

Abstract
We have examined the histological and ultrastructural features of CNS infection with Murray Valley encephalitis (MVE) virus in mice inoculated with a virulent parental strain (BH3479). Light microscopic examination revealed neuronal necrosis in the olfactory bulb and hippocampus of MVE-infected brains by 5 days post-infection (pi). Electron microscopy of these regions showed endoplasmic reticulum membrane proliferation, and tubular and spherical structures in the cisternae of the endoplasmic reticulum, Golgi complex and nuclear envelope. At seven to eight days pi, infected neurones exhibited chromatin condensation and extrusion, nuclear fragmentation, loss of segments of the nuclear envelope, reduced surface contact with adjacent cells and loss of cytoplasmic organelles. This cell injury was particularly noticeable in the proximal CA3 and distal CA1 regions of the hippocampus. The inflammatory cell profile consisted of macrophages, lymphocytes and especially neutrophils, and many of these inflammatory cells were apoptotic. High mortality rates in the BH3479-infected population of mice correlated with the intense polymorphonuclear and mononuclear leucocyte inflammatory infiltrate in the CNS.
Keywords: central nervous system, Murray Valley encephalitis virus, electron microscopy, inflammatory cells, neurones
An epidemic of severe encephalitis occurred in the Murray Valley region of New South Wales, Australia in 1950–51, and was caused by an agent which has since been named Murray Valley encephalitis (MVE) virus (Cook et al. 1970). MVE virus is a member of the family Flaviviridae. Virions in this family are approximately 50 nm in diameter, possess a lipid envelope (Brinton 1991) and their 11 kb genome consists of single-stranded positive sense RNA (Chambers et al. 1990). Members of the family that are associated with disease in humans include Japanese encephalitis (JE) virus, dengue (DEN) virus and yellow fever (YF) virus (Chambers et al. 1990). MVE virus activity is confined to the mainland of Australia and Papua New Guinea (Mackenzie et al. 1994) and infection is invariably mosquito-borne.
The main mechanisms of disease production during viral encephalitis can be accounted for by various functional abnormalities, and the accompanying immune and virus mediated cellular lesions. The immune response may prove detrimental to the host in a number of flavivirus infections. Macrophages from tick-borne encephalitis virus infected mice have been shown to produce nitric oxide which may result in a breakdown of the blood brain barrier (Kreil & Eibl 1996), therefore facilitating viral access to the brain and contributing to the severity of disease. Mori et al. (1997) demonstrated that antigen stimulated CD4+ and CD8+ T cells are responsible for producing cytokines such as IL-2 and IFN-γ that may play a critical role in the pathogenesis of dengue virus infection in humans.
There are two major forms of virus-mediated cellular damage-necrosis and apoptosis. Necrosis results from severe and sudden injury when cells are subjected to wide deviations from normal homeostasis (Cohen & Duke 1984). A key morphological characteristic of necrosis is the loss of the integrity of the cell membrane and degradation of cellular organelles such as mitochondria (Wyllie et al. 1980). In an electron microscopic study, Murphy et al. (1968) showed that neurones in St. Louis encephalitis (SLE) infected mice underwent cell lysis. Necrosis in the CNS has also been observed during other flavivirus infections such as JE (Esiri 1997) and West Nile (Camenga et al. 1974) virus infection. Apoptosis is the commonest form of eukaryotic cell death (Trauth et al. 1989). Apoptosis characteristically affects scattered individual cells. Some major morphological features of apoptosis include: (1) loss of surface contact with adjacent cells (Ormerod et al. 1994; Ying et al. 1997); (2) decrease in cell size due to cytoplasmic condensation and nuclear compaction (Fairbairn et al. 1994; Edstrom et al. 1996); and (3) fragmentation of the nucleus and cytoplasm into membrane bound apoptotic bodies with apparently normal organelles (Shinomiya et al. 1994; Adle-Biassette et al. 1995). Members of the family Flaviviridae have been reported to induce neuronal apoptosis in vitro. These include dengue virus (Despres et al. 1996) in mouse neuroblastoma cells and JE virus (Liao et al. 1997) in human neuronal progenitor cells.
In this communication, we describe histological and ultrastructural changes in the CNS of weanling Swiss mice infected with MVE virus by peripheral inoculation and characterize the CNS inflammatory cell response to this infection in the CNS. An analysis of DNA fragmentation patterns within MVE virus-infected mouse brain samples is also presented.
Methods
Virus strains and cells
MVE-BH3479 virus (Marshall et al. 1982) was passaged once in suckling mouse brain, twice in C6/36 (Aedes albopictus) cells and twice in Vero cells. Working stocks were culture supernatants of C6/36 cells. Cell culture media, cell and virus stocks were tested for the presence of endotoxin by limulus amoebocyte lysate assay (E-Toxate, Sigma, USA) before use in animal studies; measurable quantities of endotoxin were not detected in any of these reagents.
