

Abstract
Cardiotrophin-1 (CT-1) is a member of the IL-6 family of cytokines which was originally discovered as a factor which can induce hypertrophy of cardiac myocytes both in vitro and in vivo. Subsequently, CT-1 has been shown to have a wide variety of different effects on cardiac and non cardiac cells including the ability to stimulate the survival of both cardiac and neuronal cells. Interestingly, whilst activation of the p42/p44 MAP kinase pathway is necessary for the survival promoting effects of CT-1 in cardiac cells, it is not required for its hypertrophic effect which is likely to involve activation of the Jak/STAT-3 pathway. CT-1 may therefore be of use as a novel cardio-protective agent, particularly if its hypertrophic effect can be specifically inhibited.
Keywords: cardiotrophin-1, IL-6, cytokines, cardioprotection, hypertrophy
Cardiotrophin-1 (CT-1) was originally identified in a screen based on the finding that conditioned medium from differentiated embryoid bodies derived from mouse embryonic stem cells, was able to induce a hypertrophic response in neonatal cardiac myocytes. The subsequent screening of an expression cDNA library prepared from such embryoid bodies for the ability to induce hyper-trophy resulted in the identification of two clones encoding CT-1 (Pennica et al. 1995a). No DNA sequence matching these cDNA clones was identified by data base searches indicating that a novel factor capable of inducing hypertrophy had been isolated. In the four years which have elapsed since the original report of the isolation of CT-1, a number of studies of this factor have appeared. These have indicated that as well as being a hypertrophic factor, CT-1 also appears to be able to protect cardiac cells from damaging stimuli as well as to have a variety of other effects on cardiac and other cell types. The purpose of this review is to summarize these studies and to provide an overview of this factor which appears to be of critical importance in the normal functioning of the cardiovascular system and in cardiovascular pathology. A comprehensive review of the very early work on this factor in the first year following its isolation has previously been published (Pennica et al. 1996c).
CT-1 is a member of the IL-6 family. In the initial report of the cloning of CT-1 (Pennica et al. 1995a) it was shown that CT-1 showed homology to members of the interleukin-6 family of cytokines and was clearly a member of this family which includes a number of other factors such as leukamia inhibitory factor (LIF), ciliary neurotrophic factor (CNTF), oncostatin M and IL-11 (for review see Kishimoto et al. 1994,1995). The fact that CT-1 was a member of this family was subsequently confirmed by studies of its receptor (Pennica et al. 1995b). Thus, like all members of the IL-6 family, the CT-1 receptor contains a common protein chain known as gp130. Although the IL-6, CNTF and oncostatin M receptors contain in addition a specific subunit which is unique to each of these receptors (Kishimoto et al. 1994, 1995), LIF and CT-1 and in certain cells, oncostatin M share a common second receptor component which is known as LIF receptor subunit β (Pennica et al. 1995b; Wollert et al. 1996). In addition, evidence has also been presented for an additional third receptor component which may be necessary for high affinity CT-1 binding (Robledo et al. 1997).
As with all members of the IL-6 family, binding of CT-1 to its receptor sets off a cascade of signalling processes (for review see Hirano et al. 1997). These signalling pathways result in the activation of at least two types of cellular transcription factors which can then activate the expression of specific target genes. Thus, following binding of CT-1 to its receptor, activation of the p42/p44 mitogen activated protein kinase (MAPK) enzymes is observed (Sheng et al. 1997) and this results in the threonine phosphorylation of the NF-IL6 (C/EBPβ) transcription factor allowing it to activate gene transcription (Nakajima et al. 1993). Similarly, activation of Jak tyrosine kinases results in the tyrosine phosphorylation of the STAT-3 transcription factor (Sheng et al. 1997) resulting in its dimerization and transport to the nucleus where it can activate its target genes (Wegenka et al. 1993; Akira et al. 1994; for review see Horvath & Darnell 1997). It is likely that CT-1 achieves its effect via a combination of these two signalling pathways and it will be of major importance to disect which pathway(s) is of major importance in each particular effect of CT-1 (see below).
Expression of CT-1
In the adult mouse (Pennica et al. 1995a) and human (Pennica et al. 1996b) expression of CT-1 is not confined to the heart but is found in a variety of different tissues. Interestingly however, during mouse embryonic development CT-1 is first detectable in the primitive heart tube at day 8.5 whereas other tissues do not show significant expression at this time (Sheng et al. 1996). CT-1 is expressed in atrial and ventricular muscle of the developing heart but not in endocardium. Although the developing heart remains the predominant site of CT-1 expression until E10.5, at later developmental stages other organs begin to display expression of CT-1 although expression remains high in the heart (Sheng et al. 1996). The possibility that CT-1 may play an important role in cardiac development is supported by the observation that knock out mice lacking the gp130 component of the CT-1 receptor show hypoplastic ventricular myocardium leading to death in utero (Yoshida et al. 1996). This effect is likely to be dependent upon the loss of the CT-1 response rather than a loss of the response to another cytokine binding to this receptor since CT-1 is the only cytokine which is preferentially expressed in cardiac muscle cells at an early stage of development.
