

Abstract
Musculoskeletal disorders such as rheumatoid arthritis (RA) and osteoarthritis are a common cause of pain and disability. The vasculature is an important component of the musculoskeletal system, and vascularization is a key event in the development of normal cartilage and bone. By promoting the delivery of nutrients, oxygen and cells, blood vessels help maintain the structural and functional integrity of joints and soft tissue and may facilitate tissue repair and healing. The identification of pro-angiogenic mediators such as vascular endothelial growth factor (VEGF) has led to the development of antiangiogenic therapies for the treatment of neoplastic diseases. The important role of angiogenesis, and especially VEGF, in the pathogenesis of joint disorders such as RA suggests that antiangiogenic therapy may be a useful adjunct to existing approaches in RA.
Keywords: Angiogenesis, arthritis, musculoskeletal disorders, VEGF
Musculoskeletal disease and injury are estimated to account for one fifth of all disabilities and are one of the most common causes of severe long-term pain and physical disability for many millions of people across the world. The prevalence of musculoskeletal disorders exceeds that of heart disease and cancer, thus incurring a significant economic burden, at least in part due to lost earnings and reduced functional capacity (Decker 1983; Lawrence et al. 1989). The life expectancy of patients with musculoskeletal diseases such as RA is also markedly reduced (Symmons et al. 1998). The impact of bone and joint disorders on society, the healthcare system and on the individual, combined with the awareness that resources need to be more efficiently used, has culminated in the proposal that 2000–2010 be designated as the ‘Decade of the Bone and Joint’.
There are many aetiologies contributing to the development of musculoskeletal diseases. This review will focus on the role of the blood vessels in normal joint and in pathological conditions. The vasculature plays a crucial role in the development of musculoskeletal structures. Vascularization is important in the differentiation and mineralization of cartilage, which leads to formation of normal bone. By promoting the delivery of nutrients, oxygen and cells, blood vessels help maintain the structural and functional integrity of joints and soft tissue, and may facilitate tissue repair and healing. In contrast, aberrant vascular turnover is thought to contribute to the pathogenesis of disorders such as rheumatoid arthritis (RA), in which excessive vascularization may promote and maintain the inflammatory and hyperproliferative state of the joint. In the ensuing sections we will review the importance of the vasculature in the formation and function of the joint and in selected joint disorders, including inflammatory arthritides and diseases characterized by aberrant new bone formation. Finally, we will address the question of whether inhibition of new blood vessel formation should be considered as a future therapeutic strategy for the treatment of conditions such as RA.
Angiogenesis: a balance between pro- and anti-angiogenic stimuli
The formation of new blood vessels in vivo occurs in response to an increase in tissue mass and the concomitant requirement for increased delivery of oxygen and nutrients (Folkman 1995). During embryogenesis blood vessels form de novo via the assembly of endothelial precursors called angioblasts in the absence of pre-existing structures (‘vasculogenesis’), forming the cardiovascular system, which is required to sustain the metabolic needs of the developing embryo. In the adult, the normal vasculature is extremely stable, and endothelial cell turnover can often be measured in years. However the vasculature is highly adaptable and, in response to an increased tissue mass, new capillaries can rapidly arise from the existing vascular bed, during both physiological situations (such as in the female reproductive cycle and wound healing) and in many disease states. This continued expansion of the vascular tree, termed ‘angiogenesis’, is a tightly governed process, which is the net result of a balance between maintenance of mature vessels, formation of new structures and regression of newly formed blood vessels.
Many pathological conditions are characterized by overabundant or — less frequently — insufficient vascularization. Angiogenesis is crucial in the growth and maintenance of solid tumours, and in fact many advances in the understanding of angiogenesis have emerged from studies of the tumour vasculature. The pioneering work of Judah Folkman and colleagues in the early 1970s demonstrated that in order to extend beyond a volume of 1 mm3, solid tumours need to establish their own blood supply by encouraging the growth of new blood vessels into the tumour tissue. The establishment of a blood supply provides the tumour cells with nutrients that enable them to expand and ultimately to metastasize. As a consequence, many antiangiogenic therapies are currently being developed, with some promising results in clinical trials, and may in future be used as adjuncts to conventional chemotherapy (Harris 1998). A similar situation may exist in RA, where the proliferative and invasive nature of synovial pannus has frequently led to comparisons with tumour development (Paleolog 1996; Paleolog & Fava 1998; Paleolog & Miotla 1998). As in the case of tumour growth, the expansion of the synovial pannus may result in local hypoxia, thus driving the need for a compensatory neovascularization, to increase the supply of nutrients and oxygen to the joint. Conversely, some cardiovascular diseases are characterized by insufficient vascularization, leading to the concept of ‘therapeutic angiogenesis’ as a means of managing these conditions. For example, in patients with severe myocardial ischaemia, narrowing of the coronary arteries leads to a state of hypoxia in the myocardium. Recent reports have showed that gene transfer of pro-angiogenic factors restored blood supply into ischaemic myocardium and enhanced heart function (Losordo et al. 1998).
The evolution of an ‘angiogenic phenotype’ probably results not only from upregulation of endothelial growth factors, but also due to downregulation of inhibitors of angiogenesis, culminating in the continued expansion of the vascular tree (Table 1). Modulators of angiogenesis may directly affect endothelial cell proliferation, migration or apoptosis, or exert indirect effects by modulating the levels of growth factors. In response to a pro-angiogenic stimulus, destabilization of the mature blood vessel must occur, as well as loosening of the cell-cell and cell–matrix interactions. Thereafter, endothelial cells migrate into the perivascular space, assisted by the interaction of endothelial cell adhesion molecules with extracellular matrix components such as fibronectin, collagen and laminin. Subsequently endothelial cells invade the vascular stroma, through the action of proteases such as plasminogen activator, and align themselves to form capillary sprouts and loops. Proliferation of endothelial cells proximal to the sprout occurs, with concomitant migration of cells at the leading edge of the sprout. Lumen formation occurs, and pericytes associate with the abluminal surface, to generate mature capillaries and postcapillary venules. Finally, the new vessel offshoot circulates blood into the now vascularized region (Colville-Nash & Willoughby 1997). More recently, it has become apparent that postnatal vasculogenesis may also occur, in addition to the classical paradigm of vessel formation in the adult as a consequence of angiogenesis. The existence of circulating endothelial cell progenitors has been demonstrated, and putative angioblasts have been isolated from the leucocyte fraction of peripheral blood, supporting the concept of a common precursor for haematopoietic and endothelial cells. The angioblasts differentiate into endothelial cells, and are incorporated into sites of active neovascularization in vivo (Asahara et al. 1997). It is thus possible that new vessel formation in the adult may be a result of both angiogenesis and vasculogenesis. As the data concerning postnatal vasculogenesis are still relatively sparse, this review will focus on development of new vessels from pre-existing ones, i.e. angiogenesis.
Table 1.
Angiogenesis: a balance between stimulators and inhibitors. Molecules which have been demonstrated to stimulate or inhibit angiogenesis. The action of such mediators may be either direct, through effects on endothelial proliferation (e.g. VEGF), migration (e.g. HGF) or apoptosis (e.g. angiostatin, TL1), or indirect through the antagonism of the activity of other stimuli (e.g. PF-4 inhibits the binding of VEGF and FGF-2 to endothelial cells) or through the induction of other mediators (e.g. the angiostatic effects of IL-10, IL-12 and IFNγ are most likely mediated through the induction of IP-10 expression)

One of the key players in the pathogenesis of angiogenesis-dependent diseases is vascular endothelial growth factor (VEGF) (Brown et al. 1997; Achen & Stacker 1998; Neufeld et al. 1999). Many growth factors are pleiotropic in their effects, and promote proliferation of target cells other than endothelium. In contrast, VEGF appears to be the most endothelial cell-specific angiogenic factor characterized to date. The mechanisms whereby VEGF stimulates angiogenesis are both direct and indirect, in that VEGF not only stimulates microvascular endothelial cells to proliferate and migrate, but also renders these endothelial cells hyperpermeable, leading to alterations in the extracellular matrix which may favour angiogenesis. In addition to stimulating endothelial cell proliferation and chemotaxis in vitro and in vivo, VEGF induces secretion of matrix metalloproteinases (MMP) and increases urokinase-type plasminogen activator receptor expression on endothelial cells, thus enhancing the activity of urokinase-type plasminogen activator and presumably endothelial cell invasiveness. A key feature of VEGF is that it is upregulated by hypoxia, through transcriptional and post-transcriptional mechanisms (Shweiki et al. 1992). This makes VEGF production a key response in situations where excessive proliferation and hence hypoperfusion occurs (e.g. during the female reproductive cycle or in malignancies) or during tissue ischaemia (e.g. following myocardial infarction). As a consequence, VEGF is upregulated in a number of physiological and pathological conditions associated with hypoperfusion and/or hypoxia, including a range of solid tumours, atherosclerosis, RA, diabetic retinopathy, as well as during vasculogenesis in the embryo.