African green monkey kidney (Vero) cells (ATCC, CCL 81), used between passage level 124 and 128, were grown at 37 °C in 20 mm HEPES-buffered M199 supplemented with 2 mml-glutamine and 10% foetal calf serum (FCS) in 5% CO2/95% air. C6/36 cells (ATCC; CRL 1660), used between passage levels 122 and 132, were grown at 28°C in M199 supplemented with 20 mml-glutamine and 10% FCS.
Plaque assay
Virus was assayed by plaque formation on Vero cell monolayers grown in 12 well plastic trays (tissue culture grade; CoStar Scientific Inc., Cambridge, Mass.). After 1 h adsorption of serial 10-fold dilutions of virus, inoculum was removed and cells were overlaid with 1.75% methyl cellulose in 2% FCS/M199. Following five days at 37°C/5% CO2, monolayers were stained with 1% methylene blue/10% formalin and plaques counted after overnight incubation. Infectivity titres are expressed as plaque-forming units (PFU) per ml.
Origin of animals
Specific pathogen free 21 day-old Swiss outbred mice were obtained from the Animal Resources Centre, Canning Vale, Western Australia. All animal experiments were undertaken in accordance with protocols approved by the University of Western Australia Animal Experimentation Ethics Committee.
Animal inoculations
21-day-old Swiss mice were inoculated ip with 103 plaque forming units of virus in 100 μl of Hanks balanced salt solution, pH 8.0 (HBSS). At the indicated times after inoculation, mice from each group were anaesthetized with penthrane, killed by exsanguination through intracardiac puncture and perfused with sterile phosphate-buffered saline, pH 7.4 (PBS). Brains were dissected with the olfactory bulbs intact.
For viral growth studies, brains were dissected intact, snap frozen in liquid nitrogen and stored at −80°C. The brains were subsequently dispersed in Dounce homogenisers and prepared as 10% suspensions in HBSS containing 2% bovine serum albumin (BSA). Virus was titrated by plaque formation on Vero cell monolayers; the threshold for detection of virus was 100 PFU/g of tissue. The characteristic virus titres in CNS tissue during the time course of infection are published in Andrews et al. (1999).
Preparation of histological specimens
Mouse tissues were fixed in phosphate-buffered 3.8% formaldehyde (pH 7.4) for 18 h. The tissue was dehydrated in ascending grades of ethanol, washed in xylene for 1 h and treated in two wax baths at 60°C for 2 and 3 h, respectively. The tissue was then vacuum embedded in wax and placed in moulds using a Tissue-Tek III (Bayer, Australia), cut into 4 μm sections using a microtome, mounted onto glass slides and stained with haematoxylin and eosin.
Electron microscopy
Mice were terminally anaesthetized with avertin (2.5% tribromoethyl alcohol, 2.5% tertiary amyl alcohol in PBS), perfused with 50 ml of Karnovsky's fixative (4% paraformaldehyde and 2.5% glutaraldehyde in cacodylate buffer, pH 7.4) through the left ventricle and the brains dissected and placed in Karnovsky's fixative for a further 24 h. The fixed brains were further dissected to provide 1 mm3 tissue blocks of either the olfactory lobe or hippocampus; white matter surrounding the hippocampus was also gently removed. The tissue blocks were incubated in cacodylate buffer, postfixed for 1 h in 1% osmium tetroxide and stained with 2% uranyl acetate for 40 min. Specimens were dehydrated in ascending concentrations of ethanol and propylene oxide, embedded in Araldite epoxy resin and allowed to polymerize for 48 h.
Semithin (1 μm) sections were cut with a glass knife, placed onto clean glass slides, stained with 0.2% toluidine blue in 5% borax, allowed to dry at 80°C and mounted in ultramount. Ultrathin (90 nm) sections were cut with a diamond knife, placed onto 200-mesh gold grids, stained with a modified Reynolds lead citrate, pH 12.0 (Reynolds 1963) for 2 min and then washed in filtered ddH2O for 20 s. Sections were carbon coated and viewed in a Phillips 410LS microscope at 80 kV.
DNA fragmentation assay
Genomic DNA was purified from freshly dissected mouse brain tissue by homogenization in 1 ml of homogenization buffer (1 m NaCl, 0.5 m EDTA (pH 8.0), 1 m Tris HCl (pH 8.0), 1 m sucrose) using a hand held homogeniser (Dharmarajan et al. 1994). The concentration of purified DNA was determined by spectrophotometry and the solution standardized to a DNA concentration of 1 μg/μl in distilled water.