As well as its normal expression in the heart, CT-1 expression has also been shown to be upregulated in a variety of pathological states where it may contribute to ongoing disease process. Thus, for example, augmented expression of CT-1 has been detected in the ventricle of genetically hypertensive rats where it may contribute to the ongoing hypertrophic response (Ishikawa et al. 1996). Similarly, overexpression of both CT-1 and gp130 have been reported in the rat ventricle following myocardial infarction and during experimental acute Chagasic cardiomyopathy (Chandrasekar et al. 1998).
CT-1 induces cardiac myocyte hypertrophy
Although CT-1 is expressed in the normal developing and adult heart, as described above, it was first isolated as a factor capable of inducing cardiac myocyte hypertrophy. Thus, the original report on CT-1 (Pennica et al. 1995a) showed that CT-1 was a potent inducer of hypertrophy with activity being detected at concentrations of 0.1 nm or lower and the factor being more potent than other members of the IL-6 family in terms of inducing hypertrophy. As well as cell enlargement, the factor was also active in two other assays of hypertrophy namely the organization of myosin light chain 2 into sarcomeric units and the induction of atrial natriuretc peptide (ANP) secretion.
In subsequent in vitro studies (Wollert et al. 1996) it was observed that the hypertrophy induced by CT-1 was distinct from that induced, for example, by α–adrenergic stimulation both in terms of cell morphology and gene expression pattern. Thus, stimulation with CT-1 leads to an increase in cardiac cell size that is caused by an increase in cell length without a significant change in cell width. Similarly, CT-1 stimulated cells show the assembly of sarcomeric units in series rather than in parallel as is observed with α–adrenergic stimulation.
This distinct pattern of CT-1 — induced hypertrophy also applies to the pattern of gene expression which is induced during this hypertrophic response. Thus, whilst both CT-1 and phenylephrine stimulate increased ANP gene expression, only phenylephrine induces skeletal α–actin and myosin light chain-2 synthesis (Wollert et al. 1996). A comparison of these patterns of gene expression with those which occur in hypertrophy in vivo, reveals that the induction of all three genes which occurs following α–adrenergic stimulation is also characteristic of pressure overload-induced hypertrophy. In contrast, the selective induction of ANP gene expression without induction of skeletal α–actin occurs during volume overload-induced hypertrophy. Similarly, the increased addition of sarcomeric units in series rather than in parallel which is observed with CT-1 is also observed in cardiac myocytes isolated from hearts subjected to chronic volume overload. These findings led Wollert et al. (1996) to conclude that CT-1 — induced hypertrophy is closely related to the volume-overload hypertrophy which occurs during valvular insufficiency and results in irreversible loss of cardiac function in humans.
These in vitro studies suggesting a critical role for CT-1 in cardiac hypertrophy have now been supplemented by an in vivo study in which CT-1 was administered to intact mice by intraperitoneal injection (Jin et al. 1996). In this study, a dose dependent increase in both heart weight and ventricular weight was observed in the mice treated with CT-1, although total body weight was unaffected. Hence, CT-1 can clearly induce cardiac hypertrophy in vivo. Interestingly, previous studies showed that chronic stimulation of the gp130 receptor via the artificial overexpression of IL-6 and its soluble receptor chain, results in cardiac hypertrophy in transgenic mice (Hirota et al. 1995). Hence, chronic stimulation of the gp130 receptor can induce hypertrophy, mimicking the effect of CT-1.
Although a complete understanding of the role of CT-1 in normal cardiac development and hypertrophy must await the preparation of a CT-1 knock out mouse, it is possible to link the above results into a simple framework. Thus, CT-1 is likely to play a key role in the normal development of the heart and in particular in ensuring that the myocardium develops to a normal thickness. Hence, as described above, deletion of the gp130 receptor for CT-1 results in hypoplastic ventricular myocardium (Yoshida et al. 1996). Although, the continued expression of CT-1 in the adult heart suggests that it has an important role in normal cardiac function, clearly its abnormal expression may induce hypertrophy as occurs in hypertensive rats (Ishikawa et al. 1996) and potentially in humans, mimicking the abnormal effects which occur when the gp130 receptor is chronically stimulated (Hirota et al. 1995).
