinar cell regeneration (Lampel & Kern 1977; Adler et al. 1979; Jurkowska et al. 1992; Gorelick et al. 1993). Interstitial fibroblast activation and collagen deposition and removal followed the sequence reported in detail by Elsässer et al. (1989). Pancreatic weight index was reduced by 42% at 4 days compared with actual pancreatic weight losses of 50 (Gorelick et al. 1993) and 58% (Jurkowska et al. 1992) at 4.5 and 5 days, respectively, in previous studies. A reduction in pancreatic weight index consequential to acinar cell loss at 2 days was negated by stromal oedema and increased connective tissue deposition; reduction at 4 days mainly reflected reduced oedema and stromal condensation seen morphologically as closer approximation of the epithelial tissue elements. Even though the pancreas had a relatively normal histological appearance at 7 days, its weight remained markedly reduced compared with saline controls, indicating regeneration was incomplete, 10 days or so usually being required for full recovery (Gorelick et al. 1993). This model thus proved suitable for study of cellular events in caerulein-induced pancreatitis.
Aspects of acinar cell apoptosis in caerulein-induced pancreatitis have recently been reported (Kaiser et al. 1995; Sandoval et al. 1996; Fujimoto et al. 1997; Gukovskaya et al. 1997; Hahm et al. 1998; Kimura et al. 1998a,b) but the overall role of apoptosis in the sequence of morphological changes in these models has not been studied. Apoptosis here was identified histologically and ultrastructurally using standard criteria and confirmed biochemically using DNA gel electrophoresis. Acinar cell apoptotic rates of 4.6 and 8.9% were recorded at 24 and 48 h, respectively, with marked acinar cell depletion evident at the latter time. Because acinar cells depleted of zymogen and reduced in size at 48 h were sometimes difficult to identify, immunohistochemistry for amylase was used to obtain accurate counts. The trend in apoptotic rates was similar to that reported by Fujimoto et al. (1997) in caerulein controls treated similarly to our experimental animals. Given a histological half-life for apoptotic bodies of 1–2 h (Barres et al. 1992; Coles et al. 1993) and the fact that small increments in apoptotic indices of the order of 1–2% can halve the total cell population of a tissue within 24 h (Howie et al. 1994), the observed indices were sufficient to delete all or most acinar cells in a few days. Extensive acinar cell depletion is confirmed by 50% pancreatic weight loss despite inflammation and fibrosis (this study; Jurkowska et al. 1992; Gorelick et al. 1993), a 50% reduction in pancreatic DNA content (Jurkowska et al. 1992) and widespread collapse and convolution of acinar basement membranes at the margins of tubular complexes (this study). Similar pancreatic weight losses from other causes (Walker et al. 1992,1993; Kelly et al. 1999) have been associated with complete or near complete apoptotic deletion of acinar cells. In one of these, duct ligation, the exocrine pancreas is reduced to 10% of its original volume (Watanabe et al. 1995), approximating the duct cell component of the rodent pancreas (Githens 1988).
A ladder-like pattern on DNA gel electrophoresis reflecting internucleosomal DNA cleavage in pancreatic acinar cell apoptosis, seen in this study, has previously been reported after duct ligation (Kaiser et al. 1995; Wada et al. 1995; Doi et al. 1997), dietary copper depletion (Kishimoto et al. 1994; Ide et al. 1994) and caerulein administration (Fujimoto et al. 1997). Previously, faintness (Kaiser et al. 1995; Fujimoto et al. 1997) or absence of ladders (Gukovskaya et al. 1996) has been ascribed to low apoptotic counts, partial degradation of DNA to large 300–350 kb fragments only or digestion of DNA by pancreatic DNAse (Kaiser et al. 1995). The latter may provide an explanation for the faint bands seen here despite the relatively high apoptotic rates observed. Potential mediators of acinar cell apoptosis in this model are free radicals (Iovanna 1996), platelet activating factor (Sandoval et al. 1996) and tumour necrosis factor-α(Gukovskaya et al. 1997).
Some acinar cell necrosis as well as apoptosis has been reported after 6 h of caerulein infusion (Sandoval et al. 1996). While we did not see acinar cell necrosis, it is possible it occurs at shorter intervals than used in this study. It is also seen soon after pancreatic ligation, but, compared with apoptosis, it makes a negligible contribution overall to acinar cell depletion (Walker 1987).