The end labelling reaction was undertaken for 1 h at 37°C in a 50-μl volume containing the mouse brain DNA preparation, 50 mm cacodylate buffer (pH 6.8), 5 mm CoCl2, [32P] ddATP (6.25 Ci; 3.4 pm; Amersham) and 25 U of terminal deoxynucleotidyl transferase (TdT; Boehringer Mannheim). The reaction was terminated by adding 5 μl of 0.2 m EDTA (pH 8.0). Labelled DNA was precipitated by addition of 12 μl of 10 m ammonium acetate and three volumes of ice cold ethanol (−80°C for 1 h). The DNA was pelleted at 14 000 r.p.m. for 20 min (4°C). The pellet was air dried and resuspended in 40 μl of TE buffer (pH 8.0) and the samples stored at −80°C.
DNA samples were analysed by electrophoresis in a 2% agarose gel. The gel was then dried in a slab dryer (Uni Gel Dryer, 3040) under vacuum and exposed to X-ray film overnight at −80°C. Low molecular weight DNA fractions (< 15 kb) were excised from the dried gel and analysed in a liquid scintillation counter (Beckman) to provide a semiquantitative estimate of the extent of internucleosomal DNA cleavage in the samples.
Results
Histological features of MVE virus-mediated encephalitis
Groups of 21-day-old Swiss mice were infected i.p. with 103 plaque forming units of BH3479, brain tissue was collected daily between days 4 and 9 p.i. and the time course of infection was documented (Andrews et al. 1999).
The main aims of this study were to determine the extent and distribution of virus-mediated cell injury within the CNS and to characterize the CNS cellular inflammatory response to MVE virus infection.
In mice infected with BH3479, chromatolysis of occasional neurones was observed in the olfactory lobes and in the frontal cortex on the fourth day pi. By the fifth day of infection, foci of neuronal necrosis were prominent in the olfactory bulb, olfactory nuclei and piriform cortex, while the caudate-putamen, pons, midbrain and hippocampus were less affected. The occipital cortex was minimally involved, while the cerebellum was unaffected. In addition, congestion of the cerebral vasculature was present, and this was accompanied by polymorphonuclear and mononuclear leukocytic margination and perivascular accumulation. Similar changes were also present in the leptomeningeal tissues.
In the following two to three days, the foci of neuronal necrosis became more extensive and involved the thalamus, while similar changes in the dentate gyrus (Figure 1a,b) and CA3 region of the hippocampal formation became increasingly severe. Vascular congestion and infiltration of polymorphonuclear and mononuclear leucocytes were also more prominent (Figure 1e,f) and meningeal involvement increased (Figure 1c). In addition, the cerebral ventricles were dilated (Figure 1b) and leucocytes were scattered in the ventricular spaces. The majority of necrotic neurones were surrounded by microglial cells (neuronophagia) and at later times by infiltrating leucocytes (Figure 1d). Karyorrhexis was common in the necrotic regions of grey matter, but in addition, chromatin margination, nuclear and cytoplasmic condensation and the nuclear fragmentation of apoptosis were also frequently observed (Figure 1d). However, the identity of the cells undergoing apoptosis was unclear.
Figure 1.
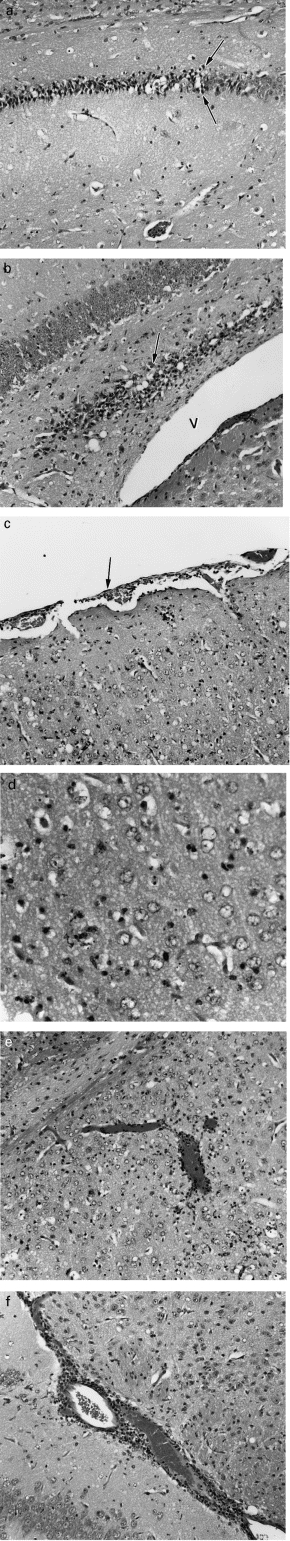
Histological sections of 21-day-old Swiss mouse brain seven days after infection with MVE virus-BH3479. Tissue was fixed in 3.8% formaldehyde for 18 h and embedded in paraffin; tissue sections (4 μm) were cut on a microtome and stained with haematoxylin and eosin. a. Dentate gyrus of the hippocampal formation, displaying a band of necrotic neurones. Note the abrupt border between necrotic and unaffected grey matter (arrows). Magnification, ×160. b. Hippocampal formation, showing a band of necrotic neurones within the dentate gyrus (arrow). The lateral ventricle (V) appears dilated. Magnification, ×160. c. Parietal cortex, showing leptomeningeal inflammation (arrow) and leukocytic infiltration of grey matter. Magnification, ×140. d. Caudate-putamen, showing leukocytic infiltration and nuclear profiles suggestive of apoptosis. Characteristics of apoptosis that are illustrated include: (1) scattered individual cells are affected; (2) decrease in cell size due to cytoplasmic condensation and nuclear compaction; and (3) fragmentation of the nucleus and cytoplasm into membrane bound apoptotic bodies. Magnification, ×350. e. Caudate-putamen, displaying vascular congestion and perivascular leukocytic infiltration. Magnification, ×140. f. Hippocampal formation, showing vascular congestion and perivascular leukocytic infiltration. Magnification, ×160.
