

Abstract
T7 RNA polymerase is known to induce bending of its promoter DNA upon binding, as evidenced by gel-shift assays and by recent end-to-end fluorescence energy transfer distance measurements. Crystal structures of promoter-bound and initially transcribing complexes, however, lack downstream DNA, providing no information on the overall path of the DNA through the protein. Crystal structures of the elongation complex do include downstream DNA and provide valuable guidance in the design of models for the complete melted bubble structure at initiation. In the current study, we test a specific structural model for the initiation complex, obtained by alignment of the C-terminal regions of the protein structures from both initiation and elongation and then simple transferal of the downstream DNA from the elongation complex onto the initiation complex. FRET measurement of distances from a point upstream on the promoter DNA to various points along the downstream helix reproduce the expected helical periodicity in the distances and support the model’s orientation and phasing of the downstream DNA. The model also makes predictions about the extent of melting downstream of the active site. By monitoring fluorescent base analogs incorporated at various positions in the DNA we have mapped the downstream edge of the bubble, confirming the model. The initially melted bubble, in the absence of substrate, encompasses 7–8 bases and is sufficient to allow synthesis of a 3 base transcript before further melting is required. The results demonstrate that despite massive changes in the N-terminal portion of the protein and in the DNA upstream of the active site, the DNA downstream of the active site is virtually identical in both initiation and elongation complexes.
Keywords: transcription, FRET, fluorescence, energy transfer, DNA, bending
INTRODUCTION
T7 RNA polymerase is the most well-characterized member of a family of single subunit RNA polymerases that share fundamental biochemical characteristics with the much larger and apparently unrelated multi-subunit RNA polymerases (1, 2). The relative simplicity of this system makes it attractive for the study of fundamental mechanistic aspects of initiation, elongation and termination of transcription.
T7 RNA polymerase is highly specific for its 17 base pair consensus promoter sequence (positions −17 to −1, relative to the start site). The formation of an initiation–competent complex between DNA and RNA polymerase is an essential first step for transcription initiation in any system. In this process, the duplex promoter DNA is sequence–specifically bound by the RNA polymerase and melts open allowing the 8 bases surrounding the cytosine (encoding guanine) at the start site (+1) to be positioned in the active site cleft of the enzyme. The templating bases placed into the enzyme active site are then ready for binding of incoming nucleoside triphosphates (3–6). The T7 RNA polymerase promoter is composed of two functional domains: the upstream binding region from position −17 to −5 is primarily responsible for binding and recognition, while the downstream region from position −5 to the start site (or beyond) is involved in initiation (7, 8). This latter domain is the region of the DNA that undergoes requisite melting prior to initiation.
The publication of high resolution structures for various RNA polymerases provides a wealth of structural information to begin to understand the process of transcription, and the structures for T7 RNA polymerase in complex with promoter DNA are consistent with the above two–domain model (9, 10). In the structures of the promoter–bound (pre–initiation) and transcribing (with a 3 base transcript) complexes of T7 RNA polymerase, the upstream end of the melted bubble is clearly defined, confirming the expected structural/functional division (9, 10). The promoter from position −17 to −5 remains duplex, while the DNA unwinds starting from position −4. However, the structures provide no information on the nature of the downstream end of the bubble, and the nature of the constructs used in crystallization ensure that the structure formed will be that of the open complex. Thus, the structures provide no information regarding the path of the downstream DNA, the extent of bubble melting, or the partitioning between open and closed forms of the enzyme-DNA complex.
It has been suggested that specific contacts of the protein with DNA template strand functional groups upstream of the start site stabilize the observed structure and guide the first two templating bases into the active site (9, 10). Some time ago, however, it was shown that the immediate connection between the upstream elements and the start site can be replaced by a flexible linkage and initiation is not substantially reduced (the precise start site does, however, become less well-defined and shifts downstream by one base) (6). Subsequently, it was shown that the templating bases can position into the active site in constructs in which the physical connection between the bulk of the upstream single-stranded region and the start site has been lost – the templating strand can be brought to the active site via downstream connections (11). This suggests a model in which the templating bases need only be tethered near the active site. Under normal conditions, specific promoter binding activates initiation by inducing a melting of the DNA and by providing a simple tethering of the melted template strand near the active site.