Other pro-angiogenic heparin-binding growth factors include members of the fibroblast growth factor (FGF) family, a group of nearly 20 heparin-binding growth factors (Burgess & Maciag 1989). The prototypic members of the FGF family — namely acidic FGF (FGF-1) and basic FGF (FGF-2) — have been studied most extensively in vitro and in vivo. These two growth factors stimulate the proliferation of cells of mesenchymal origin, including the three principal vascular cell types: fibroblasts, endothelial cells and smooth muscle cells. Angiogenin is also a heparin-binding molecule, which induces neovascularization in the chicken chorioallantoic membrane and rabbit cornea models, and promotes endothelial cell invasion of Matrigel basement membrane. There are a number of other growth factors which are pro-angiogenic, including transforming growth factor β (TGFβ), which promotes proliferation of mesenchymal cells, platelet derived growth factor (PDGF), hepatocyte growth factor (HGF) and platelet derived endothelial cell growth factor (PD-ECGF or thymidine phosphorylase) (Claesson-Welsh 1994; Cox & Maurer 1997; Rosen & Goldberg 1997). Many of the pro-angiogenic effects of these molecules may be indirect: for example, TGFβ is a potent inducer of VEGF production by fibroblasts. The same may also be true for other ‘pro-angiogenic’ mediators such as tumour necrosis factor α (TNFα), which has been shown to upregulate release of VEGF (Ryuto et al. 1996; Yoshida et al. 1997; Paleolog et al. 1998).
In terms of downregulation of angiogenesis, the finding of molecules with powerful antitumour activities in mice has produced great interest in the clinical potential of such angiogenesis inhibitors. Angiostatin and endostatin were both discovered as a result of the observation that the presence of a primary tumour can inhibit the development of metastases (O'Reilly et al. 1997). It was hypothesized that the primary tumour produced angiogenesis inhibitors, which persisted in the circulation while local angiogenic promoters were degraded and exerted no systemic effect. Angiostatin and endostatin are fragments of larger circulatory proteins with no angiogenic activity; angiostatin is a 38-kD fragment of plasminogen, and endostatin is a fragment of collagen XVIII, a type of collagen found exclusively in blood vessels. Another plasminogen-derived angiogenesis inhibitor is K1–5 (protease-activated kringles 1–5), a result of the processing of plasminogen by urokinase-activated plasmin. The inhibitory effect of K1–5 is endothelial-cell-specific and appears to be at least approximately 50-fold greater than that of angiostatin (Cao et al. 1999). Recently, a novel angiogenesis inhibitor which is a member of the TNF family has been identified from an endothelial cell cDNA library. This inhibitor, named vascular endothelial growth inhibitor (VEGI, also known as TL1), encodes a protein of 174 amino acid residues, and is expressed predominantly in endothelial cells. Local production of a secreted form of VEGI via gene transfer caused complete suppression of the growth of colon cancers in mice (Zhai et al. 1999). Interestingly, other members of the TNF family, such as TNFα itself, and the recently identified protein TWEAK, can stimulate angiogenesis under the appropriate conditions (Ryuto et al. 1996; Yoshida et al. 1997; Lynch et al. 1999).
The regulation of new blood vessel formation is thus a consequence of the balance of many factors, the concentration and relative activity of which governs whether angiogenesis is stimulated or inhibited. The subsequent sections will discuss the role of neovascularization in the context of normal joint function and disorders of the musculoskeletal system such as RA.
Angiogenesis and the normal joint
In higher organisms, a skeletal structure is fundamental for stable support, and to facilitate locomotion and protection of vital organs. In addition, tendons and ligaments transmit force and augment joint stability, whereas cartilage provides a flexible joint interface whilst the synovium lines and supports most joints. The combination of these elements forms a highly complex and co-ordinated system, which provides the speed, power, dexterity and precision of movement, to perform the tasks of life. The musculoskeletal system comprises muscles, bones, cartilage, tendons, ligaments and fascia, all of which are critical to its integrity, stability and motility. In terms of musculoskeletal disorders such as RA, the diarthrodial or synovial joints are of primary interest. Characteristic features of synovial joints include two bones, linked by a fibrous capsule, with a deeper synovium, which lines the joints, except in the areas of the articular cartilage. There is a relatively abundant blood supply, which delivers cells and nutrients to the epiphysis, joint capsule and the synovium. The synovial circulation also generates intra-articular fluid that transports glucose to chondrocytes by convection during joint movement and lubricates joints, reducing erosion of joint surfaces.
Each articular surface is composed of hyaline cartilage, which strongly adheres to the underlying subchondral bone. Normal cartilage is a complex material consisting of a matrix composed primarily of collagen and proteoglycan, which is saturated with water. Cartilage is normally resistant to vascular invasion, and supply of nutrients to the chondrocytes, which are metabolically active but not in close proximity to a capillary, is achieved by the microcirculation of the synovium. Formation of bone from cartilage (‘endochondral ossification’) occurs in the embryo and during healing of fractures, and formation of new blood vessels is an integral part of this process. During endochondral bone formation, calcified cartilage formed in the cartilaginous bone rudiment allows vascular invasion, which initiates the replacement of cartilage by bone. The normally avascular cartilage differentiates to hypertrophic cartilage, which then undergoes erosion and vascularization leading to bone deposition. Blood vessels grow into the hypertrophic cartilage (characterized by the synthesis of type X collagen and matrix calcification) and erode it to produce a scaffold on which osteoblasts settle to produce woven bone (Figure 1).
Figure 1.

The role of VEGF in angiogenesis during joint development and in the pathogenesis of arthritis. The synovial circulation delivers cells and nutrients to the epiphysis, joint capsule and the synovium, and generates synovial fluid that transports glucose to chondrocytes. Endochondral bone formation results from expression of VEGF by chondrocytes in the growth plate, allowing vascular invasion and replacement of cartilage by bone. During the development of RA, the synovial lining becomes thickened and infiltrated by cells from the blood. The additional demands for oxygen and nutrients made by the proliferating synovial cells lead to hypoperfusion and hypoxia. Expression of VEGF by synovial macrophages and fibroblasts in response to hypoxia drives angiogenesis and hence further synovial proliferation and invasion/destruction of cartilage and bone. In OA, hypoxia can occur, resulting in under-nourishment of cartilage and hence cartilage thinning. At sites of mechanical stress such as joint margins close to articular cartilage, capillary encroachment into the subchondral plate and articular cartilage may promote osteophyte formation.
The control of cartilage vascularity thus plays a key role in bone formation. It is generally believed that chondrocytes of the cartilage produce factors which inhibit endothelial cell migration and growth and prevent blood vessel invasion even in the presence of pro-angiogenesis factors. Conversely, endothelial cells of the subchondral blood vessels release factors influencing growth and differentiation of chondrocytes in the avascular growth plate, suggesting that a fine balance between chondrocyte-and endothelial-derived signals is essential for normal development and growth of bone. It has been suggested that VEGF-mediated capillary invasion regulates growth plate morphogenesis and triggers cartilage remodelling (Table 2). Hypertrophic chondrocytes in the epiphyseal growth plate express VEGF, and administration of a soluble VEGF receptor chimeric protein to 24-day-old mice was used to determine the role of VEGF in endochondral bone formation. Blood vessel invasion was almost completely suppressed, in parallel with impaired trabecular bone formation and expansion of hypertrophic chondrocyte zone. Although proliferation, differentiation and maturation of chondrocytes were apparently normal, resorption was inhibited. Cessation of the anti-VEGF treatment was followed by capillary invasion, restoration of bone growth, resorption of the hypertrophic cartilage and normalization of the growth plate architecture (Gerber et al. 1999). The importance of angiogenesis in bone formation was also demonstrated in a study in which collagen pellets containing recombinant human bone morphogenetic protein-2 (BMP-2), a potent inducer of ectopic bone formation, were implanted in mice. These mice also received an inhibitor of angiogenesis, TNP-470, a semisynthetic analogue of the antibiotic fumagillin. TNP-470 inhibited ectopic new bone formation, and histological examination revealed reduced proliferation of mesenchymal cells and chondrogenesis at the initial step of endochondral bone formation. Immunohistochemical staining with a specific antibody against the BMP receptor BMP-IA showed that TNP-470 reduced the number of receptor-positive cells surrounding the BMP pellets. This suggests that angiogenesis may play an essential role in the recruitment of BMP-receptor-positive cells that can differentiate into chondrocytes and/or osteoblasts (Mori et al. 1998). Re-establishment of the circulation is also a key early event in fracture healing. Fracture of bone has been shown to disrupt its circulation, leading to necrosis and hypoxia of adjacent bone. Changes in serum levels of a low molecular weight ‘endothelial cell stimulating angiogenesis factor’ (ESAF) have been reported in group of patients with tibial fractures.
Table 2.
Angiogenesis in the musculoskeletal system. The role of neovascularization in the musculoskeletal system, highlighting molecules demonstrated or hypothesized to play a role in modulating angiogenesis in the context of normal joint function and selected musculoskeletal disorders.