Pancreatic duct cells expressed bcl-2 (Lu et al. 1993; Bouwens & De Blay 1996; Wada et al. 1997), typical of stem and differentiating cell populations, rendering them resistant to apoptosis. As a consequence of lobular contraction and the normal continuity of ducts and acini, rapid apoptotic deletion of acinar cells resulted in closer approximation of the persistent ducts (Figure 2), culminating in tightly clustered tubular complexes associated with small numbers of residual acinar cells (Figure 2e). During regeneration, ducts again became widely separated as newly formed acinar cells accumulated (Figure 2d). Taking into account the extent of acinar cell depletion and the demonstrated persistence and condensation of ducts, it is not necessary to invoke acinar cell metaplasia or redifferentiation to duct-like cells to account for the formation of tubular complexes (Gorelick et al. 1993; Iovanna 1996; Bockman 1997) in this model. Previous accounts of acinar cells shedding cytoplasmic fragments to form simpler cells can be explained by acinar cell apoptosis involving death and fragmentation of whole cells. Moreover, during the formative stages, duct and acinar cells remained immunohistochemically discrete. While complete acinar cell loss was not demonstrated at the times studied, it may occur at an intermediate time or be impossible to demonstrate because of an overlap in occurrence of persistent and newly formed acinar cells. In models of pancreatic disease where acinar cell regeneration does not occur, tubular complexes eventually show no acinar cell differentiation; the latter has been attributed to loss of all remaining acinar cells by apoptosis and tubular complex formation solely to duct and duct cell proliferation and condensation (Pour 1988; Walker et al. 1992; Watanabe et al. 1995; Wada et al. 1997; Kelly et al. 1999). While it remains possible that duct cell proliferation caused some actual increase in duct-derived tubular complexes, no incontrovertible evidence of acinar to duct transformation to account for any such increase was found.
Although not studied in detail, the observed duct-like cell mitoses in tubular complexes at 2 and 4 days could allow partial replacement of acinar cells by duct cells or reflect attempted regeneration resulting in newly formed islet and acinar cells or apoptosis if regeneration is abortive. Possible actual increases in single islet cells at 4 days suggested duct to islet cell differentiation, also seen in other forms of pancreatic injury (Walker et al. 1992; Bonner-Weir et al. 1993; Wang et al. 1995; Rosenberg et al. 1996). During early regeneration, mitosis of newly formed (or residual) acinar cells resulted in small groupings of acinar cells more consistently than asynchronous apoptosis of the same cells during involution (Figs 2e, 2f and 3). The sequence of events, differences in the appearances and arrangement of acinar cells at 2 and 4 days, and, at 4 days, identification of intermediate duct-acinar cells ultrastructurally (Figure 4f) and immunohistochemically (Figure 3), suggest duct cells may be the cellular source for acinar cell regeneration. The main increase in acinar cell number, however, would appear to result from division of newly formed acinar cells. Similar transitional cells have previously been identified in caerulein-induced pancreatitis (Adler et al. 1979) but duct cells could not be confirmed as the source of acinar cell regeneration kineticly (Elsässer et al. 1986). Duct cells, however, have been demonstrated to give rise to acinar cells in pancreas after 90% pancreatectomy (Bonner-Weir et al. 1993) and in parotid after duct ligation (Takahashi et al. 1998).
The origin of apoptotic bodies in tubular complexes at 4 days was uncertain. Their number appeared inversely proportional to the number of acinar cells present, thus the bodies may result from early but temporarily abortive regeneration, in which early differentiating duct cells die by apoptosis after loss of bcl-2. A small proportion only displayed exocrine cell markers while islet cell markers were not examined. Despite a relatively high rate of apoptosis, tubular complexes were not rapidly depleted, presumably because there was relative balance between mitosis and apoptosis. A similar high rate of apoptosis in residual pancreatic ducts (tubular complexes) after apoptotic acinar cell deletion is seen in other models of pancreatic disease (Walker et al. 1992; Watanabe et al. 1995; Doi et al. 1997; Kelly et al. 1999) where lining cells proliferate but acinar cell regeneration does not occur; in these studies, ducts are slowly but incompletely deleted over a period of weeks and months.