Ultrastructural features of MVE virus-mediated encephalitis
Neurones
Ultrastructural evidence of viral infection was seen in several regions of BH3479-infected CNS, including the anterior olfactory nucleus, piriform cortex and the dentate gyrus, proximal CA3 and distal CA1 regions of the hippocampus. At five days pi, MVE virus-infected neurones showed endoplasmic reticulum membrane proliferation and unusual membrane aggregation characteristic of flavivirus infection (Figure 2a). This was accompanied by the appearance of tubular and spherical structures in the cisternae of the endoplasmic reticulum, the Golgi complex and the nuclear envelope (Figure 2b). The spherical structures measured 32–40 nm in diameter, possessed an electron dense nucleoid and were consistent with the appearance of newly formed flavivirus particles. The tubular structures measured 24–32 nm in diameter and displayed a distinct periodicity (5 nm), but their relationship to the spherical particles was unclear. In several of these neurones, membrane-bound structures measuring 75–145 nm in diameter, and containing electron dense material, were found within dilated segments of endoplasmic reticulum and Golgi complex, as well as in spherical vacuoles of unknown origin. A small proportion of mitochondria were swollen and the Golgi complex was occasionally whorled. Rough endoplasmic reticulum displayed extensive degranulation indicative of cell injury. These cytoplasmic changes were most prominent in the perikaryon but were also seen in the proximal segments of dendrites (Figure 2a). At seven to eight days pi, MVE virus-infected neurones showed chromatin condensation and extrusion, nuclear fragmentation, loss of segments of the nuclear envelope (Figure 3a), reduced surface contact with adjacent cells and loss of cytoplasmic organelles. The few remaining organelles in these neurones were generally aggregated within foci of swollen cytoplasm (Figure 2a and 3a). All these features were prominent in the proximal CA3 and distal CA1 neurones of the hippocampus, whereas distal CA3 and proximal CA1 neurones, although immediately adjacent, were largely unaffected. Lastly, the endoplasmic reticulum of infected neurones at these time points possessed numerous virus particles (Figure 2b).
Figure 2.
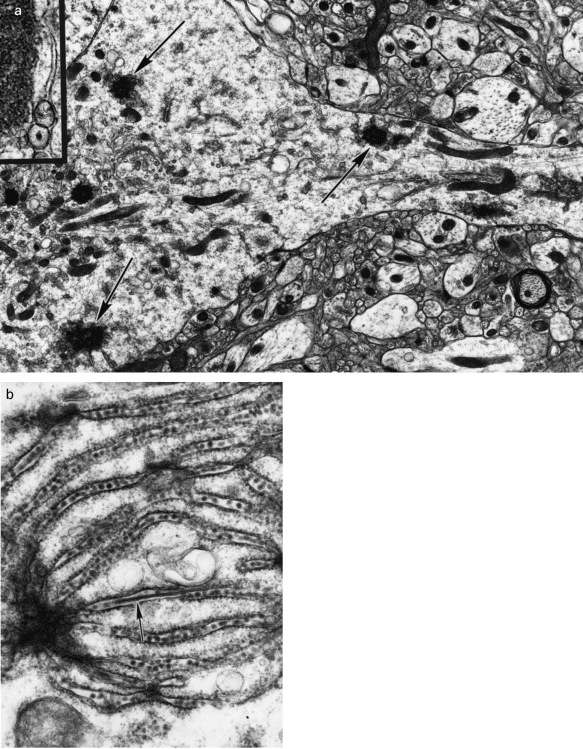
Electron micrographs of central nervous system of 21-day-old Swiss mice five days after infection with MVE virus-BH3479. Tissue preparation and staining was as outlined in Materials and Methods. a. A neurone displaying slight peripheral swelling and concentration of cytoplasmic organelles within the perikaryon. Note the complex membrane aggregation in profiles of the endoplasmic reticulum both in the dendrite and in the perikaryon (arrows). Magnification, ×13,200. Inset: High power view of the complex membrane aggregates; note also the presence of vesicular bodies. Magnification, ×58,200. b. Spherical particles (virions) are seen within cisternae of the endoplasmic reticulum of neurones; rod-like structures are also present (arrow). The rod-like structures possess a distinct periodicity (5 nm). Magnification, ×59,000.