Both fluorescence and crystal structure data for the elongation complex show that T7 RNA polymerase melts at most one base downstream of the nascent RNA transcript (12-14), suggesting an economy of energetics, but also suggesting how binding of the downstream duplex can guide the templating bases into the active site. In this work, we map precisely the downstream edge of the bubble in the initially bound and the initiating (with 3 base transcript) complexes. Downstream of the active site, the latter complex appears to be identical to an elongation complex, both in terms of the extent of melting and in terms of positioning and phasing of the downstream duplex.
RESULTS
A model for the complete initiation complex
An earlier study using circular permutation gel shift analysis provided evidence that initial binding of T7 RNA polymerase to its promoter induces an effective bend of approximately 60° in the DNA, centered near the start site of transcription (15), a result recently confirmed by FRET measurements between two probes placed at the ends of the upstream and downstream duplexes (16). Crystal structures of promoter bound complexes are consistent with this view, however, these structures contain no downstream duplex DNA and so provide no information on the path of the DNA downstream of the start site. More recent crystal structures of elongation complex models, clear of the promoter, do contain downstream duplex DNA (13, 14). Modeling of downstream DNA from the elongation complex structures onto the structure of the initiation complex provides an overall picture consistent with the DNA bending results.
We present such a model in Figure 1. Although the transition from initiation to elongation is accompanied by a large conformational change in the N-terminal domain of the protein, the C-terminal two thirds of the protein, containing the active site for phosphoryl transfer, does not appreciably change structure (13, 14). To prepare the complete model, C-terminal residues 300 to 883 in the elongation complex structure (1MSW) were aligned with the corresponding residues in the initiation complex structure (1QLN). Downstream DNA from the elongation complex was then overlayed onto the structure for the initiation complex. Although only protein residues were used in the alignment, the template strand DNA near the active site aligns very well between the two models, providing an initial validation of the model. Indeed, the overlapping backbones from positions +3 to +5 align very well (see Figure 4, in the Discussion). While coordinates for the nontemplate strand from position −3 through +1 are not available in either model, the distance is readily spanned to connect upstream and downstream elements.
Figure 1. A model for the complete initiation complex.
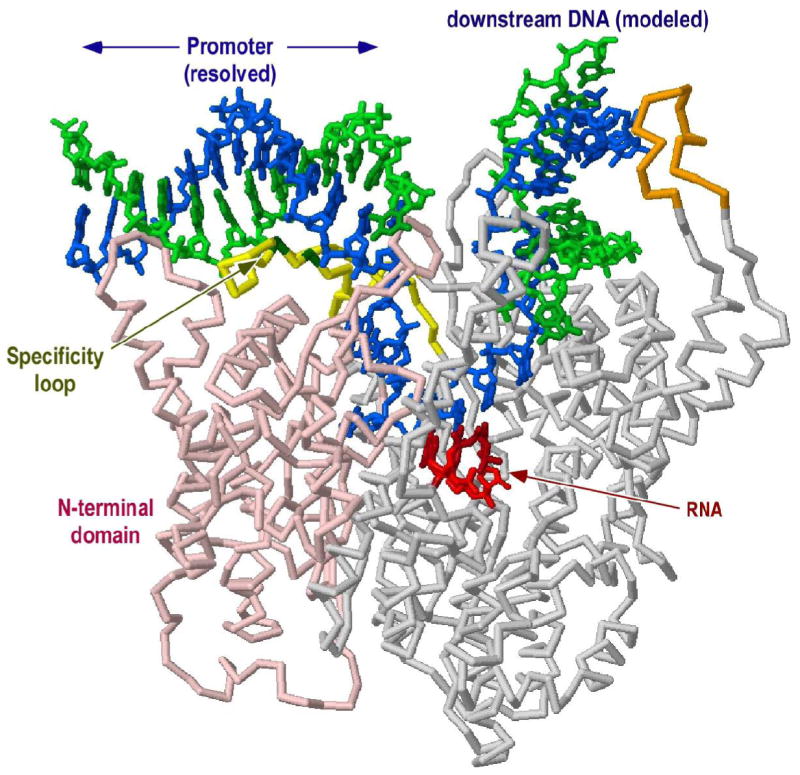
C-terminal residues 300 to 883 in the elongation complex structure (1MSW) were aligned with the corresponding residues in the initiation complex structure (1QLN). Downstream duplex DNA from 1MSW was then transferred to the 1QLN structure (shown).