Prior to an appreciation of apoptotic acinar cell death, its extent, and rapidity of dead cell removal during involution of exocrine glands (Walker 1987; Walker & Gobé 1987), duct-like structures and absence of recognizable acinar cells in the atrophic glands has usually, but not always (Pour 1988), been explained by dedifferentiation or redifferentiation of acinar cells to ‘rest’ cells with a duct cell-like phenotype (Iovanna 1996; Bockman 1997; Takahashi et al. 1998); subsequent acinar cell regeneration has been explained on the basis of redifferentiation of the rest cells to acinar cells. Here, we have demonstrated that during the evolution of caerulein-induced pancreatitis, there is acinar cell apoptosis at a rate sufficient to delete all acinar cells in a few days and that persistent ducts, resistant to apoptotic deletion, are the basis of transitory tubular complexes. Lining cells with acinar cell differentiation in the latter are likely to represent acinar cells awaiting apoptotic deletion or, during the regenerative phase, newly formed cells derived from duct or other newly formed acinar cells. Such a hypothesis has major implications for the source of acinar cell regeneration in the pancreas and is worthy of further study.
References
- Adler G, Hupp T, Kern HF. Course and spontaneous regression of acute pancreatitis in the rat. Virchows Arch. A. Pathol. Anat. 1979;382:31–47. doi: 10.1007/BF01102739. [DOI] [PubMed] [Google Scholar]
- Ansari B, Coates PJ, Greenstein BD, Hall PA. In situ end-labelling detects DNA strand breaks in apoptosis and other physiological and pathological states. J. Pathol. 1993;170:1–8. doi: 10.1002/path.1711700102. [DOI] [PubMed] [Google Scholar]
- Barres BA, Hart IK, Coles HS, et al. Cell death and control of cell survival in the oligodendrocyte lineage. Cell. 1992;70:31–46. doi: 10.1016/0092-8674(92)90531-g. [DOI] [PubMed] [Google Scholar]
- Bendayan M. Concentration of amylase along its secretory pathway in the pancreatic acinar cell as revealed by high resolution immunocytochemistry. Histochem. J. 1984;16:85–108. doi: 10.1007/BF01003438. [DOI] [PubMed] [Google Scholar]
- Bockman DE. Morphology of the exocrine pancreas related to pancreatitis. Microsc. Res. Technical. 1997;37:509–519. doi: 10.1002/(SICI)1097-0029(19970601)37:5/6<509::AID-JEMT13>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Bonner-Weir S, Baxter LA, Schuppin GT, Smith FE. A second pathway for regeneration of adult exocrine and endocrine pancreas. A possible recapitulation of embryonic development. Diabetes. 1993;42:1715–1720. doi: 10.2337/diab.42.12.1715. [DOI] [PubMed] [Google Scholar]
- Bouwens L, Braet F, Heimberg H. Identification of rat pancreatic duct cells by their expression of cytokeratins 7, 19, and 20 in vivo and after isolation and culture. J. Histochem. Cytochem. 1995;43:245–253. doi: 10.1177/43.3.7532655. [DOI] [PubMed] [Google Scholar]
- Bouwens L, De Blay E. Islet morphogenesis and stem cell markers in rat pancreas. J. Histochem. Cytochem. 1996;44:947–951. doi: 10.1177/44.9.8773559. [DOI] [PubMed] [Google Scholar]
- Coles HS, Burne JF, Raff MC. Large-scale normal cell death in the developing rat kidney and its reduction by epidermal growth factor. Development. 1993;118:777–784. doi: 10.1242/dev.118.3.777. [DOI] [PubMed] [Google Scholar]
- Doi R, Wada M, Hosotani R, et al. Role of apoptosis in duct obstruction-induced pancreatic involution in rats. Pancreas. 1997;14:39–46. doi: 10.1097/00006676-199701000-00007. [DOI] [PubMed] [Google Scholar]
- Elsässer HP, Adler G, Kern HF. Time course and cellular source of pancreatic regeneration following acute pancreatitis in the rat. Pancreas. 1986;1:421–429. doi: 10.1097/00006676-198609000-00006. [DOI] [PubMed] [Google Scholar]
- Elsässer HP, Adler G, Kern HF. Fibroblast structure and function during regeneration from hormone-induced acute pancreatitis in the rat. Pancreas. 1989;4:169–178. doi: 10.1097/00006676-198904000-00005. [DOI] [PubMed] [Google Scholar]