The identity of potential inhibitors of angiogenesis in cartilage has been the subject of many studies (Eisenstein et al. 1975; Moses et al. 1990). One such inhibitory protein may be chondromodulin-1 (ChM-1), which has been purified from foetal cartilage and reported to inhibit tube morphogenesis of cultured vascular endothelial cells in vitro and to suppress angiogenesis in chick chorioallantoic membrane in vivo. In contrast, ChM-1 stimulated DNA synthesis and proteoglycan synthesis of cultured growth plate chondrocytes, thereby stimulating cartilage growth and inhibiting replacement of cartilage by bone. In situ hybridization and immunohistochemical studies indicated that ChM-1 was specifically expressed in the avascular zone of cartilage in developing bone, but not present in calcifying cartilage (Hiraki et al. 1999). More recently, troponin I (TnI) was identified by peptide microsequencing and protein database analysis as a cartilage-derived inhibitor of neovascularization. Together with troponin-C and troponin-T, TnI is a subunit of the troponin complex, which along with tropomyosin is responsible for the calcium-dependent regulation of striated muscle contraction. TnI was found to be a potent and specific inhibitor of angiogenesis in vivo and in vitro, as well as of tumour metastasis in vivo. The mechanism by which TnI inhibits endothelial cell proliferation in vitro and angiogenesis in vivo is unknown. It is possible that TnI, could, by virtue of its affinity for heparin, bind to and compete with FGF-2 and VEGF for heparin sulphate proteoglycan on the endothelial cell surface (Moses et al. 1999). Finally, the National Cancer Institute (USA) is sponsoring clinical trials of a shark cartilage extract angiogenesis inhibitor in tumour patients, since shark cartilage contains a substance that strongly inhibits the growth of new blood vessels toward solid tumours, thereby restricting tumour growth. This inhibitor, termed U-995, is composed of two single peptides with molecular mass of 10 and 14 kD. U-995 markedly inhibited endothelial proliferation and migration, and reduced TNFα-induced angiogenesis (Sheu et al. 1998).
New blood vessel formation is thus an integral part of joint development. Suppressed synthesis of antiangiogenic factors such as ChM-1 and TnI, together with enhanced production of pro-angiogenic factors such as transferrin (Carlevaro et al. 1997), by hypertrophic chondrocytes could promote the invasion of hypertrophic cartilage by blood vessels. Under certain conditions angiogenesis in the joint becomes disregulated, contributing to the pathology of joint disorders. Defective chondrocyte hypertrophy and angiogenesis may underlie impairment of skeletal growth, such as that observed in rickets. For example, rickets is induced clinically and experimentally by a deficiency of vitamin D, metabolites of which, such as 1, 25-dihydroxy vitamin D3, induce production of an angiogenic molecule by hypertrophic chondrocytes (Wang et al. 1996). In adults, angiogenesis may occur in the synovium, cartilage and entheses, during the course of the development of diseases such as RA, osteoarthritis (OA) and ankylosing spondylitis.
Angiogenesis in musculoskeletal disorders
Angiogenesis and rheumatoid arthritis
RA is a chronic autoimmune inflammatory disease, most frequently involving the small joints of hands, wrists and feet. The prevalence of RA in the populations of Western Europe and the US is approximately 1%, with a female to male ratio of 3:1. The peak onset age occurs between the fifth and sixth decades of life. In spite of many years of intensive investigation, the cause of RA still remains unknown, although current thinking favours the concept of a multifactorial disease, in which contributory genetic factors (in particular, an association with HLA-DR4) combine with environmental and possibly infectious influences to initiate disease. The primary site of inflammation in RA is the synovium, which lines the closed spaces of articular joints (Feldmann et al. 1996). Under normal conditions, the synovium is 1–2 cell layers thick, but in RA the lining increases in thickness up to 6–8 cell layers, and becomes infiltrated by cells of lympho-haematopoietic origin (chiefly activated memory CD4+T cells and macrophages). This infiltration is a result of an increase both in the recruitment of cells from the circulation by the vascular endothelium and in the retention of cells within the synovial membrane. The additional demands for oxygen and nutrients made by the infiltrating leucocytes and by the proliferating synovial lining cells would thus result in areas of hypoxia within the synovium. There have been many reports of reduced synovial fluid pO2 measurements in human arthritic joints. In a study of patients with a variety of joint disorders, the lowest synovial fluid pO2 values were found in individuals with RA. Several studies have also demonstrated that the oxygen consumption of the RA synovium (per unit weight of tissue) is increased, and that glucose is oxidized through the anaerobic, as opposed to aerobic, pathway (Blake et al. 1989; Stevens et al. 1991b). Moreover, the large volume of synovial fluid within the joints may increase intra-articular pressure and temporarily obliterate capillary flow in the synovium, compounding the hypoxic state. The resting intra-articular pressure in inflamed joints has been found to be elevated relative to normal joints, and this effect would be exarcebated during movement of joints. Such a situation of increased requirement for nutrients and oxygen is an ideal environment for enhanced angiogenesis. Endothelial cells lining the small blood vessels within RA synovium have been shown to express cell cycle-associated antigens such as PCNA and Ki67, as well as markers of angiogenesis such as integrin αVβ3 (Ceponis et al. 1998; Walsh et al. 1998). However, despite this pro-angiogenic phenotype, it may well be that the process of new blood vessel formation does not actually keep pace with synovial tissue proliferation, leading to areas of hypoperfusion, thus further perpetuating the hypoxic state in RA (Figure 1).
The presence of mRNA and protein for many factors capable of inducing angiogenesis in RA synovium is well-documented, and it is generally thought that an accumulation of signals (e.g. recurrent hypoxia, release of low/moderate levels of pro-angiogenic cytokines) may eventually breach the angiogenic ‘threshold’, leading to disregulated vascular proliferation (Paleolog & Fava 1998; Paleolog & Miotla 1998). In the context of the human arthritic joint, Brown and colleagues reported as long ago as 1980 that synovial fluids from patients with RA contained a low-molecular-weight angiogenesis factor apparently identical with that derived from tumours (Brown et al. 1980). Subsequently it was demonstrated that RA synovial fluids caused early morphological changes in endothelial cell cultures, including the formation of tubular networks morphologically resembling capillaries (Semble et al. 1985). The key molecules which may play a role in modulating angiogenesis in RA are listed in Table 2. Many of the broadly acting growth factors have been detected in RA synovial tissue (Sano et al. 1990; Sano et al. 1993; Qu et al. 1995). Both FGF-1 and FGF-2 have been demonstrated to be present in human disease and in animal models of arthritis. Synovial tissue from patients with RA and degenerative joint diseases express FGF-1, with staining present in the lining synoviocytes and at the pannus–cartilage interface, suggesting that FGF-1 may play a role in synovial hyperplasia and joint destruction. FGF-2 staining correlated with the extent and intensity of synovial mononuclear cell infiltration. Scatter factor/HGF has been found at significant levels in RA synovial fluids in a biologically active form, and anti-HGF partially neutralized the chemotactic activity for endothelial cells found in RA synovial fluids. Expression of PDGF and PD-ECGF has also been reported (Remmers et al. 1991). Immunostaining of RA synovium with anti-PDGF antibody showed predominant localization of this mitogen to macrophage-like cells, and the extent and intensity of staining correlated with mononuclear cell infiltration. PDGF receptors are expressed by synovial lining cells of the fibroblastic type, suggesting that PDGF may play a more important role in stimulating fibroblast rather than endothelial proliferation. Like HGF, PD-ECGF has been found to be present in rheumatoid synovial fluids and tissues. TGFβ has been shown to immunolocalize to the macrophages and fibroblasts in the RA synovial lining layer (Chu et al. 1992). The potential role of this growth factor in angiogenesis during the course of RA is complex, in that TGFβ exerts diverse effects on different cell types. Neutralizing anti-TGFβ antibodies have been shown to both enhance experimental arthritis (when given systemically), or to ameliorate arthritis (when given locally). In vitro, TGFβ generally exerts inhibitory rather than stimulatory effects on endothelial cell proliferation, although paradoxically in vivo TGFβ is a powerful pro-angiogenic agent. This may be due to the ability of TGFβ to induce VEGF gene expression and secretion in fibroblast and epithelial cell lines, and indeed TGFβ has been found to be by far the most powerful inducer of VEGF secretion by human synovial fibroblasts. Thus, it seems likely that in RA, TGFβ exerts its angiogenic effects largely through stimulation of VEGF secretion by fibroblasts.
VEGF is one of the more recent additions to the list of heparin-binding pro-angiogenic stimuli which are expressed in RA (Fava et al. 1994; Koch et al. 1994; Nagashima et al. 1995; Paleolog et al. 1998). The induction of VEGF expression by hypoxia makes this growth factor one of the more likely players in the generation of a pro-angiogenic phenotype in RA. Studies from our group have shown that incubation of RA synovial membrane cells in hypoxic conditions selectively upregulated release of VEGF, indicating that a component of the new blood vessel formation observed in RA may result from hypoxia-driven induction of VEGF expression (Paleolog et al. 1998). In addition, the dual role of VEGF as both an endothelial cell mitogen and vascular permeability factor is of particular relevance in RA, in which angiogenesis and joint oedema are important. VEGF has been detected in synovial fluids from RA patients, and incubation of RA synovial fluids with neutralizing anti-VEGF antibody significantly inhibited endothelial cell chemotaxis (Koch et al. 1994). RA synovial tissue has been shown to express VEGF, predominantly in macrophages and lining cells, as well as endothelial cells lining small blood vessels within the pannus. Microvascular endothelial cells in the vicinity of VEGF-positive cells express mRNA for both VEGF receptors (Fava et al. 1994; Koch et al. 1994). We have also reported that serum levels of VEGF were markedly elevated in patients with RA, and modulated by therapies such as anti-TNFα monoclonal antibody (Paleolog et al. 1998). For example, in 30 patients with established RA, serum VEGF levels were found to be 0.59 ± 0.07 ng/ml (mean ± SEM), compared with 0.22 ± 0.02 ng/ml in 53 age-matched individuals without arthritis ( P < 0.001 vs. patients with RA; Figure 2). Treatment of RA patients with anti-TNFα antibody (REMICADE) reduced serum VEGF levels to 0.38 ± 0.05 ng/ml after 2 weeks ( P < 0.001 vs. pretreatment serum VEGF); however, theses values were still significantly ( P < 0.01) higher than in normal controls (Figure 2).