Cardioprotective effect of CT-1
As noted above, the continued expression of CT-1 in the adult heart suggests that it continues to have some function following the completion of cardiac development. One possibility in this regard, is that CT-1 has a cardioprotective effect. Thus, Sheng et al. (1996) observed that treatment with CT-1 was able to enhance the survival of neonatal rat cardiac myocytes cultured in serum free medium. Moreover, we showed (Stephanou et al. 1998) that pretreatment with CT-1 was able to protect cultured neonatal cardiac myocytes against subsequent exposure to either elevated temperature (heat shock) or simulated ischaemia/hypoxia. (Figure 1). This effect was associated with the ability of CT-1 to induce enhanced levels of the heat shock protein hsp70 and hsp90 whose overexpression had previously been shown to protect cardiac myocytes against both thermal and ischaemic stress (Heads et al. 1994, 1995; Cumming et al. 1996).
Figure 1.
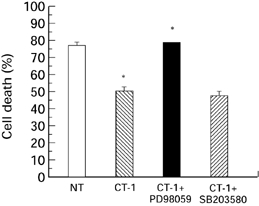
CT-1 was cultured with cardiac myocytes for 30min under normoxic conditions in the absence ( ) and presence of 50μm PD98059 (▪) or 10μm SB203580 (
) and presence of 50μm PD98059 (▪) or 10μm SB203580 ( ). No treatment (□). The inhibitors were added to the cells 30 min before the addition of CT-1. The CT-1 and inhibitors were removed from the cells which were then exposed to a 6-h simulated lethal hypoxic/ischaemic stress. The cells were harvested and cell death was assessed by trypan blue exclusion. *P < 0.05.
). No treatment (□). The inhibitors were added to the cells 30 min before the addition of CT-1. The CT-1 and inhibitors were removed from the cells which were then exposed to a 6-h simulated lethal hypoxic/ischaemic stress. The cells were harvested and cell death was assessed by trypan blue exclusion. *P < 0.05.
This finding that CT-1 protected cardiac cells against ischaemia in vitro raised the possibility that this factor might be used to minimize the effect of ischaemic damage in the human heart. This possibility was supported by the finding that CT-1 appears to exert its survival promoting effects by minimizing the degree of programmed cell death (apoptosis) which is induced by serum removal (Sheng et al. 1997) or by thermal and ischaemic stress (Stephanou et al. 1998; Brar et al. unpublished observation). In these different studies, the protective effect of CT-1 against apoptotic death was observed in a variety of assays for apoptosis including DNA gel electrophoresis (Sheng et al. 1997) TUNEL labelling (Sheng et al. 1997; Stephanou et al. 1998) FACS analysis (Stephanou et al. 1998) and surface binding of annexin V (Brar et al. unpublished observation) which unlike the other assays does not measure DNA fragmentation but rather the translocation of phosphatidyl serine to the outside of the cell membrane which is an early event of apoptosis (Figure 2).
Figure 2.
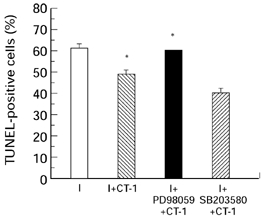
Percentage of TUNEL positive cells in the cardiac myocytes treated with CT-1 and the various signal transduction inhibitors 30 min prior to exposure to simulated lethal hypoxia/ischaemia (I). I + CT-1 ( ); I + CT-1 in the presence of 50μm PD98059 (▪) or 10μm SB203580 (
); I + CT-1 in the presence of 50μm PD98059 (▪) or 10μm SB203580 ( ). Untreated cardiac myocytes (□). The cells were fixed using 4% paraformaldehyde and the percentage of TUNEL positive nuclei were analysed using fluorescein-12 dUTP and terminal transferase. Values are the means of three determinations whose S.E.M. is shown by the bars. *P < 0.05.
). Untreated cardiac myocytes (□). The cells were fixed using 4% paraformaldehyde and the percentage of TUNEL positive nuclei were analysed using fluorescein-12 dUTP and terminal transferase. Values are the means of three determinations whose S.E.M. is shown by the bars. *P < 0.05.
These findings are of particular importance in that a number of studies have suggested that apoptotic cell death may be of particular importance following myocardial infarction in both animal models (Cheng et al. 1996; Kajstura et al. 1996) and humans (Olivetti et al. 1994,1997). Interestingly, in several studies, such apoptotic cell death of cardiac myocytes has been observed during reperfusion following the ischaemic injury (Gottlieb et al. 1994; Umansky et al. 1995). We therefore tested whether CT-1 was able to protect cardiac myocytes against the damaging effect of ischaemia/reperfusion when added to the cells in vitro after the ischaemic period at the time of reoxygenation. In these experiments (Figure 3) we were able to observe a clear protective effect of CT-1 when added after the ischaemic period at the onset of reoxygenation (Brar et al. unpublished observation). This effect was observed both in assays of total cell death (Figure 3) and in assays specifically measuring apoptotic cell death such as TUNEL labelling or annexin V staining.
Figure 3.