- Fujimoto K, Hosotani R, Doi R, et al. Role of neutrophils in cerulein-induced pancreatitis in rats: possible involvement of apoptosis. Digestion. 1997;58:421–430. doi: 10.1159/000201478. [DOI] [PubMed] [Google Scholar]
- Githens S. The pancreatic duct cell: proliferative capabilities, specific characteristics, metaplasia, isolation, and culture. J. Pediatr. Gastroenterol. Nutr. 1988;7:486–506. [PubMed] [Google Scholar]
- Gobé GC, Axelsen RA. Genesis of renal tubular atrophy in experimental hydronephrosis in the rat. Lab. Invest. 1987;56:273–281. [PubMed] [Google Scholar]
- Gorelick FS, Adler G, Kern HF. Cerulein-induced pancreatitis. In: Go VLW, et al., editors. The Pancreas: Biology, Pathobiology and Disease. New York: Raven Press Ltd.; 1993. pp. 501–526. [Google Scholar]
- Grasl-Kraupp B, Ruttkay Nedecky B, Koudelka H, Bukowska K, Bursch W, Schulte Hermann R. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology. 1995;21:1465–1468. doi: 10.1002/hep.1840210534. [DOI] [PubMed] [Google Scholar]
- Gukovskaya AS, Perkins P, Zaninovic V, et al. Mechanisms of cell death after pancreatic duct obstruction in the opossum and the rat. Gastroenterology. 1996;110:875–884. doi: 10.1053/gast.1996.v110.pm8608898. [DOI] [PubMed] [Google Scholar]
- Gukovskaya AS, Gukovsky I, Zaninovic V, Song M, Sandoval D, Gukosvsky S. Pancreatic acinar cells produce, release and respond to tumor necrosis factor-β. Roles in regulating cell death and pancreatitis. J. Clin. Invest. 1997;100:1853–1862. doi: 10.1172/JCI119714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahm KB, Kim JH, You BM, et al. Induction of apoptosis with an extract of Artemisia asiatica attenuates the severity of cerulein-induced pancreatitis in rats. Pancreas. 1998;17:153–157. doi: 10.1097/00006676-199808000-00007. [DOI] [PubMed] [Google Scholar]
- Howie SE, Harrison DJ, Wyllie AH. Lymphocyte apoptosis — mechanisms and implications in disease. Immunol. Rev. 1994;142:141–156. doi: 10.1111/j.1600-065x.1994.tb00887.x. [DOI] [PubMed] [Google Scholar]
- Ide H, Yeldandi AV, Reddy JK, Rao MS. Increased expression of sulfated glycoprotein-2 and DNA fragmentation in the pancreas of copper-deficient rats. Toxicol. Appl. Pharmacol. 1994;126:174–177. doi: 10.1006/taap.1994.1104. [DOI] [PubMed] [Google Scholar]
- Iovanna JL. Redifferentiation and apoptosis of pancreatic cells during acute pancreatitis. Int. J. Pancreatol. 1996;20:77–84. doi: 10.1007/BF02825505. [DOI] [PubMed] [Google Scholar]
- Jurkowska G, Grondin G, Masse S, Morisset J. Soybean trypsin inhibitor and cerulein accelerate recovery of cerulein-induced pancreatitis in rats. Gastroenterology. 1992;102:550–562. doi: 10.1016/0016-5085(92)90103-6. [DOI] [PubMed] [Google Scholar]
- Kaiser AM, Saluja AK, Sengupta A, Saluja M, Steer ML. Relationship between severity, necrosis, and apoptosis in five models of experimental acute pancreatitis. Am. J. Physiol. 1995;269:C1295–C1304. doi: 10.1152/ajpcell.1995.269.5.C1295. [DOI] [PubMed] [Google Scholar]
- Kelly L, Reid L, Walker NI. Massive acinar cell apoptosis with secondary necrosis, origin of ducts in atrophic lobules and failure to regenerate in cyanohydroxybutene pancreatophy in rats. Int. J. Exp. Pathol. 1999:217–226. doi: 10.1046/j.1365-2613.1999.00117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, Shimosegawa T, Sasano H, et al. Endogenous glucocorticoids decrease the acinar cell sensitivity to apoptosis during cerulein pancreatitis in rats. Gastroenterology. 1998a;114:372–381. doi: 10.1016/s0016-5085(98)70490-1. [DOI] [PubMed] [Google Scholar]
- Kimura K, Shimosegawa T, Abe R, et al. Low doses of lipopolysaccharide upregulate acinar cell apoptosis in cerulein pancreatitis. Pancreas. 1998b;17:120–126. doi: 10.1097/00006676-199808000-00002. [DOI] [PubMed] [Google Scholar]