Figure 4. Modeling the downstream edge of the initiation bubble.
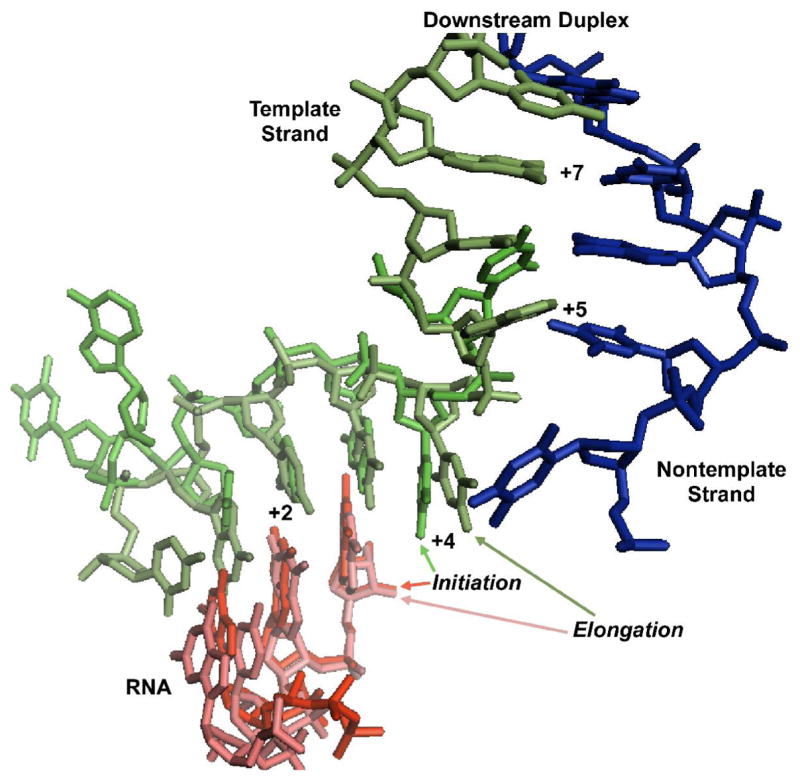
Alignment of the C-terminal domains of the initiating complex stalled at position +3 (1QLN) and the elongation complex clear of the promoter (1MSW) yields excellent alignment of the DNA template strand backbones (the source of the coordinates is indicated above: Initiation/Elongation). The model predicts a return to base pairing at position +5, as observed in the current fluorescence study. The coincidence of the template strand backbones also suggests that the downstream duplex in the initiation complex will take the same overall path as seen in the elongation complex, a result confirmed by the observed FRET distance phasing.
Distance measurements by DNA-to-DNA FRET
To more directly test this model, we have measured specific distances between a single position in the upstream DNA and a variety of positions in the downstream DNA, as illustrated in Figure 2. The model predicts not only that the downstream DNA will come closer to the upstream DNA on bending, but also predicts a specific helical pattern in the distances. Fluorescence resonance energy transfer (FRET) is well suited for these measurements and DNA is readily prepared with labels at specific positions.
Figure 2. FRET measurements test the model.
