

An increasing number of enzymes of eukaryotic and prokaryotic origin have been shown to contain novel cofactors derived from endogenous amino acids.1 Many of these protein derived cofactors are formed autocatalytically.1 The tryptophan tryptophylquinone (TTQ) cofactor of methylamine dehydrogenase (MADH) is different in that formation of active enzyme requires co-expression of four processing genes2, 3 including mauG, which encodes a di-heme c-type cytochrome with unusual properties.4, 5
TTQ is derived from two β subunit residues of MADH, βW57 and βW108 in P. denitrificans.6 Two oxygens are added to βW57 at positions C6 and C7, and a cross-link is formed between C4 of βW57 and C2 of βW108 (Figure 1). X-ray crystallographic studies have demonstrated that the C6 position of βW57 is the site of nucleophilic attack by substrate during MADH turnover, displacing the oxygen at C6 to form a Schiff's base.7
Figure 1.
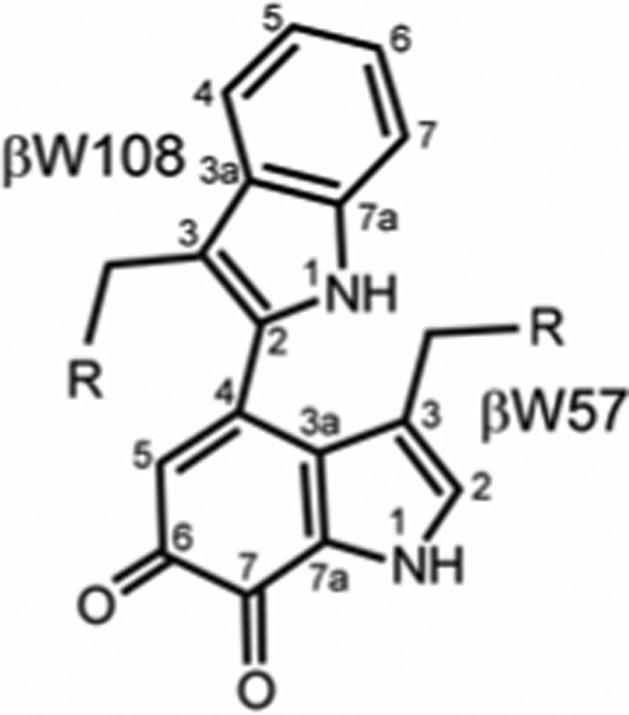
Structure of mature TTQ.
Previous studies have revealed that the biogenesis of TTQ is a multistep process that occurs after the MADH β subunit is translocated into the periplasm.2, 3, 8-10 The earliest intermediate in TTQ synthesis so far observed is a species lacking any modifications to either βW57 or βW108, but with a full complement of six intrasubunit disulfide bonds.10 The second intermediate that has been isolated is monohydroxylated at βW57 and lacks a cross-link to βW108.9 The mechanism of this first hydroxylation is poorly understood, however it is known to require the MADH active site residue βD76.10 As βD76 is located close to the C6 position of TTQ in the mature enzyme, it seemed likely that the first oxygen was incorporated into βW57 at this position.10 The second hydroxylation, cross-link formation and oxidation to quinone are catalyzed in an O2 and reducing equivalent dependent manner by MauG.
In order to further probe oxygen incorporation into TTQ, the monohydroxylated MADH biosynthetic intermediate lacking the cross-link was incubated in vitro with purified MauG, NADH and either isotopically labeled water (H218O, 95 %, Cambridge Isotope Laboratories) or O2 (18O2, 99 %, Isotec Inc.) for 24 hours.11 Mass spectrometry was used to assess the formation of TTQ as well as determine whether 18O had been incorporated.
Initial experiments, in which mature MADH was incubated in buffer containing H218O, confirmed previous studies12 which reported that MADH contains only one exchangeable carbonyl (Figure 2, green trace), consistent with the crystal structure in which the C6 carbonyl is exposed to solvent while the C7 carbonyl is hydrogen bonded to a backbone amide.6 Further incubation of singly 18O-labelled and unlabelled MADH with hydrazine, which displaces the oxygen at C6 of βW57 forming a covalent bond,7 results in loss of the 18O label (Figure 2, orange trace). This means that any oxygen atom added to the C6 position of TTQ during synthesis will exchange with solvent upon cofactor maturation.
Figure 2.