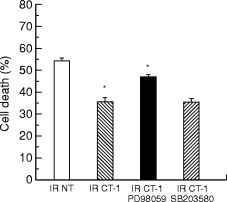
Cardiac myocytes were exposed to simulated lethal hypoxic/ischaemic injury for 6 h. Immediately after this period, the stimulated ischaemic buffer was removed and replaced with normal growth medium supplemented with CT-1 (2 ng/ml) only ( ) and in the presence of 50μm PD98059 (▪) or 10μm SB203580 (
) and in the presence of 50μm PD98059 (▪) or 10μm SB203580 ( ). IR NT, cardiac myocytes reoxygenated in the absence of CT-1 (□). The inhibitors were added to the cells 10 min before the addition of CT-1. The cells were incubated under normoxic, oxygenated conditions for 2 h. Subsequent to this period the cells were harvested and cell death following ischaemia/reoxygenation (IR) was assessed by trypan blue exclusion. *P < 0.05.
). IR NT, cardiac myocytes reoxygenated in the absence of CT-1 (□). The inhibitors were added to the cells 10 min before the addition of CT-1. The cells were incubated under normoxic, oxygenated conditions for 2 h. Subsequent to this period the cells were harvested and cell death following ischaemia/reoxygenation (IR) was assessed by trypan blue exclusion. *P < 0.05.
These studies therefore establish that CT-1 can have a protective effect in cultured cardiac myocytes in vitro regardless of whether it is added prior to the period of ischaemia or at the period of reperfusion. Although these observations suggest a potential therapeutic role for CT-1, it is obviously necessary to test whether CT-1 has a protective effect in the intact heart. For this reason we tested the protective effect of CT-1 against an ischaemic episode in the isolated rat heart on a Langendorff perfusion aparatus. In these experiments, we were able to demonstrate a protective effect of CT-1 (as measured by a reduction in the infarct size to zone at risk ratio) when CT-1 was added either prior to or during the ischaemic episode. Most importantly, we were also able to demonstrate such a protective effect when CT-1 was added at the time of reperfusion (Brar et al. unpublished observation).
Although it is obviously necessary to repeat these experiments in the intact heart in vivo our experiments represent the first demonstration that CT-1 can have a protective effect in the isolated heart as opposed to cultured cardiac myocytes and demonstrate that it is possible to produce a protective effect when the CT-1 is added at the time of reperfusion as would be necessary in a therapeutic situation.
Signalling pathways in CT-1 induced hypertrophy and protection
Clearly, any therapeutic use of CT-1 would be greatly facilitated if its protective effect could be retained whilst eliminating the potentially damaging hypertrophic effect which was the original cause of CT-1 being identified. Progress towards this end would be greatly facilitated by an understanding of the signalling pathways which are involved, respectively, in the ability of CT-1 to stimulate hypertrophy and to produce protection. Most interestingly, there is evidence that of the two signalling pathways for CT-1 described above, one is used predominantly to induce hypertrophy whilst the other is involved in the protective effect. Thus, Sheng et al. 1997 showed that PD98059, an inhibitor of the p42/p44 MAPK pathway was able to block the protective effect of CT-1 whilst having no effect on its ability to induce hypertrophy. Similarly, a dominant negative mutant of the MAP kinase kinase 1 which is an upstream activator of p42/p44 MAPK was similarly able to block the protective effect of CT-1 without affecting the ability to induce hypertrophy.
Although all these experiments are based on the serum withdrawal model, we have obtained similar results in cardiac cells exposed to ischaemia. Thus, the p42/p44 MAPK inhibitor PD98059 was able to block the protective effect of CT-1 on cardiac cells in vitro regardless of whether the CT-1 was added prior to the ischaemic period (Figure 1 and 2) or after this period at the time of reperfusion (Figure 3). In contrast, a specific inhibitor of the p38 MAPK pathway SB203580 was unable to produce a significant inhibitory effect on the CT-1 protection in either situation (Figure 1–3). A similar effect was also observed in the ex vivo experiments with the intact heart where PD98059 was able to block the effect of CT-1 added prior to ischaemia or after ischaemia at the time of reperfusion (Brar et al. unpublished observation).
These experiments therefore indicate that the protective effect of CT-1 is likely to be dependent upon its ability to activate the p42/p44 MAPK pathway. Interestingly, we have previously shown that the NF-IL6 transcription factor which is activated by the p42/p44 pathway (Nakajima et al. 1993) is able to stimulate the synthesis of the heat shock proteins (Stephanou et al. 1997). It is therefore possible that the activation of p42/p44 MAPK by CT-1 leads to the activation of NF-IL6 which in turn induces the synthesis of the protective heat shock proteins hsp70 and hsp90 which we previously observed in our studies of CT-1 (Stephanou et al. 1998). However, more recent data from our laboratory (Brar et al. unpublished observation) have suggested that CT-1 can have a protective effect even in the absence of new protein synthesis. It is therefore possible that the protective effect of p42/p44 MAPK activation is achieved via direct phosphorylation of a protective protein, rather than the phosphorylation of a transcription factor which then stimulates de novo gene transcription. One obvious candidate in this regard is the 27 kD heat shock protein (hsp27) which has been shown to be phosphorylated following exposure to stressful stimuli with such phosphorylation enhancing its protective effect against such stimuli (Martin et al. 1997). Whatever the precise nature of the phosphorylated protein which mediates the protective effect of CT-1, it is clear that this protective effect involves the ability of CT-1 to activate the p42/p44 MAPK pathway. In contrast, the phosphorylation of the STAT-3 transcription factor was entirely unaffected by inhibition of the MAPK pathway and is therefore likely to mediate the hypertrophic effect of CT-1 which was similarly unaffected by inhibition of this pathway (Sheng et al. 1997).