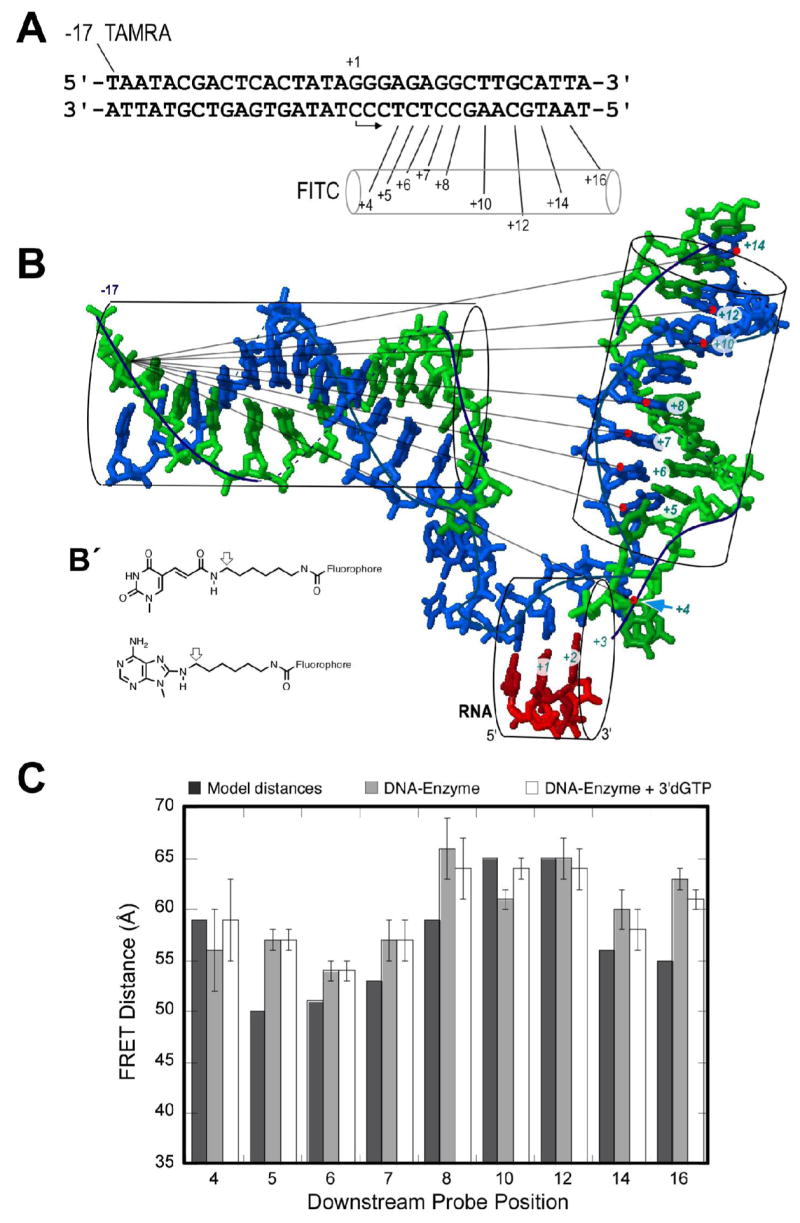
A) The sequence of the DNA constructs. B) Modeled structure (protein omitted) showing expected distances. The FITC probe is conjugated to bases in the downstream template strand (positions +4, +5, +6, +7, +8, +10, +12, +14 and +16). TAMRA is coupled to the thymidine base at position -17 of the upstream nontemplate strand. Although the model shows DNA only to position +14, the constructs used were fully duplex to position +17. B′) Fluorophore coupling to A, C, or T is as shown. Modeled distances are approximated from the positions of the arrows, assuming base geometries from the model in (B). C) FRET distance data for each labeled DNA construct were obtained from an average of 4 measurements with standard deviations shown as error bars. From left to right within a set are: 1) model distances; 2) FRET distance on addition of enzyme at 4:1 ratio over DNA; and 3) FRET distances after addition of 100 μM 3′dGTP to (2).
In this study, at position −17, we incorporated a dT analog coupled at the 5 position with a spacer arm terminating in a primary amine. This allows coupling with the succinimidyl ester derivative of 5-carboxytetramethylrhodamine (TAMRA), to place a FRET acceptor at a fixed position upstream. We then prepared a series of complementary template strands containing dT, dC, or dA analogs with similar amino-terminated spacers at a variety of positions downstream of the active site. Coupling of fluorescein-5-isothiocyanate (FITC) with these primary amines to form a thiourea linkage then provides FRET donors at various positions, for measurement of a progression of donor-acceptor distances. The strands are designed such that the nearest guanine base is at least 6 base pairs away from the TAMRA label to avoid quenching of TAMRA by guanine (17). All constructs are fully duplex and have the same DNA sequence and length.