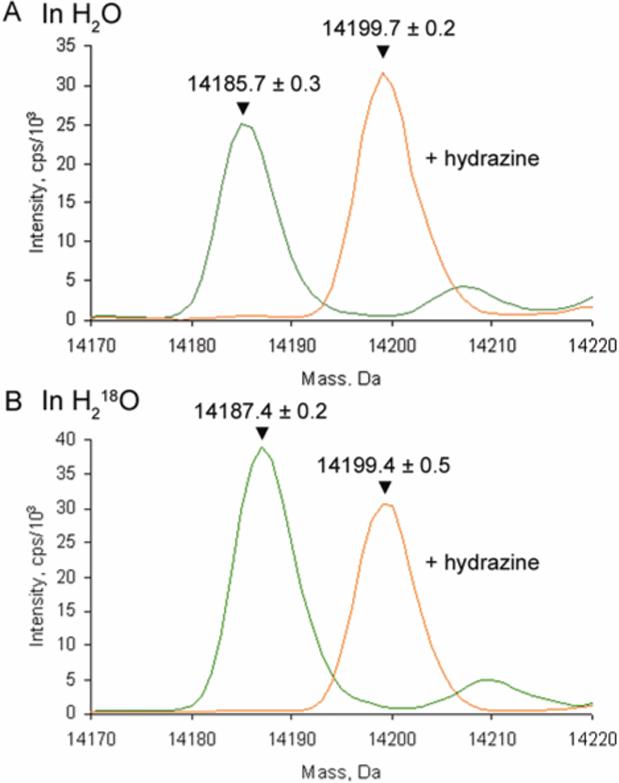
Mature MADH β subunit contains one exchangeable carbonyl at the C6 position of βW57. Sample preparation and mass spectrometry analysis were as previously described.9 Shown are deconvoluted mass spectra of the mature MADH β subunit. Green lines are after overnight incubation in buffer containing (A) H216O and (B) H218O, orange lines are after the further addition of a five-fold excess of hydrazine to each sample.
The MauG-dependent TTQ biosynthetic reaction requires O2.9, 11 When the reaction was carried out in the presence of unlabelled water and 18O2, mass spectrometry showed that no label was present in the mature TTQ cofactor (Figure 3A). This observation indicates that the MauG-dependent addition of oxygen is not to the non-exchangeable C7 position (Figure 4). If it were then 18O would be present in the mature TTQ. This result strongly suggests that the second oxygen is added at C6 and then rapidly exchanged with H2O. The only other possibility is that the second oxygen could be derived from solvent following O2 dependent substrate activation. To test this, the MauG-dependent biosynthetic reaction was carried out in the presence of H218O. Mass spectrometry showed that a single 18O had been incorporated into the resulting TTQ (Figure 3B). If the MauG-dependent second oxygen is derived from solvent and added to the non-exchangeable C7 position, then two 18O atoms should be present, since the oxygen at position C6 of βW57 would rapidly exchange with solvent upon cofactor maturation. Thus, these results confirm that the first oxygen is added to C7 during TTQ biogenesis, and that the second MauG-dependent oxygenation occurs at C6.
Figure 3.
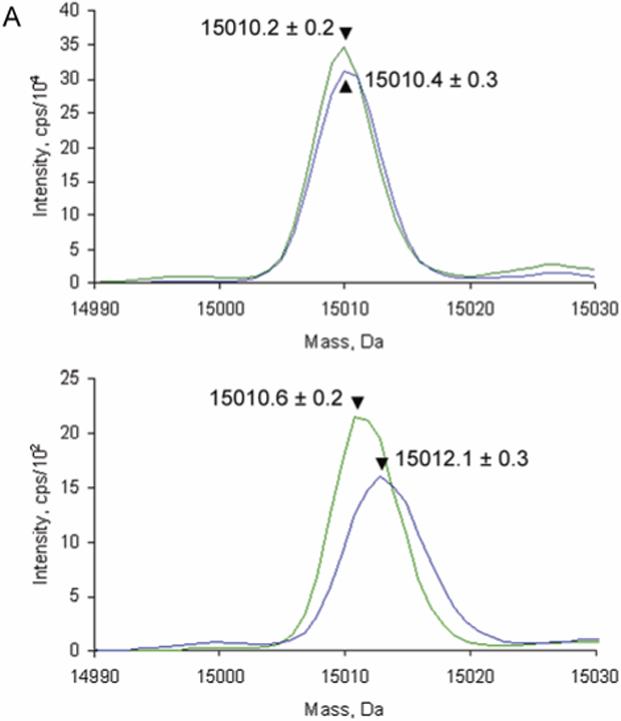
MauG catalyzes insertion of the second oxygen at the C6 position of βW57. Reaction mixtures contained 10 μM MADH β subunit monohydroxylated biosynthetic intermediate,9 1 μM MauG and 60 μM NADH. (A) The reaction was carried out in the presence of 16O2 (green) or 18O2 (blue). (B) The reaction was carried out in buffer containing either H216O (green) or H218O (blue). Sample preparation and mass spectrometry analysis were as previously described.9 The larger mass of the mature subunit relative to that in Figure 2 is because the biosynthetic intermediate used as substrate in this experiment included a 6xhistidine C-terminal tag.
Figure 4.
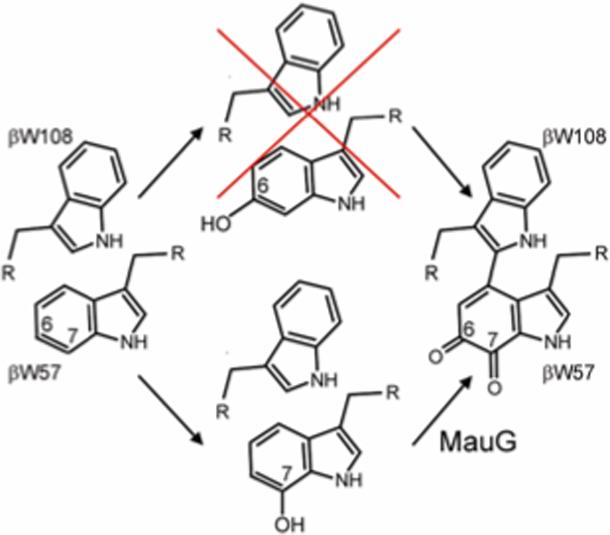
Refined mechanism of TTQ maturation.