These findings therefore suggest that the two signalling pathways activated by CT-1 may mediate, respectively, its protective and its hypertrophic effects. These findings therefore offer the potential that in future it may be possible to either design analogues of CT-1 which only stimulate the protective pathway or to administer CT-1 together with a specific inhibitor of the hypertrophic pathway. Further studies are clearly required in order to develop these possibilities and hence take advantage of the cardioprotective effect of CT-1.
Other effects of CT-1
Effects on the heart
Although the hypertrophic and cardioprotective effects of CT-1 have been extensively characterized, other effects of CT-1 on the heart have also been documented. Thus, it has been shown (Jin et al. 1998) that intravenous administration of CT-1 to rats results in significantly elevated cardiac output and heart rate as well as a decreased mean arterial pressure and systemic vascular resistance. As no alteration was observed in stroke volume, it is likely that the increased cardiac output was secondary to the increased heart rate. Interestingly both the depressor effect of CT-1 on blood pressure and the tachycardic effect could be significantly reduced by treatment with a nitric oxide synthase inhibitor suggesting that the haemodynamic effects of CT-1 may be mediated by nitric oxide. It is evidently possible that these effects of CT-1 may improve cardiac function via a reduction in afterload adding to the potentially beneficial effects of CT-1.
Interestingly, CT-1 has also been shown to reduce the serum and heart levels of tumour necrosis factor (TNF) in mice treated with lipopolysaccharide (Benigni et al. 1996b). Again this effect may be beneficial since TNF has been implicated as a pathogenic mediator in a number of different diseases with elevated levels being observed in patients with myocardial infarction or chronic heart failure and having been shown to correlate with the severity of the disease (Latini et al. 1994). In this case however, it is unlikely that CT-1 acts directly on the heart to reduce TNF levels since reduced TNF production could also be observed when CT-1 was added to mouse blood cells cultured in vitro with lipopolysaccharide (Benigni et al. 1996b).
In addition to these other in vivo effects of CT-1 on cardiac function, several genes have now been identi-fied whose expression is stimulated by treatment of cardiac cells with CT-1. One of these, that encoding ANP was shown to be induced as part of the studies on the hypertrophic response to CT-1 (Pennica et al. 1995a) and has been shown to be transcriptionally induced by CT-1 treatment (Wollert et al. 1996). Similarly, brain natriuretic peptide (BNP) which is induced in response to the hypertrophic agent endothelin-1 (ET-1) is also induced by treatment with CT-1 (Kuwahara et al. 1998). Moreover, in our studies of the protective effect of CT-1, we showed that this factor is able to induce the expression of the genes encoding the protective heat shock proteins hsp70 and hsp90 (Stephanou et al. 1998).
Although a number of genes induced by CT-1 have thus been defined, it is of interest that in no case has the CT-1 response element been mapped in the gene promoter. Thus, Wollert et al. (1996) found that a 3 kilobase promoter fragment of the ANP gene conferred inducibility by phenylephrine upon the reporter gene but did not confer inducibility by CT-1. Similarly, almost two kilobases of the BNP promoter was insufficient to confer a response to CT-1, although it did confer responsiveness to ET-1 (Kuwahara et al. 1998). These findings may indicate that the response element conferring inducibility by CT-1 is not located in the immediate 5′ flanking region of the inducible gene. Alternatively, it is possible that in some cases CT-1 may induce enhanced mRNA and protein levels by post-transcriptional processes, although it should be noted that at least in the case of the ANP gene transcription regulation has apparantly been demonstrated by nuclear run on assays (Wollert et al. 1996).
Other tissues
As indicated above, the receptor for CT-1 contains the ubiquitously expressed gp130 protein and the LIF receptor subunit β which is also widely distributed. It is not surprising therefore that many of the effects that have been documented for other members of the IL-6 family, particularly IL-6 itself have also been observed for CT-1 (for review see Pennica et al. 1996c). Thus, for example, CT-1 in common with other members of the IL-6 family is able to induce the liver acute phase response (Peters et al. 1995) and to potentiate the elevation of serum cortiocosterone which is induced by IL-1 (Benigni et al. 1996a). Moreover, administration of CT-1 in vivo also stimulates the growth of the liver, kidney and spleen whilst causing atrophy of the thymus and increasing platelet and red blood cell counts (Jin et al. 1996).