The donor and acceptor fluorophores are attached to the DNA by linkages that have both rigid and flexible components. For consistency in the following analysis, model distances are calculated from the point farthest out on the rigid component, indicated by an arrow in Figure 2B′. Thus model distances are not expected to match exactly distances derived from experiment, but periodic trends arising from the positioning of the downstream helix should remain.
The DNA sequence used and the positions labeled are shown in Figure 2A. In model linear duplex DNA, the closest downstream probe (+4) is expected to lie >80 Å from the upstream probe at position –17. Given that Ro for this FRET pair is 45 Å, this closest pair (and all others) should show no appreciable energy transfer in the absence of DNA bending (18). Indeed, we observe that the double stranded forms of all of the constructs show no detectable energy transfer (data not shown).
For the polymerase-bound complex, bending of the DNA will reduce all of these distances. Moreover, as illustrated in Figure 2, the model predicts a specific helical periodicity in the distances. The data presented in Figure 2C compare the distances predicted by modeling with the inter-probe distances calculated from measured FRET efficiencies. Addition of RNA polymerase to each DNA construct yields distances within the range of these FRET probes and consistent with the predictions of the model (the probe at position +16 lies very near the end of the DNA at position +17 and so may not position according to simple models). The general agreement of the distances with the model both verifies that a bent and open complex forms in the absence of substrate NTPs and confirms the model for positioning of the downstream DNA.
In order to insure complete complex formation and melting of the DNA, one can exploit mass action to drive these processes forward. Addition of GTP to polymerase poised on a template encoding RNA of sequence GGGA… yields a steady state distribution of species synthesizing RNA slippage products (19). All of these species are poised at position +3 of the template. Alternatively, addition of 3′dGTP to the above complexes allows binding of the first two substrates into positions +1 and +2 of the active site (without phosphodiester bond formation) and has been shown to drive binding and melting to completion (20). Crystal structures with and without a three-base transcript show no significant change in the complex structure near the active site and the fluorescence measurements following addition of 3′dGTP, shown in Figure 2C, confirm this prediction (similar results are obtained for the addition of GTP, not shown). No substantial change in distances or shift in the phase of the periodicity is detectable, suggesting that the initial binding of DNA to the polymerase poises the system for synthesis of the initial transcript; no additional DNA is drawn into the active site. The fact that addition of 3′dGTP leads to no detectable change in FRET efficiency also supports our earlier arguments that near-complete open complex formation does not require binding of the initial substrate (21).
Fluorescence mapping of the melted bubble
The model presented in Figures 1 and 2B makes three general predictions: 1) that there is a substantial bend in the DNA, 2) that the downstream DNA adopts a specific rotational position, and 3) that the base pairs are melted from position -5 through about position +4. The above results clearly confirm a substantial bending of the DNA on binding of the RNA polymerase and are consistent with the positioning of the DNA predicted by the model. These results are consistent with a local melting of the DNA at the start site and are not consistent with extensive melting well beyond the active site; the observed helical periodicity is consistent only with positioning of duplex DNA downstream of about position +4.
In earlier studies, we used fluorescent base analogs to directly map the melted bubble in both initiation and elongation complexes (12, 21). Fluorescence mapping of the elongation complex showed that the enzyme melts at most one base downstream of the last base incorporated, later confirmed by x-ray crystallography (9, 10). Mapping of the initial complex defined the upstream edge of the bubble at position −4, also later confirmed by crystallography (9, 10, 21). However, mapping of the initially bound complex did not probe the downstream edge of the bubble. Similarly, the constructs used in crystallography lacked downstream duplex DNA.
In order to more precisely map the extent of the open bubble at initiation, we have incorporated fluorescent analogs of dC, dA, and dG into the consensus promoter sequence. In particular, in order to maintain a consensus promoter sequence, the guanine analog 6–methylisoxanthopterin (6–MI) was incorporated at positions +1, +3, and +5, as shown in Figure 3. 6–MI has an emission maximum at 430 nm with excitation at 340 nm, far from protein absorbances (22). The cytosine analog pyrrolo–dC (pyr-dC) is well-suited to pair with guanine, allowing for two Watson-Crick hydrogen bonds in an isosteric pair (23). The spectral properties of this analog are also superior to those of 2–aminopurine (2–AP) in that excitation and emission maxima (350 nm and 460 nm, respectively) are also shifted away from those of the protein. Fluorescent base analogs introduce alterations in the major or minor grooves of the DNA duplex, but appear not to dramatically alter helix stability. In the application of these probes in the current study, subtle changes to the major or minor grooves are not expected to have a substantial impact on function.