Figure 2.
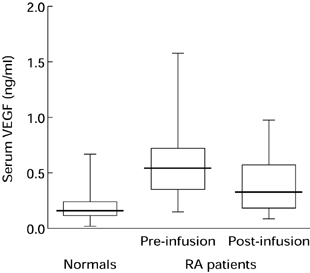
Serum VEGF levels are elevated in patients with RA: effect of anti-TNFα antibody therapy. Serum VEGF levels were measured in 30 patients with active RA, which was defined as the presence of 6 swollen joints plus at least three of four secondary criteria (duration of morning stiffness > 45 min, > 6 tender or painful joints, erythrocyte sedimentation rate > 28 mm/h, C-reactive protein > 20 mg/l). Patients were enrolled in clinical trials of a human/murine chimeric monoclonal IgG1k anti-TNFα antibody (cA2, REMICADE), produced by Centocor, USA. Serum samples were obtained before and 2 weeks after a single infusion of cA2. For comparison, serum VEGF was measured in 53 age- and sex-matched individuals without evidence of arthritis. VEGF levels were assayed by ELISA. Boxes are interquartile ranges; bars indicate median VEGF levels.
There are many other factors which may regulate new blood vessel formation in RA, including thrombospondin-1 (TSP-1) and TNFα (Brennan et al. 1989). TSP-1 is generally found in the matrix of synovial vessels, although immunoreactivity for TSP-1 was also seen associated with macrophages in synovium and in the synovial lining layer. It is possible that TSP-1 plays both an inductive and inhibitory role on angiogenesis in RA, depending upon the context within which the endothelium is exposed to TSP-1. TNFα, which plays a pivotal role in the pathogenesis of RA, exerts both stimulatory and inhibitory effects on the angiogenic process, and it is likely that this cytokine will exert either direct or indirect effects on blood vessel formation in RA. Certainly production by synovial joint cells of angiogenic cytokines such as VEGF is at least in part induced by TNFα (Paleolog et al. 1998). Synovial fluids of patients with RA were shown to induce angiogenesis in a mouse model. This effect could be neutralized using monoclonal antibodies against TNFα, suggesting that angiogenesis observed in rheumatoid synovitis may be due, at least in part, to the angiogenic effect of locally produced TNFα. The beneficial effects of anti-TNFα in both animal models of arthritis and in human RA have also led to the hypothesis that modulation of the activation status of vascular endothelium, in terms of either leucocyte extravasation or proliferation, is an important consequence of anti-TNFα therapy (Paleolog et al. 1996). Finally, the heparin-binding protein angiogenin has been found to be present at relatively high levels in synovial fluids from RA patients, and colocalized with FGF-2 in synovial membranes (Hosaka et al. 1995). Synovial lining cells, macrophages, endothelial cells and vascular smooth muscle cells were immunopositive for angiogenin.
In summary, many growth factors and cytokines are potential inducers of angiogenesis in RA. The most likely scenario is that an accumulation of suboptimal levels of signals, such as VEGF, chemokines or growth factors, will eventually propel the synovial microvasculature towards disregulated angiogenesis, and hence increased synovial tissue mass. Although angiogenesis has always been considered to be a detrimental process in RA, in that it is thought to maintain the destructive pannus, it is also possible that new vessel formation may be part of a reparative process, to reconstruct the damaged bone/cartilage. Arguing against this concept is the demonstration of the effectiveness of angiogenesis inhibition in animal models of arthritis. In a rat model of arthritis, in which disease is induced by injection of heterologous collagen, leading to synovitis, joint erosion and associated neovascularization, the antiproliferative agent TNP-470 was found to suppress established disease, in parallel with marked inhibition of pannus formation and neovascularization (Peacock et al. 1992; Oliver et al. 1994; Oliver et al. 1995). More recently, specific antagonists of αVβ3 were used in a rabbit model resembling human RA, in which arthritis was induced by injection of ovalbumin and FGF-2. Intra-articular injections of αVβ3 antibody decreased vascularity in the arthritic synovium, associated with a significant decrease in all arthritic parameters including joint swelling and synovitis. More importantly, the beneficial effects of angiogenesis inhibition were evident even in chronic, well established disease (Storgard et al. 1999). It therefore seems likely that like psoriasis and tumours, RA can also be classified as an ‘angiogenesis-dependent disease’.
Angiogenesis and osteoarthritis
OA is the commonest joint disorder, and affects the majority of people over the age of 65. Genetic and systemic factors predispose to the condition, whereas local biomechanical factors dictate the site and severity of joint disease. In contrast to RA, which is a systemic disease with predilection for the joints, OA is predominantly characterized by pathological changes at one or more of the large, weight-bearing joints of the lower extremities. Although traditionally viewed as failure of the joint to withstand acute or chronic mechanical insult, it is now widely believed that the osteoarthritic process is not solely a degenerative one, but rather that age-related reparative responses occur, the net result of which is to potentiate instead of repairing joint damage. Key pathological features include degeneration of cartilage, irregular thickening and remodelling of subchondral bone with sclerosis and cystic change, together with marginal soft tissue changes and osteophyte growth. Advanced cases also show moderate patchy synovitis and joint capsule thickening. Disregulated tissue turnover in weight-bearing cartilage and subchondral bone are thought to be responsible. The pathological changes are mediated by local production of cytokines and proteases from cells of the cartilage, bone and synovium.
Although relatively little is known about vascular responses in OA, nevertheless several lines of evidence suggest that a pro-angiogenic state exists in osteoarthritic joints (Table 2). Firstly, as in RA, the intra-articular milieu in OA is potently pro-angiogenic. Hypoxia can occur, and indeed it has been suggested that hypoxia caused by reduced subchondral blood flow plays a role in the development of tissue damage in OA. Certainly in patients with late stage unilateral OA, the subchondral pO2 in the diseased hip was significantly lower than the pO2 in the normal hip (Kiaer et al. 1988). Inflammatory synovitis is observed, particularly in late stages of OA, thereby providing a potential source of pro-angiogenic molecules such as TNFα (Westacott & Sharif 1996). The ability of RA synovial fluids to induce capillary-like structure formation in human endothelial cell cultures were described earlier, and similar effects of OA fluids have been demonstrated by the same authors (Semble et al. 1985). Synovial fluid white cell counts or serum rheumatoid factor titres were not associated with appearance or time of onset of capillary-like structure formation, implying the relative lack of importance of joint inflammation for these effects. A number of pro-angiogenic factors can be isolated from osteoarthritic joints, one of which is the poorly characterized 600 D endothelial-specific growth factor ESAF. In two-thirds of patients with OA analysed, there were sufficient levels of ESAF in the synovial fluid to induce angiogenesis in a chick chorioallantoic membrane assay. This compared with only 15% of RA joint effusions (Brown et al. 1980). Far fewer inflammatory cells were present in the OA effusions compared with either sero-positive RA or sero-negative arthritis groups, again implying that the production of angiogenic activity as measured by the presence of ESAF could not be solely attributed to inflammatory cells. Our own studies have demonstrated that VEGF may play a role in OA. We observed that serum levels of VEGF in patients with OA were higher than in age-matched controls, but lower than in patients with RA (Ballara et al. unpublished observation), in agreement with published data (Harada et al. 1998). VEGF is expressed in lining cells and fibroblasts in OA synovial tissue, albeit rather weakly relative to RA (Koch et al. 1994; Nagashima et al. 1995; Jackson et al. 1997). Other pro-angiogenic molecules expressed in OA synovium include HGF, PD-ECGF, FGF-2 and angiogenin. The net effect appears to be increased vascularity in OA synovium and invasion of cartilage by new vessels from underlying bone (Stephens et al. 1979).
Neovascularization may be important in three processes implicated in the pathogenesis of OA — changes in the subchondral plate, joint remodelling and osteophyte formation (Brown & Weiss 1988) (Figure 1). The subchondral bone is likely to be crucial to the pathogenesis of OA. Until recently it was accepted that OA was primarily a disorder of cartilage with reactive subchondral changes. There is now some evidence that subchondral pathology may be equally important in generation of joint changes in OA, although at present it is unclear whether subchondral vascular responses represent healing phenomena or contribute to joint pathology. Subchondral bone changes in OA include redistribution of blood supply, marrow hypertension, oedema, micronecrosis and microfractures. Vascular invasion, an essential element of fracture repair, may reflect ongoing fracture healing at the joint margin. The subchondral region appears to be more sensitive to mechanical stress and impairment of the microvascular supply (for example due to subchondral sclerosis), resulting in under-nourishment of cartilage. Cartilage changes similar to human OA can be precipitated by the induction of prolonged ischaemia of underlying subchondral bone in rabbits (Graf et al. 1992). In addition to vascular ischaemia or shunting, cartilage thinning may be induced by direct extension of the subchondral growth plate by calcification of deep-zone articular cartilage. This process is well documented in OA and requires vascular invasion prior to ossification. Such encroachment has been associated with observations of unremodelled cartilage islands in the bone beneath the plate. Incomplete remodelling of this kind may result in failure of the joint to adapt to changing mechanical stresses or impair load carrying, causing eventual cartilage degeneration by mechanical failure.