Of these widespread biological effects of CT-1, perhaps those of the greatest interest concern its effects on the nervous system. Thus, CT-1 produced by differentiated muscle cells has been shown to support the long-term survival of spinal motor neurones (Pennica et al. 1996a) as well as of the dopaminergic neurones which are lost in Parkinson's disease and ciliary ganglion neurones (Pennica et al. 1995b). Although the effect on ciliary ganglion neurones can be mimicked by the CNTF member of the IL-6 family, the protective effect on dopaminergic neurones is not produced by CNTF. Therefore, CT-1 has survival promoting effects in the nervous system which are not shared by other members of the IL-6 family. Interestingly, the protective effect of CT-1 on motor neurone survival requires an additional receptor component apart from gp130 and the LIF receptor β subunit (Pennica et al. 1996c) and an additional receptor component of molecular mass 80 kD has been identified biochemically on neuronal cells (Robledo et al. 1997).
In addition to these protective effects on a variety of neuronal cells, CT-1 is also able to induce a switch in the transmitter phenotype of cultured sympathetic neurones. This results in their conversion to a cholinergic phenotype producing choline acetyl transferase and vasoactive intestinal peptide with a corresponding loss of the enzymes of catecholamine biosynthesis such as tyrosine hydroxylase (Habecker et al. 1995). These findings suggest that the effects of CT-1 in the nervous system may ultimately prove to be as important as its effect in the cardiovascular system and render of still greater importance an analysis of the phenotype of knock out mice lacking CT-1.
Conclusion
A considerable amount of information has accumulated in the four years since the existence of CT-1 was reported. These studies have indicated a wide range of activities of CT-1 on a number of different cell types and organ systems. This pleiotropic activity of CT-1 indicates that it could equally well have been discovered in several different biological systems other than on the basis of its effect on cardiac cell hypertrophy.
Nonetheless the effect on cardiac hypertrophy remains the most extensively analysed action of CT-1, along with its subsequently discovered cardioprotective effect. Both these effects of CT-1 on cardiac cells have now also been demonstrated in the intact heart and it appears that they arise from two distinct signalling pathways induced by CT-1, one of which is important for hypertrophy whilst the other is of importance for cardioprotection. These findings offer the possibility that CT-1 may make the transition from being discovered as an agent which induces the pathological hypertrophic response to being of ultimate therapeutic use in the treatment of ischaemic damage in the heart.
Acknowledgments
I am most grateful to the members of my laboratory working in the area of CT-1 and in particular to Anastasis Stephanou, Bervan Brar and Julia Sargeant for performing the work from our laboratory described in this review. I am also most grateful to Diane Pennica and Genetech Inc for the generous supply of CT-1. The work in my laboratory on CT-1 which is described in this review has been most generously supported from its inception by the British Heart Foundation.
References
- Akira S, Nishio Y, Inoue M, et al. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91 related transcription factor involved in the gp130 mediated signalling pathway. Cell. 1994;77:63–71. doi: 10.1016/0092-8674(94)90235-6. [DOI] [PubMed] [Google Scholar]
- Benigni F, Fantuzzi G, Sacco S, et al. Six different cytokines that share gp130 as a receptor subunit, induce serum amyloid A and potentiate the induction of the interleukin-6 and the activation of the hypothalamus-pituitary-adrenal axis by interleukin-1. Blood. 1996;87:1851–1854. [PubMed] [Google Scholar]
- Benigni F, Sacco S, Pennica D, Ghezzi P. Cardiotrophin-1 inhibits tumor necrosis factor production in the heart and serum of lipoplysaccharide-treated mice and in vitro in mouse blood cells. Am. J. Pathol. 1996;149:1847–1850. [PMC free article] [PubMed] [Google Scholar]
- Chandrasekar B, Melby PC, Pennica D, Freeman GL. Over expression of Cardiotrophin-1 and gp130 during experimental acute chagasic cardiomyopathy. Immunol. Lett. 1998;61:89–95. doi: 10.1016/s0165-2478(97)00167-3. [DOI] [PubMed] [Google Scholar]