Figure 3. Fluorescence measurements from nine different constructs incorporating pyr–dC, 2–AP, or 6–MI at individual positions both upstream and downstream of the start site for the initiation of transcription.
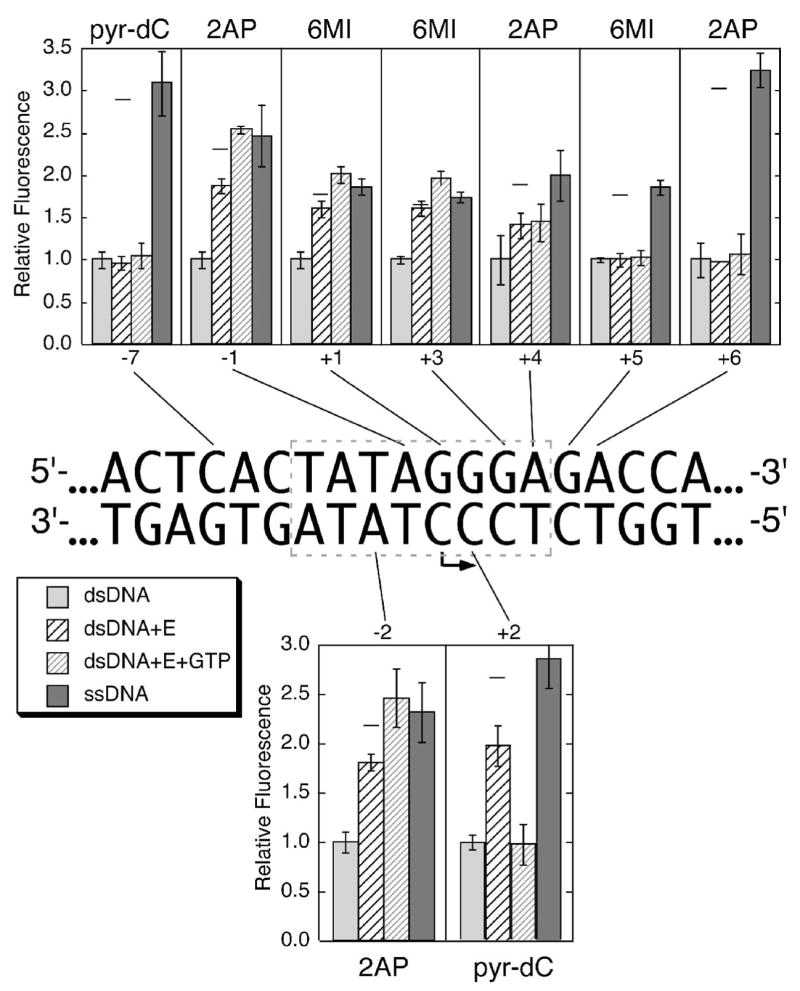
Fluorescence measurements for each construct were taken (in order, from left to right within a set) as follows: 1) double stranded DNA in transcription buffer, followed by 2) addition of enzyme to an equimolar concentration, then 3) addition of GTP to 1.0 mM (allows initiation and cycling to position +3). Concentrations of polymerase and promoter were each 1.0 μM, except for constructs with labels at positions +4 and +6, which were at concentrations of 2.0 μM. Lane 4 measures fluorescence of the single (nontemplate) strand containing the indicated fluorophore. Relative fluorescence values for each group are normalized to the fluorescence in double stranded DNA for that construct. Each value represents the average of three separate measurements (with standard deviation as indicated).