In addition to its role in subchondral bone, neovascularization may potentiate cartilage damage directly in a number of ways. The disruption of net antiangiogenic activity and resultant cartilage vascularization can be expected to compromise cartilage biomechanical properties. Failure of cartilage to produce protease inhibitors to block neovascularization has been put forward as a basis for initiation of cartilage loss in response to mechanical loading in OA (Stephens et al. 1979). Alternatively, cartilage and bone may be directly degraded by disturbance in the control of endogenous enzymes normally involved in remodelling. Connective tissue degradation is dependent on three MMPs, namely interstitial collagenase (MMP-1), stromelysin-1 (MMP-3) and gelatinase A (MMP-2). These enzymes are also involved in breakdown of the vascular basement membrane and therefore are an important feature of angiogenesis. ESAF has been shown to activate all three enzymes and is also the only physiological molecule able to reactivate each MMP when it is complexed with tissue inhibitor of metalloproteinase (TIMP) (McLaughlin & Weiss 1996). ESAF may therefore be an important molecule both in terms of angiogenic responses and also of promotion of metalloproteinase directed connective tissue degradative effects within arthritic joints. The presence of ESAF in other conditions characterized by noninflammatory angiogenesis (e.g. bone growth, diabetic retinopathy) (Odedra & Weiss 1991) suggests a role for this factor in noninflammatory as opposed to inflammatory angiogenesis.
Another integral feature of OA is the formation of bony outgrowths called osteophytes, which occur at sites of mechanical stress such as insertion points of ligaments and tendons (entheses) and at joint margins close to articular cartilage. Osteophyte formation starts from activation of periosteal layers to an initial cartilaginous structure, which subsequently ossifies. A prerequisite for this process, akin to endochondral ossification, is the invasion of cartilage by new vessels. Capillary encroachment into the subchondral plate and deep zone of articular cartilage has been observed at an early stage of osteophyte formation. Since normal cartilage contains inhibitors of angiogenesis and of MMP activity, this natural barrier to vascular invasion must be breached for osteophyte formation to occur. Similar phenomena called syndesmophytes occur in other debilitating rheumatological conditions such as ankylosing spondylitis. This disease is characterized by inflammatory sacro-iliitis and spondylitis. Subsequent bony fusion of the sacro-iliac joints occurs and the vertebral spine becomes fused as a result of bridging of the syndesmophytes. The resulting spinal rigidity is a significant cause of pain, disability and mortality. As in OA, ESAF activity is found in synovial fluid in 66% of patients with ankylosing spondylitis (Brown et al. 1980). ESAF was also raised in the sera of patients with ankylosing spondylitis compared with controls and higher levels correlated with degree of enthesitis as assessed by a validated enthesitis index (Jones et al. 1994). This index correlated with degree of radiological sacro-iliitis and syndesmophyte formation. No correlations between serum ESAF levels and markers of inflammatory disease activity were found. In vitro, ESAF activity appears to be associated with osteogenesis, since cultured growth plate chondrocytes synthesize a cartilage-like matrix when ESAF is added (McFarland et al. 1990). Angiogenesis may therefore play an important role in the excessive new bone formation that characterizes both these OA and ankylosing spondylitis.
At present no agent can effectively control new bone formation. As discussed earlier, the angiogenesis inhibitor TNP-470 inhibits ectopic bone formation following implantation of pellets containing recombinant human BMP-2 in mice (Mori et al. 1998). This raises the possibility that antiangiogenic agents may have a therapeutic role in conditions such as OA and ankylosing spondylitis, where they may be useful in inhibiting both excessive bone formation and bone destruction by inflammatory tissues.
Angiogenesis in vertebral disc and soft tissue disease
Other musculoskeletal conditions include back pain, which in particular is a major clinical problem, with a prevalence of 14% at any one time and a lifetime prevalence of 60–80%. A common cause of acute back pain is posterior herniation of the intervertebral disc following a minor mechanical stress such as bending or lifting. There is clear evidence that neovascularization is associated with this condition. The intervertebral disc consists of a central gelatinous nucleus pulposus, composed predominantly of proteoglycan, surrounded by a fibrous annulus constructed of concentric layers of collagen fibres. It is vascularized in early life but the nucleus pulposus becomes avascular after adolescence. Nutritional conditions are highly critical in the normal intervertebral disc. Blood vessels are found only at the margin of the annulus fibrosus and in the vertebral marrow spaces, where permeability and diffusion coefficients for glucose are low (Maroudas et al. 1975). Histological and angiographic studies of lumbar spines show increasing vascularity of the annulus with advancing age and degeneration of the disc (Kauppila 1995). Herniated lumbar discs excised at surgery have shown a high frequency of microvessels at the superficial edges of herniated nucleus pulposus, a finding not mirrored in control unherniated age matched specimens.
Little is known about whether angiogenic responses predispose to disc prolapse or represent an attempt to heal a degenerating disc. Vascularization has been shown to occur prior to degenerative changes in intervertebral discs, suggesting that disturbances in the nutritional supply may precede degenerative change (Kauppila 1995). The prevalence of blood vessels in herniated discs is very high (> 90%), and these vessels predominate in close proximity to disc cells, suggesting a role in the neovascularization process after disc injury. VEGF-and PDGF-immunopositive capillaries were identified in over 70% of disc herniation samples, whereas FGF-2 was found in 81% of samples in the same study (Tolonen et al. 1997). In this study FGF-2 was predominantly localized to small blood vessels and scattered disc cells. In addition to being a potent pro-angiogenic mediator, FGF-2 regulates extracellular proteolysis and may have effects on proteolytic activity and resorption of the herniated disc. Resorption occurs in the majority of herniated discs. Once prolapsed, it is likely that neovascularization augments resorption by promoting the delivery of phagocytic cells.
Other components of the musculoskeletal system where vascularity and angiogenesis are likely to be important are the peri-articular structures such as tendons, ligaments, entheses and joint capsules. Mechanical damage to these structures is extremely common, resulting in complete or partial tears and inflammatory responses from overuse. The vascular supply to many of these regions is precarious. For example, in the rotator cuff of the shoulder, a critical zone of low blood flow and vascularity occurs in the supraspinatus tendon at a common site for tendonitis and traumatic damage (Determe et al. 1996). Poor vascularity has been found in studies of a number of ruptured tendons including Achilles, posterior tibial and calcaneal tendons (Carr & Norris 1989). As with wound healing, repair of damaged tendons or ligaments seems to require pro-angiogenic responses. FGF-2 enhanced the rate of repair and correlated with increased neovascularization and granulation tissue formation in surgically induced lacerations in canine anterior cruciate ligaments. Pro-angiogenic mediators have also been used in other systems to promote healing — for example angiogenin implantation into experimentally injured menisci (King & Vallee 1991). Physical training — a standard therapeutic strategy — has also been shown to promote vascularization and repair at the myotendinous junction in an experimental system, whereas only limited post-traumatic healing occurs in tissues in which vascularization and angiogenic responses are poor, such as the ligaments in and around the knee (Kvist et al. 1995).
In contrast to acute injuries to soft tissues, chronic repetitive strain often leads to hypervascularity. Severe recalcitrant soft tissue disease requiring surgical treatment, such as chronic lateral epicondylitis or Achilles/patellar tendonitis is associated with prominent angiogenesis (Astrom & Rausing 1995). The mechanisms underlying these processes are unclear but varying degrees of chronic inflammatory, fibrotic and degenerative change are observed. In certain conditions such as adhesive capsulitis of the shoulder, a number of pro-inflammatory cytokines with angiogenic properties have been demonstrated (e.g. PDGF, TNFα, HGF and TGFβ) at a higher frequency than patients with synovitis without fibrotic change (Rodeo et al. 1997).
Angiogenesis inhibition: a therapeutic strategy for the treatment of RA?