- Cheng W, Kajstura J, Nitahara JA, et al. Programmed myocyte cell death affects the viable myocardium after infarction in rats. Exp Cell Res. 1996;226:316–327. doi: 10.1006/excr.1996.0232. [DOI] [PubMed] [Google Scholar]
- Cumming DVE, Heads RJ, Watson A, Latchman DS, Yellon DM. Differential protection of primary rat cardiocytes by transfection of specific heat stress proteins. J. Mol. Cell. Cardiol. 1996;28:2343–2349. doi: 10.1006/jmcc.1996.0227. [DOI] [PubMed] [Google Scholar]
- Gottlieb RA, Burleson KO, Kloner RA, Babior BM, Engler RL. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J. Clin. Invest. 1994;94:1621–1628. doi: 10.1172/JCI117504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habecker BA, Pennica D, Lando D. Cardiotrophin-1 is not the sweat gland-derived differentiation factor. Neuroreport. 1995;7:41–44. [PubMed] [Google Scholar]
- Heads RJ, Latchman DS, Yellon DM. Stable high level expression of a transfected human HSP70 gene protects a heart-derived muscle cell line against thermal stress. J. Mol. Cell. Cardiol. 1994;26:695–699. doi: 10.1006/jmcc.1994.1084. [DOI] [PubMed] [Google Scholar]
- Heads RJ, Yellon DM, Latchman DS. Differential cytoprotection against heat stress or hypoxia following expression of specific stress protein goes in myogenic cells. J. Mol. Cell. Cardiol. 1995;27:1669–1678. doi: 10.1016/s0022-2828(95)90722-x. [DOI] [PubMed] [Google Scholar]
- Hirano T, Nakajima K, Hibi M. Signaling mechanisms through gp130: a model of the cytokine system. Cytokine Growth Factor Rev. 1997;8:241–252. doi: 10.1016/s1359-6101(98)80005-1. [DOI] [PubMed] [Google Scholar]
- Hirota H, Yoshida K, Kishimoto T, Taga T. Continuous activation of gp130, a signal transducing receptor component for interleukin 6 related cytokines causes myocardial hypertrophy in mice. Proc. Natl. Acad. Sci. USA. 1995;92:4862–4866. doi: 10.1073/pnas.92.11.4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath CM, Darnell JE. The state of the STATs: recent developments in the study of signal transduction to the nucleus. Current Opinion Cell Biol. 1997;9:233–239. doi: 10.1016/s0955-0674(97)80067-1. [DOI] [PubMed] [Google Scholar]
- Ishikawa M, Saito Y, Miyamoto Y, et al. cDNA cloning of rat cardiotrophin-1 (CT-1): augmented expression of CT-1 gene in ventricle of genetically hypertensive rats. Biochem. Biophys. Res. Comm. 1996;219:377–381. doi: 10.1006/bbrc.1996.0241. [DOI] [PubMed] [Google Scholar]
- Jin H, Yang R, Keller GA, et al. In vivo effects of cardiotrophin-1. Cytokine. 1996;8:920–926. doi: 10.1006/cyto.1996.0123. [DOI] [PubMed] [Google Scholar]
- Jin H, Yang R, Ko A, Pennica D, Wood WI, Paoni NF. Effects of cardiotrophin-1 on haemodynamics cardiac function in conscious rats. Cytokine. 1998;10:19–25. doi: 10.1006/cyto.1997.0241. [DOI] [PubMed] [Google Scholar]
- Kajstura J, Cheng W, Reiss K, et al. Apoptotic and necrotic myocyte cell death are independent contributing variables of infarct size in rats. Lab. Invest. 1996;74:86–107. [PubMed] [Google Scholar]
- Kishimoto T, Taga T, Akira S. Cytokine signal transduction. Cell. 1994;76:253–262. doi: 10.1016/0092-8674(94)90333-6. [DOI] [PubMed] [Google Scholar]
- Kishimoto T, Akira S, Narazaki M, Taga T. Interleukin-6 family of cytokines and gp130. Blood. 1995;86:1243–1254. [PubMed] [Google Scholar]
- Kuwahara K, Saito Y, Ogawa Y, et al. Endothelin-1 and Cardiotrophin-1 induce brain natriuretic peptide gene expression by distinct transcriptional mechanisms. J. Clin. Pharmacol. 1998;31:S354–S356. doi: 10.1097/00005344-199800001-00099. [DOI] [PubMed] [Google Scholar]
- Latini R, Bianchi M, Correale E, et al. Cytokines in acute myocardial infarction: selective increase in circulating tumor necrosis factor, its soluble receptor, and interleukin-1 receptor antagonist. J. Cardiovasc. Pharmacol. 1994;23:1–6. [PubMed] [Google Scholar]
- Martin JL, Mestril R, Hilal-Dandan R, Brunton LL, Dilmann WH. Small heat-shock proteins and protection against ischaemic injury in cardiac myocytes. Circulation. 1997;96:4343–4348. doi: 10.1161/01.cir.96.12.4343. [DOI] [PubMed] [Google Scholar]