Each construct studied contains only a single 2–AP, 6–MI or pyr–dC substitution, base-paired to the corresponding unmodified base, enabling us to monitor the environment at a unique position in the DNA. Base pair opening at the location of the probe is expected to lead to increases in fluorescence intensities, since in all cases, fluorescence is higher for a probe in a single stranded context than for the same probe base paired. The analog 2–AP was substituted into various positions on both the template and nontemplate strands, both immediately upstream of the start site, as in our previous study, and farther out into the templating DNA, to measure the extent of downstream opening. To probe melting of the initially templating bases at positions +1, and +2, 6–MI was incorporated. Finally, incorporation of pyr–dC in place of C in the template strand allows us not only to confirm the results with 6–MI, but also allows observation of the formation of the initial heteroduplex.
In the previous fluorescence study (21), the DNA duplex extended downstream only to position +5, so it is possible that the construct presented an artificially low barrier to melting. In the current work, duplex constructs extend to position +15, ensuring a near-native barrier to melting. In order to confirm the previous results that the TATA region immediately upstream of the start site is unwound upon enzyme binding (21, 24, 25), we substituted 2–AP at position −2 on the template strand, and at position −1 on the nontemplate strand. The DNA sequence from position +1 downstream is GGGAGA…, allowing the use of 2–AP in the template strand at positions +4 and +6, and the placement of 6–MI in the nontemplate strand at the actual start site and beyond. The analog pyr–dC provides a complement in the template strand at position −2. As a control, pyr–dC was also placed at position −7 of the nontemplate strand, in a region of the promoter known to remain duplex throughout the initiation of transcription. Combined with the earlier study, this range of probes provides a complete picture of the open complex (21).
Mapping the melted region in pre–initiation and initiation complexes
The results summarized in Figure 3 compare the average relative fluorescence of each probe substituted at various positions. In this plot, the label “ssDNA” represents the fluorescence from a probe in the corresponding single stranded DNA, approximating the melted state, while “dsDNA” represents fluorescence from the same DNA annealed to its complementary strand. The label “dsDNA+E” represents fluorescence from a probe in the enzyme-DNA complex. Finally, the bars labeled “dsDNA+E+GTP” represent fluorescence from the enzyme-DNA complex in the presence of saturating GTP (1 μM each of dsDNA and enzyme, plus 800 μM GTP), with the fluorescence measurement complete within four minutes, to avoid substantial depletion of the substrate GTP. In all experiments, the appropriate background subtraction was carried out, as previously reported (21).
As expected from previous studies, the fluorescence from 2–AP at positions −2 of the template strand and −1 of the nontemplate strand increases on the addition of enzyme. At the concentrations used (1 μM each), we expect about 90% complex formation, assuming a Kd of about 5 nM (8, 26). The fluorescence signals from 6–MI at positions +1 and +3 and from pyr–dC at position +2, all increase substantially upon addition of enzyme, indicating that melting extends to at least position +3 in the absence of substrate. The signal from 2–AP at position +4 of the nontemplate strand also increases, but to a lesser extent, while the fluorescence signals from 6–MI and 2–AP at positions +5 and +6, respectively, do not change substantially. The bubble extends clearly to position +3, and perhaps to position +4, but no further.
Subsequent addition of GTP drives the synthesis of a 3 base transcript, which is then extended slowly by slippage synthesis (19, 27). The slight increase in fluorescence at most melted positions reflects the expected mass action-driven formation of 100% of an open complex under these conditions (the varying lengths of poly-G RNA should not, to a first approximation, influence the fluorescence). The control containing pyr–dC at position −7 in the upstream binding region shows no increase in fluorescence, as expected.
Throughout slippage synthesis, the enzyme never needs to translocate beyond position +3. The unchanged fluorescence from probes at positions +4, +5 and +6 indicates that these bases remain duplex in the steady state average complex, demonstrating that the downstream edge of the bubble has not moved at all on progression of the active site to position +3. At this point, the active site must now be positioned very close to the downstream edge of the bubble, as observed in elongation complex crystal structures (13, 14).
Interestingly, the fluorescence signal from pyr–dC at position +2 of the template strand returns to the equivalent signal of the probe in a duplex environment, consistent with the expected presence of a 3 base pair RNA hybridized to the template strand. This result confirms that at steady state, RNA product is bound (release of product is slower than reinitiation). This result is also consistent with previous studies which indicate that formation of an A–form duplex also provides an appropriate environment for fluorescence quenching (28–30).