It has been recognized for over 20 years that the vasculature plays a key role in both the development of the normal joint and in the pathogenesis of musculoskeletal disorders such as RA, OA and ankylosing spondylitis. The general consensus is that angiogenesis is a detrimental component of the disease process. It is interesting that in RA, the disease in which neovascularization has been most extensively studied, it is still unclear whether the density of blood vessels in the arthritic synovium per unit area is increased, unchanged or even reduced. It is thought that although angiogenesis occurs during the development of the arthritic pannus, the process of new blood vessel formation may not actually keep pace with synovial tissue proliferation, and hence the vessel density may, if anything, appear to be reduced. A study in 1991 suggested that capillaries in RA synovium are distributed more deeply, and are less densely arranged, than in normal synovial tissue (Stevens et al. 1991a). Whether or not the density of blood vessels per unit area is increased, unchanged or decreased in the RA synovium, there is undoubtedly formation of new blood vessels in RA, as demonstrated by the expression of many pro-angiogenic mediators and molecules associated with angiogenic vasculature, such as integrin αVβ3. It has been demonstrated that in RA, 1% of endothelial cells lining the small blood vessels of the synovium express cell cycle-associated antigens such as PCNA and Ki67 (Ceponis et al. 1998; Walsh et al. 1998). These values are higher than in control samples, suggesting that pro-angiogenic status is favoured in RA. The number of synovial blood vessels has been found to correlate with synovial cell hyperplasia, mononuclear cell infiltration and indices of joint tenderness (Rooney et al. 1988).
In terms of antiangiogenic therapy in RA, it is interesting that some of the antirheumatic drugs currently in clinical use or in trials may exert effects on the vasculature. Studies from our group at the Kennedy Institute of Rheumatology have demonstrated that anti-TNFα monoclonal antibody, which results in amelioration of disease symptoms, modulates VEGF levels. Serum VEGF concentrations are significantly elevated in the sera of RA patients relative to nonarthritic individuals, and correlate with disease activity. Treatment of RA patients with anti-TNFα markedly decreased serum VEGF (Figure 2), and this reduction has been found to correlate with alterations in disease parameters such as C-reactive protein levels and swollen joint scores. These observations suggested that the clinical benefits of anti-TNFα antibody therapy are associated with a reduction in circulating VEGF levels, which may lead to decreased joint vascularity and swelling (possibly due to reduced vascular permeability) (Paleolog et al. 1998). Other antirheumatic drugs, such as indomethacin, methotrexate and corticosteroids inhibit angiogenesis. Methotrexate, at doses which are attained in the serum of RA patients treated with this drug, inhibited both basal and stimulated endothelial proliferation in vitro. Thiol containing and gold compounds, also used therapeutically in RA, may modulate neovascularization indirectly by inhibiting the production of angiogenic activity by macrophages.
Anti-angiogenic therapies, such as TNP-470 and Taxol, have been demonstrated to be effective in animal models of arthritis (Peacock et al. 1992; Oliver et al. 1994, 1995). In addition, thalidomide, as well as an endogenous metabolite of estradiol, 2-methoxyestradiol, have also been found to suppress disease in collagen-induced arthritis via antiangiogenic effects (Josefsson & Tarkowski 1997; Oliver et al. 1998). More specifically, antagonists of the neovascular integrin αVβ3 reduced joint swelling and synovitis in a rabbit model of arthritis. Based on the close association of angiogenesis and VEGF in arthritic disease, a hypothesis could also be made that direct suppression of VEGF activity should be effective in RA. However, the benefits of VEGF-targeted therapy in arthritis have yet to be proven.
Conclusions
The pathogenesis of most musculoskeletal problems remains obscure, and it is therefore not surprising that treatment is unsatisfactory and is based largely on the control of symptoms, until damage is of sufficient severity to warrant surgical intervention. In inflammatory joint disease, early events initiating and maintaining the immune reaction are unresolved. Standard treatments are largely empirical, with little knowledge of how they work, or with nonspecific anti-inflammatory effects. In recent years our understanding of the inflammatory process in disease such as RA has increased, and this has led to the introduction of more rational therapies targeting specific elements in the pro-inflammatory response — for example anti-TNFα monoclonal antibody (REMICADE) (Elliott et al. 1994; Paleolog et al. 1996, 1998) or the TNF p75 receptor:Fc fusion protein (Etanercept) (Moreland et al. 1997, 1999). These have been highly successful in alleviating symptoms and signs of inflammatory joint disease but like more established treatments, prevention of joint damage and disability have only been demonstrated in the short to medium term. Long-term benefits are limited and a considerable proportion of patients are resistant to standard therapies. Moreover, monitoring of response to treatment rests on surrogate markers of joint inflammation, such as acute phase proteins. Even effective control of inflammation does not necessarily prevent deterioration of joint damage in RA or progressive spinal fusion in ankylosing spondylitis. Therefore, a better understanding of the processes contributing to the disease process is essential if better disease monitoring and therapeutic strategies are to be developed.
The microvascular supply and processes controlling blood vessel density and distribution may play an important role in many diseases of the musculoskeletal system. In some processes such as an acutely injured tendon, promotion of the pro-angiogenic drive may be beneficial for repair of damaged tissue. In diseases such as RA, the vasculature promotes delivery of inflammatory mediators, cells and nutrients and therefore potentiates ongoing disease. Anti-angiogenic therapy may be a useful adjunct to existing anti-inflammatory approaches in this setting. The angiogenic drive is also likely to be an important component of OA and in diseases characterized by new bone formation. As the complex processes governing neovascularization are clarified, modulation of vascular growth is fast becoming a reality. Having demonstrated efficacy in animal models of disease, including inflammatory arthritis (Storgard et al. 1999), angiogenic modulators now in trials in a variety of human diseases (Brem 1998). Apart from wound healing and the female reproductive cycle, there is minimal requirement for angiogenesis in adults and undesirable side-effects of such treatment are likely to be modest. As we have outlined in this review, therapy directed against blood vessels has enormous clinical potential in musculoskeletal disease. However, further elucidation of the diverse roles of vascular growth and redistribution in the joints remain an essential prerequisite to realize this potential.
Acknowledgments
The authors are grateful to Professor R.N. Maini and Professor Marc Feldmann for their advice and encouragement. We also gratefully acknowledge the support of the Arthritis Research Campaign of Great Britain, AstraZeneca Pharmaceuticals (J.M.M.) and the Charing Cross Hospital Trustees (S.B.).
References
- Achen MG, Stacker SA. The vascular endothelial growth factor family; proteins which guide the development of the vasculature. Int. J. Exp. Pathol. 1998;79:255–265. doi: 10.1046/j.1365-2613.1998.700404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahara T, Murohara T, Sullivan A, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- Astrom M, Rausing A. Chronic Achilles tendinopathy. A survey of surgical and histopathologic findings. Clin. Orthop. 1995;316:151–164. [PubMed] [Google Scholar]
- Blake DR, Merry P, Unsworth J, et al. Hypoxic-reperfusion injury in the inflamed human joint. Lancet. 1989;1:289–293. doi: 10.1016/s0140-6736(89)91305-6. 8633. [DOI] [PubMed] [Google Scholar]
- Brem S. Angiogenesis antagonists: current clinical trials. Angiogenesis. 1998;2:9–20. doi: 10.1023/a:1009068807898. [DOI] [PubMed] [Google Scholar]
- Brennan FM, Chantry D, Jackson A, Maini R, Feldmann M. Inhibitory effect of TNF alpha antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet. 1989;2:244–247. doi: 10.1016/s0140-6736(89)90430-3. 8657. [DOI] [PubMed] [Google Scholar]