- Nakajima T, Kinoshita S, Sasagawa T, et al. Phosphorylation at threonine-235 by a ras-dependent mitogen activated protein kinase cascade is essential for transcription factor NF-IL6. Proc. Natl. Acad. Sci. USA. 1993;90:2207–2211. doi: 10.1073/pnas.90.6.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivetti G, Quani F, Sala R, et al. Acute myocardial infarction in humans is associated with activation of programmed myocyte cell death in the surviving portion of the heart. J. Mol Cellular Cardiol. 1994;28:2005–2016. doi: 10.1006/jmcc.1996.0193. [DOI] [PubMed] [Google Scholar]
- Olivetti G, Abbi R, Quani F, et al. Apoptosis in the failing human heart. New Engl. J. Med. 1997;336:1131–1141. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- Pennica D, King KL, Shaw KJ, et al. Expression cloning of cardiotrophin 1 a cytokine that induces cardiac myocyte hypertrophy. Proc. Natl. Acad. Sci. USA. 1995;92:1142–1146. doi: 10.1073/pnas.92.4.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennica D, Shaw KJ, Swanson TA, et al. Cardiotrophin-1: Biological activities and binding to the leukemia inhibitory factor receptor/gp130 signaling complex. J. Biol. Chem. 1995b;270:10915–10922. doi: 10.1074/jbc.270.18.10915. [DOI] [PubMed] [Google Scholar]
- Pennica D, Arce V, Swanson A, et al. Cardiotrophin-1 a cytokine present in embryonic muscle, supports long term survival of spinal motoneurons. Neuron. 1996a;17:63–74. doi: 10.1016/s0896-6273(00)80281-0. [DOI] [PubMed] [Google Scholar]
- Pennica D, Swanson TA, Shaw KJ, et al. Human cardiotrophin-1: protein and gene structure, biological and binding activities and chromosomal localizations. Cytokine. 1996b. pp. 183–189. [DOI] [PubMed]
- Pennica D, Wood WI, Chien KR. Cardiotrophin-1: a multifunctional cytokine that signals via LIF receptor gp130 Dependent pathways. Cytokine Growth Factor Rev. 1996c;7:81–91. doi: 10.1016/1359-6101(96)00007-x. [DOI] [PubMed] [Google Scholar]
- Peters M, Roeb E, Pennica D, Meyer zum Buschenfelde K.-H. & Rose-John S. A new hepatocyte stimulating factor: cardiotrophin-1 (CT-1) FEBS Lett. 1995;372:177–180. doi: 10.1016/0014-5793(95)00972-c. [DOI] [PubMed] [Google Scholar]
- Robledo O, Fourcin M, Chevalier S, et al. Signaling of the Cardiotrophin-1 Receptor. J. Biol. Chem. 1997;272:4855–4863. doi: 10.1074/jbc.272.8.4855. [DOI] [PubMed] [Google Scholar]
- Sheng Z, Pennica D, Wood WI, Chien KR. Cardiotrophin-1 displays early expression in the murine heart tube and promotes cardiac myocyte survival. Development. 1996;122:419–428. doi: 10.1242/dev.122.2.419. [DOI] [PubMed] [Google Scholar]
- Sheng Z, Knowlton K, Chen J, Hoshijima M, Brown H, Chein KR. Cardiotrophin 1 (CT-1) inhibition of cardiac myocyte apoptosis via a mitogen activated protein kinase dependent pathway. J. Biol. Chem. 1997;272:5783–5791. doi: 10.1074/jbc.272.9.5783. [DOI] [PubMed] [Google Scholar]
- Stephanou A, Amin V, Isenberg DA, Akira S, Kishimoto T, Latchman DS. Interleukin-6 activates heat shock protein 90 B gene expression. Biochem. J. 1997;321:103–106. doi: 10.1042/bj3210103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephanou A, Brar B, Heads R, et al. Cardiotrophin-1 induces heat shock protein accumulation in cultured cardiac cells and protects them from stressful stimuli. J. Mol. Cell. Cardiol. 1998;30:849–855. doi: 10.1006/jmcc.1998.0651. [DOI] [PubMed] [Google Scholar]
- Umansky SR, Cuenco GM, Khutzian SS, Barr PJ, Tomei LD. Post-ischemic apoptotic death of rat neonatal cardiomyocytes. Cell Death Differentiation. 1995;2:235–241. [PubMed] [Google Scholar]
- Wegenka UM, Buschmann J, Lutticken C, Heinrich PC, Horn F. Acute-phase response factor, a nuclear factor binding to acute-phase response elements, is rapidly acivated by interleukin-6 at the posttranslational level. Mol. Cell. Biol. 1993;13:276–288. doi: 10.1128/mcb.13.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollert KC, Taga T, Saito M, et al. Cardiotrophin-1 activates a distinct form of cardiac muscle cell hypertrophy. J. Biol. Chem. 1996;271:9535–9545. doi: 10.1074/jbc.271.16.9535. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Taga T, Saito M, et al. Targeted disruption of gp130, a common signal transducer for the interleukin 6 family of cytokines, leads to myocardial and hematological disorders. Proc. Natl. Acad. Sci. USA. 1996;93:407–411. doi: 10.1073/pnas.93.1.407. [DOI] [PMC free article] [PubMed] [Google Scholar]