- Kishimoto S, Iwamoto S, Masutani S, et al. Apoptosis of acinar cells in the pancreas of rats fed on a copper-depleted diet. Exp. Toxicol. Pathol. 1994;45:489–495. doi: 10.1016/S0940-2993(11)80510-5. [DOI] [PubMed] [Google Scholar]
- Lampel M, Kern HF. Acute interstitial pancreatitis in the rat induced by excessive doses of a pancreatic secretagogue. Virchows. Arch. A. Pathol. Anat. 1977;373:97–117. doi: 10.1007/BF00432156. [DOI] [PubMed] [Google Scholar]
- Lu QL, Poulsom R, Wong L, Hanby AM. Bcl-2 expression in adult and embryonic non-haematopoietic tissues. J. Pathol. 1993;169:431–437. doi: 10.1002/path.1711690408. [DOI] [PubMed] [Google Scholar]
- Pour PM. Mechanism of pseudoductular (tubular) formation during pancreatic carcinogenesis in the hamster model. An electron-microscopic and immunohistochemical study. Am. J. Pathol. 1988;130:335–344. [PMC free article] [PubMed] [Google Scholar]
- Rosenberg L, Rafaeloff R, Clas D, et al. Induction of islet cell differentiation and new islet formation in the hamster. Further support for a ductular origin. Pancreas. 1996;13:38–46. doi: 10.1097/00006676-199607000-00005. [DOI] [PubMed] [Google Scholar]
- Sandoval D, Gukovskaya A, Reavey P, et al. The role of neutrophils and platelet-activating factor in mediating experimental pancreatitis. Gastroenterology. 1996;111:1081–1091. doi: 10.1016/s0016-5085(96)70077-x. [DOI] [PubMed] [Google Scholar]
- Schüssler MH, Skoudy A, Ramaekers F, Real FX. Intermediate filaments as differentiation markers of normal pancreas and pancreas cancer. Am. J. Pathol. 1992;140:559–568. [PMC free article] [PubMed] [Google Scholar]
- Sun T, Tseng SC, Huang AJ-W, et al. Monoclonal antibody studies of mammalian epithelial keratins. A Rev. Ann. New York Acad. Sci. 1985;455:307–329. doi: 10.1111/j.1749-6632.1985.tb50419.x. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Schoch E, Walker NI. Origin of acinar cell regeneration after atrophy of the rat parotid induced by duct obstruction. Int. J. Exp. Pathol. 1998;79:293–301. doi: 10.1046/j.1365-2613.1998.710405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada M, Doi R, Hosotani R, et al. Expression of Bcl-2 and PCNA in duct cells after pancreatic duct ligation in rats. Pancreas. 1997;15:176–182. doi: 10.1097/00006676-199708000-00010. [DOI] [PubMed] [Google Scholar]
- Wada M, Doi R, Hosotani R, Lee JU, Imamura M. Apoptosis of acinar cells in rat pancreatic duct ligation. J. Gastroenterol. 1995;30:813–814. doi: 10.1007/BF02349655. [DOI] [PubMed] [Google Scholar]
- Walker NI. Ultrastructure of the rat pancreas after experimental duct ligation. I. The role of apoptosis and intraepithelial macrophages in acinar cell deletion. Am. J. Pathol. 1987;126:439–451. [PMC free article] [PubMed] [Google Scholar]
- Walker NI, Gobé GC. Cell death and cell proliferation during atrophy of the rat parotid gland induced by duct obstruction. J. Pathol. 1987;153:333–344. doi: 10.1002/path.1711530407. [DOI] [PubMed] [Google Scholar]
- Walker NI, Winterford CM, Kerr JF. Ultrastructure of the rat pancreas after experimental duct ligation. II. Duct and stromal cell proliferation, differentiation, and deletion. Pancreas. 1992;7:420–434. doi: 10.1097/00006676-199207000-00002. [DOI] [PubMed] [Google Scholar]
- Walker NI, Winterford CM, Williamson RM, Kerr JF. Ethionine-induced atrophy of rat pancreas involves apoptosis of acinar cells. Pancreas. 1993;8:443–449. doi: 10.1097/00006676-199307000-00007. [DOI] [PubMed] [Google Scholar]
- Wang RN, Kloppel G, Bouwens L. Duct- to islet-cell differentiation and islet growth in the pancreas of duct-ligated adult rats. Diabetologia. 1995;38:1405–1411. doi: 10.1007/BF00400600. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Abe K, Anbo Y, Katoh H. Changes in the mouse exocrine pancreas after pancreatic duct ligation: a qualitative and quantitative histological study. Arch. Histol. Cytol. 1995;58:365–374. doi: 10.1679/aohc.58.365. [DOI] [PubMed] [Google Scholar]