- Brown LF, Detmar M, Claffey K, et al. Vascular permeability factor/vascular endothelial growth factor: a multifunctional angiogenic cytokine. EXS. 1997;79:233–269. doi: 10.1007/978-3-0348-9006-9_10. [DOI] [PubMed] [Google Scholar]
- Brown RA, Weiss JB, Tomlinson IW, Phillips P, Kumar S. Angiogenic factor from synovial fluid resembling that from tumours. Lancet. 1980;1:682–685. [PubMed] [Google Scholar]
- Brown RA, Weiss JB. Neovascularisation and its role in the osteoarthritic process. Ann. Rheum. Dis. 1988;47:881–885. doi: 10.1136/ard.47.11.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess WH, Maciag T. The heparin-binding (fibroblast) growth factor family of proteins. Annu. Rev. Biochem. 1989;58:575–606. doi: 10.1146/annurev.bi.58.070189.003043. [DOI] [PubMed] [Google Scholar]
- Cao R, Wu HL, Veitonmaki N, et al. Suppression of angiogenesis and tumor growth by the inhibitor K1–5 generated by plasmin-mediated proteolysis. Proc. Natl. Acad. Sci. USA. 1999;96:5728–5733. doi: 10.1073/pnas.96.10.5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlevaro MF, Albini A, Ribatti D, et al. Transferrin promotes endothelial cell migration and invasion: implication in cartilage neovascularization. J. Cell Biol. 1997;136:1375–1384. doi: 10.1083/jcb.136.6.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr AJ, Norris SH. The blood supply of the calcaneal tendon. J. Bone Joint Surg. 1989;71:100–101. doi: 10.1302/0301-620X.71B1.2914976. [DOI] [PubMed] [Google Scholar]
- Ceponis A, Konttinen YT, Imai S, et al. Synovial lining, endothelial and inflammatory mononuclear cell proliferation in synovial membranes in psoriatic and reactive arthritis: a comparative quantitative morphometric study. Br. J. Rheumatol. 1998;37:170–178. doi: 10.1093/rheumatology/37.2.170. [DOI] [PubMed] [Google Scholar]
- Chu CQ, Field M, Allard S, Abney E, Feldmann M, Maini RN. Detection of cytokines at the cartilage/pannus junction in patients with rheumatoid arthritis: implications for the role of cytokines in cartilage destruction and repair. Br. J. Rheumatol. 1992;31:653–661. doi: 10.1093/rheumatology/31.10.653. [DOI] [PubMed] [Google Scholar]
- Claesson-Welsh L. Platelet-derived growth factor receptor signals. J. Biol. Chem. 1994;269:32023–32026. [PubMed] [Google Scholar]
- Colville-Nash PR, Willoughby DA. Growth factors in angiogenesis: current interest and therapeutic potential. Mol. Med. Today. 1997;3:14–23. doi: 10.1016/S1357-4310(96)10048-4. [DOI] [PubMed] [Google Scholar]
- Cox DA, Maurer T. Transforming growth factor-beta. Clin. Immunol. Immunopathol. 1997;83:25–30. doi: 10.1006/clin.1996.4308. [DOI] [PubMed] [Google Scholar]
- Decker JL. American Rheumatism Association nomenclature and classification of arthritis and rheumatism (1983) Arthritis Rheum. 1983;26:1029–1032. doi: 10.1002/art.1780260813. [DOI] [PubMed] [Google Scholar]
- Determe D, Rongieres M, Kany J, et al. Anatomic study of the tendinous rotator cuff of the shoulder. Surg. Radiol. Anat. 1996;18:195–200. doi: 10.1007/BF02346127. [DOI] [PubMed] [Google Scholar]
- Eisenstein R, Kuettner KE, Neapolitan C, Soble LW, Sorgente N. The resistance of certain tissues to invasion. III. Cartilage extracts inhibit the growth of fibroblasts and endothelial cells in culture. Am. J. Pathol. 1975;81:337–347. [PMC free article] [PubMed] [Google Scholar]
- Elliott MJ, Maini RN, Feldmann M, et al. Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet. 1994;344:1105–1110. doi: 10.1016/s0140-6736(94)90628-9. [DOI] [PubMed] [Google Scholar]
- Fava RA, Olsen NJ, Spencer-Green G, et al. Vascular permeability factor/endothelial growth factor (VPF/VEGF): accumulation and expression in human synovial fluids and rheumatoid synovial tissue. J. Exp. Med. 1994;180:341–346. doi: 10.1084/jem.180.1.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann M, Brennan FM, Maini RN. Rheumatoid arthritis. Cell. 1996;85:307–310. doi: 10.1016/s0092-8674(00)81109-5. [DOI] [PubMed] [Google Scholar]
- Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat. Med. 1999;5:623–628. doi: 10.1038/9467. [DOI] [PubMed] [Google Scholar]
- Graf J, Neusel E, Freese U, Simank HG, Niethard FU. Subchondral vascularisation and osteoarthritis. Int. Orthop. 1992;16:113–117. [PubMed] [Google Scholar]
- Harada M, Mitsuyama K, Yoshida H, et al. Vascular endothelial growth factor in patients with rheumatoid arthritis. Scand. J. Rheumatol. 1998;27:377–380. doi: 10.1080/03009749850154429. [DOI] [PubMed] [Google Scholar]
- Harris AL. Anti-angiogenesis therapy and strategies for integrating it with adjuvant therapy. Recent Results Cancer Res. 1998;152:341–352. doi: 10.1007/978-3-642-45769-2_33. [DOI] [PubMed] [Google Scholar]
- Hiraki Y, Mitsui K, Endo N, et al. Molecular cloning of human chondromodulin-I, a cartilage-derived growth modulating factor, and its expression in Chinese hamster ovary cells. Eur J. Biochem. 1999;260:869–878. doi: 10.1046/j.1432-1327.1999.00227.x. [DOI] [PubMed] [Google Scholar]
- Hosaka S, Shah MR, Barquin N, Haines GK, Koch AE. Expression of basic fibroblast growth factor and angiogenin in arthritis. Pathobiology. 1995;63:249–256. doi: 10.1159/000163957. [DOI] [PubMed] [Google Scholar]
- Jackson JR, Minton JA, Ho ML, Wei N, Winkler JD. Expression of vascular endothelial growth factor in synovial fibroblasts is induced by hypoxia and interleukin 1beta. J. Rheumatol. 1997;24:1253–1259. [PubMed] [Google Scholar]
- Jones PB, Makki RJ, Weiss JB. Endothelial cell stimulating angiogenesis factor — a new biological marker for disease activity in ankylosing spondylitis? Br. J. Rheumatol. 1994;33:332–335. doi: 10.1093/rheumatology/33.4.332. [DOI] [PubMed] [Google Scholar]
- Josefsson E, Tarkowski A. Suppression of type II collagen-induced arthritis by the endogenous estrogen metabolite 2-methoxyestradiol. Arthritis Rheum. 1997;40:154–163. doi: 10.1002/art.1780400120. [DOI] [PubMed] [Google Scholar]
- Kauppila LI. Ingrowth of blood vessels in disc degeneration. Angiographic and histological studies of cadaveric spines. J. Bone Joint Surg. Am. 1995;77:26–31. doi: 10.2106/00004623-199501000-00004. [DOI] [PubMed] [Google Scholar]
- Kiaer T, Gronlund J, Sorensen KH. Subchondral pO2, pCO2, pressure, pH, and lactate in human osteoarthritis of the hip. Clin. Orthop. 1988;229:149–155. [PubMed] [Google Scholar]
- King TV, Vallee BL. Neovascularisation of the meniscus with angiogenin. An experimental study in rabbits. J. Bone Joint Surg. 1991;73:587–590. doi: 10.1302/0301-620X.73B4.1712788. [DOI] [PubMed] [Google Scholar]
- Koch AE, Harlow LA, Haines GK, et al. Vascular endothelial growth factor. A cytokine modulating endothelial function in rheumatoid arthritis. J. Immunol. 1994;152:4149–4156. [PubMed] [Google Scholar]
- Kvist M, Hurme T, Kannus P, et al. Vascular density at the myotendinous junction of the rat gastrocnemius muscle after immobilization and remobilization. Am. J. Sports Med. 1995;23:359–364. doi: 10.1177/036354659502300320. [DOI] [PubMed] [Google Scholar]
- Lawrence RC, Hochberg MC, Kelsey JL, et al. Estimates of the prevalence of selected arthritic and musculoskeletal diseases in the United States. J. Rheumatol. 1989;16:427–441. [PubMed] [Google Scholar]
- Losordo DW, Vale PR, Symes JF, et al. Gene therapy for myocardial angiogenesis: initial clinical results with direct myocardial injection of phVEGF165 as sole therapy for myocardial ischemia. Circulation. 1998;98:2800–2804. doi: 10.1161/01.cir.98.25.2800. [DOI] [PubMed] [Google Scholar]
- Lynch CN, Wang YC, Lund JK, Chen YW, Leal JA, Wiley SR. TWEAK induces angiogenesis and proliferation of endothelial cells. J. Biol. Chem. 1999;274:8455–8459. doi: 10.1074/jbc.274.13.8455. [DOI] [PubMed] [Google Scholar]
- Maroudas A, Stockwell RA, Nachemson A, Urban J. Factors involved in the nutrition of the human lumbar intervertebral disc: cellularity and diffusion of glucose in vitro. J. Anat. 1975;120:113–130. [PMC free article] [PubMed] [Google Scholar]
- McFarland CD, Brown RA, McLaughlin B, Ali SY, Weiss JB. Production of endothelial cell stimulating angiogenesis factor (ESAF) by chondrocytes during in vitro cartilage calcification. Bone Miner. 1990;11:319–333. doi: 10.1016/0169-6009(90)90028-e. [DOI] [PubMed] [Google Scholar]
- McLaughlin B, Weiss JB. Endothelial-cell-stimulating angiogenesis factor (ESAF) activates progelatinase A (72 kDa type IV collagenase), prostromelysin 1 and procollagenase and reactivates their complexes with tissue inhibitors of metalloproteinases: a role for ESAF in non-inflammatory angiogenesis. Biochem. J. 1996;317:739–745. doi: 10.1042/bj3170739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreland LW, Baumgartner SW, Schiff MH, et al. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75) -Fc fusion protein. N Engl. J. Med. 1997;337:141–147. doi: 10.1056/NEJM199707173370301. [DOI] [PubMed] [Google Scholar]