Figure 3.
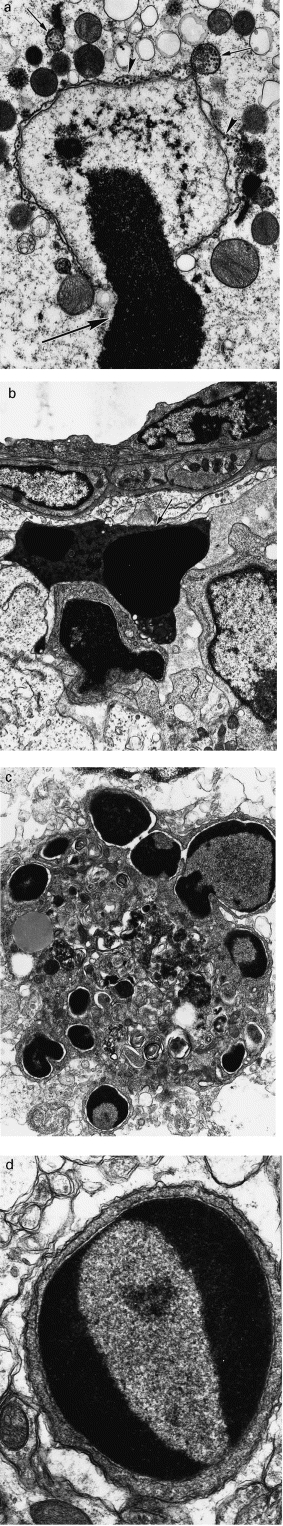
Electron micrographs of the murine hippocampal formation seven days after infection with MVE virus-BH3479. Tissue preparation and staining was as outlined in Materials and Methods. a. A neurone showing the extrusion of a heterochomatin mass from the nucleus (large arrow). Mitochondria are enlarged and the endoplasmic reticulum is vacuolated. Virions and vesicles are seen within these vacuoles (arrows) and within the swollen nuclear envelope (arrowheads). Magnification, ×19,200. b. Apoptotic polymorphonuclear leucocyte (arrow) in the perivascular zone. Magnification, ×9,000. c. Mononuclear phagocyte showing nuclear fragmentation characteristic of apoptosis. Magnification, ×10,500. d. Lymphocyte displaying heterochromatin aggregates characteristic of apoptosis. Magnification, ×26,000.
Inflammatory cells
At 7 days p.i., inflammatory cells were seen to infiltrate the CNS parenchyma of BH3479-infected mice. These infiltrates were composed of neutrophils (Figure 3b), macrophages (Figure 3c) and lymphocytes (Figure 3d), and several abutted onto neurones (neuronophagia). In addition, many of these inflammatory cells displayed the characteristic features of apoptosis, such as nuclear chromatin condensation, cytoplasmic condensation and apoptotic body formation (Figure 3c). Such apoptotic cells were most frequent in the CNS parenchyma and were less commonly seen in perivascular regions. Macrophages were most prominent in the inflammatory infiltrate at seven days pi, whilst polymorphonuclear cells were more numerous at eight and nine days pi. There was no ultrastructural evidence of viral infection of infiltrating inflammatory cells in infected mice.
Time course for development of DNA fragmentation within MVE-infected brain
Further evidence for the development of apoptosis in murine CNS cells during MVE infection was provided by examination of the fragmentation patterns of 32P end-labelled, purified DNA samples from MVE-infected mouse brain.
All of the BH3479-infected mouse brain DNA samples showed characteristic DNA ‘laddering’ in multiples of 185 bp (Figure 4). Interestingly, the intensity of the DNA signal increased markedly over the time course of infection. Counting of radioactivity (using a liquid scintillation counter) in the 32P labelled, low molecular weight DNA (< 15kb) excised from the gel, showed that the amount of DNA fragmentation at days seven and eight pi was 10 fold greater than that observed at days five and six pi (data not shown). DNA fragmentation was not observed in the mock-infected samples. The marked increase in apoptotic DNA fragmentation between days six and seven pi correlated with the infiltration of inflammatory cells in BH3479-infected mouse brain. Many of these inflammatory cells also showed ultrastructural (Figure 3b-d) evidence of apoptosis.
Figure 4.