These results suggest that the structure of the initial biosynthetic intermediate of MADH with no oxygens added to βW57 differs from that of the mature enzyme, despite the fact that the six disulfide cross-links of the β subunit are already formed. In particular it seems likely that the position of βD76 relative to βW57 is altered as this residue is required for the first hydroxylation,10 shown here to be at the C7 position of βW57 rather than C6 which is much closer to βD76 in mature MADH. A different pre-TTQ maturation β subunit conformation is also consistent with our previous finding that the αβ subunit interactions of MADH are relatively weak until TTQ biosynthesis is complete.9, 13
These data have enabled us to refine our model of TTQ biogenesis (Figure 4). They indicate that after translocation into the periplasm and formation of the six disulfide linkages, the first MauG-independent hydroxylation of βW57 occurs specifically at the C7 position in a βD76-dependent manner.6 MauG then catalyzes insertion of the second oxygen at the C6 position of βW57, the formation of a cross-link to βW108 and oxidation to a quinone.8 Upon completion of TTQ maturation the quinone carbonyl at position C6 of βW57 exchanges rapidly with solvent. Therefore, it is not possible from these data to determine the source of the oxygen incorporated by MauG, although other recent work favors a P450-like mechanism in which MauG inserts an oxygen derived from O2.11
ACKNOWLEDGMENT
This work was supported by NIH Grants GM-41574 (V.L.D.) and GM-66569 (C.M.W.). Mass spectrometry studies were carried out at the Center for Mass Spectrometry and Proteomics at The University of Minnesota.
References
- 1.Okeley NM, van der Donk WA. Chem. Biol. 2000;7:R159–171. doi: 10.1016/s1074-5521(00)00140-x. [DOI] [PubMed] [Google Scholar]
- 2.van der Palen CJ, Slotboom DJ, Jongejan JA, Reijnders WN, Harms N, Duine JA, van Spanning RJ. Eur. J. Biochem. 1995;230:860–871. doi: 10.1111/j.1432-1033.1995.tb20629.x. [DOI] [PubMed] [Google Scholar]
- 3.van der Palen CJ, Reijnders WN, de Vries S, Duine JA, van Spanning RJ. Antonie Van Leeuwenhoek. 1997;72:219–28. doi: 10.1023/a:1000441925796. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Li X, Jones LH, Pearson AR, Wilmot CM, Davidson VL. J. Am. Chem. Soc. 2005;127:8258–8259. doi: 10.1021/ja051734k. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Graichen ME, Liu A, Pearson AR, Wilmot CM, Davidson VL. Biochemistry. 2003;42:7318–7325. doi: 10.1021/bi034243q. [DOI] [PubMed] [Google Scholar]
- 6.Chen L, Doi M, Durley RC, Chistoserdov AY, Lidstrom ME, Davidson VL, Mathews FS. J. Mol. Biol. 1998;276:131–149. doi: 10.1006/jmbi.1997.1511. [DOI] [PubMed] [Google Scholar]
- 7.Huizinga EG, van Zanten BA, Duine JA, Jongejan JA, Huitema F, Wilson KS, Hol WG. Biochemistry. 1992;31:9789–9795. doi: 10.1021/bi00155a036. [DOI] [PubMed] [Google Scholar]
- 8.Chistoserdov AY, Chistoserdova LV, McIntire WS, Lidstrom ME. J. Bacteriol. 1994;176:4052–65. doi: 10.1128/jb.176.13.4052-4065.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pearson AR, De la Mora-Rey T, Graichen ME, Wang Y, Jones LH, Marimanikkupam S, Agger SA, Grimsrud PA, Davidson VL, Wilmot CM. Biochemistry. 2004;43:5494–5502. doi: 10.1021/bi049863l. [DOI] [PubMed] [Google Scholar]
- 10.Jones LH, Pearson AR, Tang Y, Wilmot CM, Davidson VL. J. Biol. Chem. 2005;280:17392–17396. doi: 10.1074/jbc.M500943200. [DOI] [PubMed] [Google Scholar]
- 11.Li X, Jones LH, Pearson AR, Wilmot CM, Davidson VL. Unpublished.
- 12.Backes G, Davidson VL, Huitema F, Duine JA, Sanders-Loehr J. Biochemistry. 1991;30:9201–9210. doi: 10.1021/bi00102a011. [DOI] [PubMed] [Google Scholar]
- 13.Pearson AR, Jones LH, Higgins L, Ashcroft AE, Wilmot CM, Davidson VL. Biochemistry. 2003;42:3224–3230. doi: 10.1021/bi027073a. [DOI] [PubMed] [Google Scholar]