- Moreland LW, Schiff MH, Baumgartner SW, et al. Etanercept therapy in rheumatoid arthritis. A randomized, controlled trial. Ann. Intern. Med. 1999;130:478–486. doi: 10.7326/0003-4819-130-6-199903160-00004. [DOI] [PubMed] [Google Scholar]
- Mori S, Yoshikawa H, Hashimoto J, et al. Antiangiogenic agent (TNP-470) inhibition of ectopic bone formation induced by bone morphogenetic protein-2. Bone. 1998;22:99–105. doi: 10.1016/s8756-3282(97)00248-2. [DOI] [PubMed] [Google Scholar]
- Moses MA, Sudhalter J, Langer R. Identification of an inhibitor of neovascularization from cartilage. Science. 1990;248:1408–1410. doi: 10.1126/science.1694043. [DOI] [PubMed] [Google Scholar]
- Moses MA, Wiederschain D, Wu I, et al. Troponin I is present in human cartilage and inhibits angiogenesis. Proc. Natl. Acad. Sci. USA. 1999;96:2645–2650. doi: 10.1073/pnas.96.6.2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashima M, Yoshino S, Ishiwata T, Asano G. Role of vascular endothelial growth factor in angiogenesis of rheumatoid arthritis. J. Rheumatol. 1995;22:1624–1630. [PubMed] [Google Scholar]
- Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13:9–22. [PubMed] [Google Scholar]
- Odedra R, Weiss JB. Low molecular weight angiogenesis factors. Pharmacol. Ther. 1991;49:111–124. doi: 10.1016/0163-7258(91)90025-h. [DOI] [PubMed] [Google Scholar]
- Oliver SJ, Banquerigo ML, Brahn E. Suppression of collagen-induced arthritis using an angiogenesis inhibitor, AGM-1470, and a microtubule stabilizer, taxol. Cell Immunol. 1994;157:291–299. doi: 10.1006/cimm.1994.1223. [DOI] [PubMed] [Google Scholar]
- Oliver SJ, Cheng TP, Banquerigo ML, Brahn E. Suppression of collagen-induced arthritis by an angiogenesis inhibitor, AGM-1470, in combination with cyclosporin: reduction of vascular endothelial growth factor (VEGF) Cell Immunol. 1995;166:196–206. doi: 10.1006/cimm.1995.9978. [DOI] [PubMed] [Google Scholar]
- Oliver SJ, Cheng TP, Banquerigo ML, Brahn E. The effect of thalidomide and 2 analogs on collagen induced arthritis. J. Rheumatol. 1998;25:964–969. [PubMed] [Google Scholar]
- O'Reilly MS, Boehm T, Shing Y, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- Paleolog EM. Angiogenesis: a critical process in the pathogenesis of RA — a role for VEGF? Br. J. Rheumatol. 1996;35:917–919. doi: 10.1093/rheumatology/35.10.917. [DOI] [PubMed] [Google Scholar]
- Paleolog EM, Hunt M, Elliott MJ, Feldmann M, Maini RN, Woody JN. Deactivation of vascular endothelium by monoclonal anti-tumor necrosis factor alpha antibody in rheumatoid arthritis. Arthritis Rheum. 1996;39:1082–1091. doi: 10.1002/art.1780390703. [DOI] [PubMed] [Google Scholar]
- Paleolog EM, Fava RA. Angiogenesis in rheumatoid arthritis. Implications for Future Therapeutic Strategies. Springer Seminars in Immunopathology. 1998;20:73–94. doi: 10.1007/BF00832000. [DOI] [PubMed] [Google Scholar]
- Paleolog EM, Miotla JM. Angiogenesis in arthritis: role in disease pathogenesis and as a potential therapeutic target. Angiogenesis. 1998;2:295–307. doi: 10.1023/a:1009229508096. [DOI] [PubMed] [Google Scholar]
- Paleolog EM, Young S, Stark AC, McCloskey RV, Feldmann M, Maini RN. Modulation of angiogenic vascular endothelial growth factor (VEGF) by TNFα and IL-1 in rheumatoid arthritis. Arthritis Rheum. 1998;41:1258–1265. doi: 10.1002/1529-0131(199807)41:7<1258::AID-ART17>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Peacock DJ, Banquerigo ML, Brahn E. Angiogenesis inhibition suppresses collagen arthritis. J. Exp. Med. 1992;175:1135–1138. doi: 10.1084/jem.175.4.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Z, Huang XN, Ahmadi P, et al. Expression of basic fibroblast growth factor in synovial tissue from patients with rheumatoid arthritis and degenerative joint disease. Lab. Invest. 1995;73:339–346. [PubMed] [Google Scholar]
- Remmers EF, Sano H, Lafyatis R, et al. Production of platelet derived growth factor B chain (PDGF-B/c-sis) mRNA and immunoreactive PDGF B-like polypeptide by rheumatoid synovium: coexpression with heparin binding acidic fibroblast growth factor-1. J. Rheumatol. 1991;18:7–13. [PubMed] [Google Scholar]
- Rodeo SA, Hannafin JA, Tom J, Warren RF, Wickiewicz TL. Immunolocalization of cytokines and their receptors in adhesive capsulitis of the shoulder. J. Orthop. Res. 1997;15:427–436. doi: 10.1002/jor.1100150316. [DOI] [PubMed] [Google Scholar]
- Rooney M, Condell D, Quinlan W, et al. Analysis of the histologic variation of synovitis in rheumatoid arthritis. Arthritis Rheum. 1988;31:956–963. doi: 10.1002/art.1780310803. [DOI] [PubMed] [Google Scholar]
- Rosen EM, Goldberg ID. Regulation of angiogenesis by scatter factor. EXS. 1997;79:193–208. doi: 10.1007/978-3-0348-9006-9_8. [DOI] [PubMed] [Google Scholar]
- Ryuto M, Ono M, Izumi H, et al. Induction of vascular endothelial growth factor by tumor necrosis factor alpha in human glioma cells. Possible roles of SP-1. J. Biol. Chem. 1996;271:28220–28228. doi: 10.1074/jbc.271.45.28220. [DOI] [PubMed] [Google Scholar]
- Sano H, Forough R, Maier JA, et al. Detection of high levels of heparin binding growth factor-1 (acidic fibroblast growth factor) in inflammatory arthritic joints. J. Cell Biol. 1990;110:1417–1426. doi: 10.1083/jcb.110.4.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano H, Engleka K, Mathern P, et al. Coexpression of phosphotyrosine-containing proteins, platelet-derived growth factor-B, and fibroblast growth factor-1 in situ in synovial tissues of patients with rheumatoid arthritis and Lewis rats with adjuvant or streptococcal cell wall arthritis. J. Clin. Invest. 1993;91:553–565. doi: 10.1172/JCI116235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semble EL, Turner RA, McCrickard EL. Rheumatoid arthritis and osteoarthritis synovial fluid effects on primary human endothelial cell cultures. J. Rheumatol. 1985;12:237–241. [PubMed] [Google Scholar]
- Sheu JR, Fu CC, Tsai ML, Chung WJ. Effect of U-995, a potent shark cartilage-derived angiogenesis inhibitor, on anti-angiogenesis and anti-tumor activities. Anticancer Res. 1998;18:4435–4441. [PubMed] [Google Scholar]
- Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- Stephens RW, Ghosh P, Taylor TK. The pathogenesis of osteoarthrosis. Med. Hypotheses. 1979;5:809–816. doi: 10.1016/0306-9877(79)90041-0. [DOI] [PubMed] [Google Scholar]
- Stevens CR, Blake DR, Merry P, Revell PA, Levick JR. A comparative study by morphometry of the microvasculature in normal and rheumatoid synovium. Arthritis Rheum. 1991a;34:1508–1513. doi: 10.1002/art.1780341206. [DOI] [PubMed] [Google Scholar]
- Stevens CR, Williams RB, Farrell AJ, Blake DR. Hypoxia and inflammatory synovitis: observations and speculation. Ann. Rheum. Dis. 1991b;50:124–132. doi: 10.1136/ard.50.2.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storgard CM, Stupack DG, Jonczyk A, Goodman SL, Fox RI, Cheresh DA. Decreased angiogenesis and arthritic disease in rabbits treated with an alphavbeta3 antagonist. J. Clin. Invest. 1999;103:47–54. doi: 10.1172/JCI3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symmons DP, Jones MA, Scott DL, Prior P. Longterm mortality outcome in patients with rheumatoid arthritis: early presenters continue to do well. J. Rheumatol. 1998;25:1072–1077. [PubMed] [Google Scholar]
- Tolonen J, Gronblad M, Virri J, et al. Platelet-derived growth factor and vascular endothelial growth factor expression in disc herniation tissue: an immunohistochemical study. Eur. Spine J. 1997;6:63–69. doi: 10.1007/BF01676576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DA, Wade M, Mapp PI, Blake DR. Focally regulated endothelial proliferation and cell death in human synovium. Am. J. Pathol. 1998;152:691–702. [PMC free article] [PubMed] [Google Scholar]
- Wang DS, Yamazaki K, Nohtomi K, et al. Increase of vascular endothelial growth factor mRNA expression by 1,25- dihydroxyvitamin D3 in human osteoblast-like cells. J. Bone Miner. Res. 1996;11:472–479. doi: 10.1002/jbmr.5650110408. [DOI] [PubMed] [Google Scholar]
- Westacott CI, Sharif M. Cytokines in osteoarthritis: mediators or markers of joint destruction? Semin. Arthritis Rheum. 1996;25:254–272. doi: 10.1016/s0049-0172(96)80036-9. [DOI] [PubMed] [Google Scholar]
- Yoshida S, Ono M, Shono T, et al. Involvement of interleukin-8, vascular endothelial growth factor, and basic fibroblast growth factor in tumor necrosis factor alpha-dependent angiogenesis. Mol. Cell Biol. 1997;17:4015–4023. doi: 10.1128/mcb.17.7.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Y, Ni J, Jiang GW, et al. VEGI, a novel cytokine of the tumor necrosis factor family, is an angiogenesis inhibitor that suppresses the growth of colon carcinomas in vivo. FASEB J. 1999;13:181–189. doi: 10.1096/fasebj.13.1.181. [DOI] [PubMed] [Google Scholar]