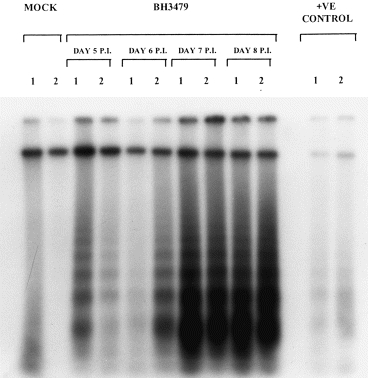
An examination of [32P]-ddATP, 3′ end labelled DNA purified from MVE virus-BH3479-infected Swiss mouse brain at increasing times pi. Autoradiograph of DNA (1 μg) isolated from virus infected mouse brain on days 5, 6, 7 and 8 pi, showing internucleosomal DNA fragmentation in multiples of approximately 185 bp; two mice per time point (1 and 2) are shown. The negative control (MOCK) consists of DNA from mock-infected Swiss mouse brain. The positive control consists of DNA from Sindbis virus-infected (MRM39 strain) newborn mouse brain; Sindbis virus infection is known to induce apoptotic DNA fragmentation in newborn mouse brain (Lewis et al. 1996).
Discussion
In this study, we have shown that MVE virus preferentially induces neuronal cell injury in the olfactory lobe and structures of the hippocampal formation as early as five days pi. BH3479-infected mice developed a uniformly fatal encephalitis from five days pi, and this was associated with development of a mixed cell inflammatory infiltrate, including both polymorphonuclear and mononuclear leucocytes.
The ultrastructural features of MVE virus infection in CNS neurones seen in this study, including endoplasmic reticulum and Golgi complex membrane proliferation and the appearance of membrane bound spherical vesicles (75–145 nm) are similar to those observed by previous authors who have studied the related flaviviruses SLE virus (Murphy et al. 1968) and JE virus (Hase et al. 1990a, b). A fine reticular web was often seen within the large vesicles which has been postulated to be progeny viral RNA (Ng & Hong 1989). An ultrastructural study by Ng & Hong (1989) demonstrated that at an advanced stage of Kunjin virus infection, membrane bound vesicles became empty which coincided with the accumulation of intracellular and extracellular viruses. There is a strong possibility that the vesicles were produced by budding from endoplasmic reticulum or Golgi complex membranes. Rod-like structures with a diameter of 24–32 nm in diameter, similar to those seen scattered in the cytoplasm of Kunjin virus-infected Vero cells in vitro (Ng & Hong 1989), were observed within cisternae of the endoplasmic reticulum of MVE-infected mouse neurones. Ng & Hong (1989) showed that at late time-points the tubules in Kunjin virus-infected Vero cells appeared to have the viral nonstructural protein NS3 associated with them. However, it has been demonstrated that microtubule disrupting drugs such as vinblastine sulphate caused drastic decreases in Kunjin virus yields (Ng & Hong 1989), suggesting that cytoskeletal microtubules are necessary for replication of Kunjin virus. In the present study, the close association of the tubule-like structures with newly formed MVE virus particles suggests that they may represent aggregates of nucleocapsid material.
Andrews et al. (1999) studied the development of apoptosis within MVE-infected neurones by double labelling for MVE viral RNA by in situ hybridization, and for apoptosis by in situ terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling (TUNEL) assay. This study showed that limited virally induced neuronal apoptosis was observed in heavily infected regions of the CNS such as the hippocampus from 5 to 6 days pi. The DNA fragmentation seen at days five and six pi (Figure 4) is thought to be partly due to CNS cell programmed cell death. Examination of MVE virus-infected mouse hippocampal sections indicated preferential infection of the proximal CA3 and distal CA1 regions. In a murine study of dengue virus infection, it was shown that apoptotic cell death and dengue virus replication was restricted to the neurones of the cortical and hippocampal regions (Despres et al. 1998). Thus, it appears that the neurones of the hippocampus are particularly vulnerable to apoptotic cell death during flavivirus infection. Interestingly, the unrelated Borna disease virus has also been shown to infect specific regions of the hippocampus, including the dentate gyrus and CA3 region (Mu Zheng et al. 1993).
At five days pi onwards, much of the inflammatory infiltrate observed in BH3479-infected mouse brain showed evidence of apoptosis (Figure 3b,c,d). Therefore the high levels of DNA fragmentation seen at days seven and eight pi (Figure 4) is believed to be largely due to apoptotic inflammatory cells. At late time-points during MVE virus-infection, the inflammatory response consisted mainly of neutrophils. Andrews et al. (1999) showed that neutrophils contributed to the severity of disease due to MVE infection of mice. Neutrophils are the most abundant and shortest lived of all leucocytes (Squier et al. 1995). Ageing neutrophils have been shown to undergo apoptosis spontaneously, a process that is believed to limit neutrophil-mediated tissue injury in inflamed sites (Ohta et al. 1994). This postulated mechanism does not appear to be efficacious during MVE virus-infection as disease due to BH3479 continues to progress despite the programmed cell death of polymorphonuclear cells.
In conclusion, the results from this study suggest that the significant neuronal necrosis observed from days 5–9 pi is likely to have an adverse effect on CNS function. The pathogenesis of MVE is compounded by inflammatory cell infiltration and release of inflammatory mediators, from 7 days pi which appear to be associated with the eventual death of the host (Andrews et al. 1999). Future studies should address the mechanisms underlying the high levels of apoptotic inflammatory cells observed in MVE virus-infected brains.
Acknowledgments
We thank Ms. Nichole Dutton for excellent technical assistance with electron microscopy and Associate Professor Dharmarajan for his kind assistance with the DNA fragmentation assay. This work was undertaken with the support of grants from the Arthur Yeldham and Mary Raine Medical Research Foundation of Western Australia, the Australian Research Council (04/15/412/288) and the National Health and Medical Research Council (970106).
References
- Adle-Biassette H, Levy M, Colombel M, et al. Neuronal apoptosis in HIV infection in adults. Neuropathol. Appl. Neurobiol. 1995;21:218–227. doi: 10.1111/j.1365-2990.1995.tb01053.x. [DOI] [PubMed] [Google Scholar]
- Andrews DM, Matthews VB, Sammels LM, Carrello AC, McMinn PC. The severity of Murray Valley Encephalitis in mice is linked to neutrophil infiltration and inducible nitric oxide synthase activity in the central nervous system. J. Virol. 1999;73:8781–8790. doi: 10.1128/jvi.73.10.8781-8790.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinton MA. Flaviviruses. In: MCKEndall R R, STroop W G, editors. Handbook of Neurovirology. London: Marcel Dekker Inc; 1991. pp. 379–389. [Google Scholar]
- Camenga DL, Nathanson N, Cole GA. Cyclophosphamide-potentiated West Nile viral encephalitis-Relative influence of cellular and humoral factors. J. Infect. Dis. 1974;130:634–641. doi: 10.1093/infdis/130.6.634. [DOI] [PubMed] [Google Scholar]
- Chambers TJ, Hahn CS, Galler R, Rice CM. Flavivirus genome organisation, expression, and replication. Ann. Rev. Microbiol. 1990;44:649–688. doi: 10.1146/annurev.mi.44.100190.003245. [DOI] [PubMed] [Google Scholar]
- Cohen JJ, Duke RC. Glucocorticoid activation of a calcium-dependent endonuclease in thymocyte nuclei leads to cell death. J. Immunol. 1984;132:38–42. [PubMed] [Google Scholar]
- Cook I, Allan BC, Horsfall WR, Flanagan JE. A fatal case of Murray Valley Encephalitis. Med. J. Aust. 1970;1:1110–1112. doi: 10.5694/j.1326-5377.1970.tb84449.x. [DOI] [PubMed] [Google Scholar]
- Despres P, Flamand M, Ceccaldl P, Deubel V. Human isolates of dengue type 1 virus induce apoptosis in mouse neuroblastoma cells. J. Virol. 1996;70:4090–4096. doi: 10.1128/jvi.70.6.4090-4096.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despres P, Frenkel M-P, Ceccaldi P-E, Dos Santos CD, Deubel V. Apoptosis in the mouse central nervous system in response to infection with mouse-neurovirulent dengue viruses. J. Virol. 1998;72:823–829. doi: 10.1128/jvi.72.1.823-829.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmarajan AM, Goodman SB, Tilly KI, Tilly JL. Apoptosis during functional corpus luteum regression: Evidence of a role for chorionic gonadotrophin in promoting luteal cell survival. Endocrine J. 1994;2:295–303. [Google Scholar]
- Edstrom A, Ekstrom PA.R, Tonge D. Axonal outgrowth and neuronal apoptosis in cultured adult mouse dorsal root ganglion preparations: effects of neurotrophins, of inhibition of neurotrophin actions and of prior axotomy. Neuroscience. 1996;75:1165–1174. doi: 10.1016/0306-4522(96)00324-7. [DOI] [PubMed] [Google Scholar]
- Esiri MM. Viruses and Rickettsiae. Brain Path. 1997;7:695–709. doi: 10.1111/j.1750-3639.1997.tb01084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbairn DW, Carnahan KG, Thwaits RN, Grigsby RV, Holyoak GR, O'neill KL. Detection of apoptosis induced DNA cleavage in Scrapie-infected sheep brain. FEMS Microbiol. Lett. 1994;115:341–346. doi: 10.1111/j.1574-6968.1994.tb06661.x. [DOI] [PubMed] [Google Scholar]
- Hase T, Dubois DR, Summers PL. Comparative study of mouse brains infected with Japanese encephalitis virus by intracerebral or intraperitoneal inoculation. Int. J. Exp. Pathol. 1990a;71:857–869. [PMC free article] [PubMed] [Google Scholar]
- Hase T, Summers PL, Dubois DR. Ultrastructural changes of mouse brain neurons infected with Japanese encephalitis virus. Int. J. Exp. Pathol. 1990b;71:493–505. [PMC free article] [PubMed] [Google Scholar]
- Kreil TR, Eibl MM. Nitric oxide and viral infection-No antiviral activity against a flavivirus in vitro, and evidence for contribution to pathogenesis in experimental infection in vivo. Virology. 1996;219:304–330. doi: 10.1006/viro.1996.0252. [DOI] [PubMed] [Google Scholar]
- Lewis J, Wesselingh SL, Griffin DE, Hardwick JM. Alphavirus-induced apoptosis in mouse brains correlates with neurovirulence. J. Virol. 1996;70:1828–1835. doi: 10.1128/jvi.70.3.1828-1835.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao CL, Lin YL, Wang JJ, et al. Effect of enforced expression of human Bcl-2 on Japanese encephalitis virus-induced apoptosis in cultured cells. J. Virol. 1997;71:5963–5971. doi: 10.1128/jvi.71.8.5963-5971.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie JS, Lindsay MD, Coelen RJ, Broom AK, Hall RA, Smith DW. Arboviruses causing human disease in the Australasian zoogeographic region. Arch. Virol. 1994;136:447–461. doi: 10.1007/BF01321074. [DOI] [PubMed] [Google Scholar]
- Marshall ID, Woodroofe GM, Hirsch S. Viruses recovered from mosquitoes and wildlife serum collected in the Murray Valley of south-eastern Australia, February 1974, during an epidemic of encephalitis. Aust. J. Exp Biol. Med. Sci. 1982;60:457–470. doi: 10.1038/icb.1982.51. [DOI] [PubMed] [Google Scholar]
- Mori M, Kurane I, Janus J, Ennis FA. Cytokine production by Dengue virus antigen-responsive human T lymphocytes In Vitro examined using a double immunocytochemical technique. J. Leukoc. Biol. 1997;61:338–345. doi: 10.1002/jlb.61.3.338. [DOI] [PubMed] [Google Scholar]
- Murphy FA, Harrison AK, Gary GW, Whitfield MS, Forrester FT. St. Louis encephalitis virus infection of mice. Lab. Invest. 1968;19:652–662. [PubMed] [Google Scholar]
- Mu Zheng Y, Schafer MK.-H, Weihe E, et al. Severity of neurological signs and degree of inflammatory lesions in the brains of rats with Borna Disease correlate with the induction of nitric oxide synthase. J. Virol. 1993;67:5786–5791. doi: 10.1128/jvi.67.10.5786-5791.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng ML, Hong SS. Flavivirus infection-Essential ultrastructural changes and association of Kunjin virus NS3 protein with microtubules. Arch. Virol. 1989;106:103–120. doi: 10.1007/BF01311042. [DOI] [PubMed] [Google Scholar]
- Ohta H, Yatomi Y, Sweeney EA, Hakomori S, Igarashi Y. A possible role of sphingosine in induction of apoptosis by tumor necrosis factor-α in human neutrophils. FEBS Lett. 1994;355:267–270. doi: 10.1016/0014-5793(94)01218-0. [DOI] [PubMed] [Google Scholar]
- Ormerod MG, O'neill CF, Robertson D, Harrap KR. Cisplatin induces apoptosis in a human ovarian carcinoma cell line without concomitant internucleosomal degradation of DNA. Exp. Cell Res. 1994;211:231–237. doi: 10.1006/excr.1994.1082. [DOI] [PubMed] [Google Scholar]
- Reynolds ES. The use of lead citrate at high pH as an electron opaque stain in electron microscopy. J. Cell Biol. 1963;17:208–212. doi: 10.1083/jcb.17.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinomiya N, Shinomiya M, Wakiyama H, Katsura Y, Rokutanda M. Enhancement of CDDP cytotoxicity by caffeine is characterised by apoptotic cell death. Exp. Cell Res. 1994;210:236–242. doi: 10.1006/excr.1994.1035. [DOI] [PubMed] [Google Scholar]
- Squier MK.T, Sehnert AJ, Cohen JJ. Apoptosis in leucocytes. J. Leukoc Biol. 1995;57:2–10. doi: 10.1002/jlb.57.1.2. [DOI] [PubMed] [Google Scholar]
- Trauth BC, Klas C, Peters AM.J, et al. Monoclonal antibody-mediated tumor regression by induction of apoptosis. Science. 1989;245:301–305. doi: 10.1126/science.2787530. [DOI] [PubMed] [Google Scholar]
- Wyllie AH, Kerr JF.R, Currie AR. Cell death-the significance of apoptosis. Int. Rev. Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- Ying S, Meng Q, Taborda-Barata L, Kay AB. Association of apoptosis of neutrophils and eosinophils and their ingestion by macrophages with resolution of the allergen-induced cutaneous late-phase response in atopic human subjects. Proc. Assoc Am. Physicians. 1997;109:42–50. [PubMed] [Google Scholar]