Assessments of the extent and energetics of melting
In principle, RNA polymerases can bind promoter DNA in both closed and open complexes, and should partition between these two states (31). The fluorescent probes used here should allow us to quantitatively assess the extent of melting. An accurate assessment of the extent of melting requires having the proper controls for the fluorescence signal from fully duplex and fully melted DNA, but also requires an independent measure of the (overall) binding constant, as residual unbound DNA will, of course, remain unmelted. As a control for fluorescence from the fully melted state, one can use either single stranded DNA, a protein-DNA complex containing DNA lacking the nontemplate strand in the melted region, or one can drive melting by the addition of GTP or 3′dGTP. The dissociation constant for binding of the consensus promoter to T7 RNA polymerase under conditions similar to those used here is approximately 5 nM (8, 26). As noted above, under the concentrations of enzyme and DNA used in these studies, this predicts that about 90% of the DNA should be bound by protein. The fluorescence change expected for fully melted complexes should then be 90% of that for fully melted controls: single stranded DNA or the complex with added GTP (driving ≈100% complex formation). For each probe position, a horizontal bar in Figure 3 indicates this level.
For most positions melted by the binding of RNA polymerase (in the absence of GTP), the change in fluorescence is less than expected for a completely open complex. Assuming 90% overall DNA binding and using single stranded DNA as a control for fully melted DNA, the calculated fractional melting for positions −2 through +3 ranges from 66–92%, measured at different positions along the bubble. If instead, the complex with added GTP (producing a ladder of products from a stall at position +3) is used as the control, the data suggest about 62–71% of the complexes are melted in the absence of NTPs.
DISCUSSION
Numerous recent crystal structures of RNA polymerases with and without DNA bound have set the stage for a deeper understanding of mechanisms in initiation and elongation in transcription. Structures of the multisubunit enzymes from both prokaryotes and eukaryotes demonstrate the complexity of highly regulated transcription (32–38). In contrast, the single subunit bacteriophage RNA polymerases present a model of simplicity – for an inherently complex process (9, 10, 13, 14, 39). Despite very different overall structures, the multi- and single-subunit RNA polymerases share substantial functional homology (40).
The current study completes the structural picture of the initial transcription bubble in the model single subunit RNA polymerase from bacteriophage T7. Consistent with earlier studies and with recent crystal structures, the results demonstrate melting within the sequence TATA, adjacent to the start site, and confirm that the region upstream of position −5 remains duplex (9, 10, 21). The use of new fluorescent base analogs allows us to extend previous analysis into and downstream of the start site, a region where high resolution structural information is lacking (13, 14, 21).
Fluorescence defines the complete bubble
The results presented in Figure 3 show that in the absence of GTP, the transcription bubble extends downstream to position +3 or +4. The ambiguity arises from the fact that the enzyme-induced increase in fluorescence from a probe placed at position +4 is only one half that of the single stranded control. There are two potential explanations for this result. It cannot be completely ruled out that at steady state, there are two populations of the complex, with different translocational states (one in which the DNA is open a single base farther downstream than the other). This is unlikely in that the same intermediate fluorescence is observed at position +4, in the absence of substrate GTP and under saturating concentrations of GTP with a 3 base heteroduplex present – very different conditions. Alternatively, the intermediate fluorescence might reflect a probe environment that is intermediate between that of double stranded and single stranded DNA (with respect to the stacking interactions that lead to quenching). This might be expected for a base sitting at the edge of an open bubble, as predicted by the model.
Addition of GTP as the sole substrate allows formation of a 3 base heteroduplex, with reiterative synthesis of poly–G RNA (19). Interestingly, the fluorescence results indicate that the downstream edge of the bubble shows no sign of movement on addition of GTP. This suggests that the crystal structure of a similar ternary complex, with a resolved three base RNA fragment and no downstream duplex, forms a good foundation on which to build a model of a complete open complex, in the presence or absence of substrate (10). It seems that initial DNA melting in the absence of substrate primes the system for synthesis of up to 3 bases of RNA without movement of the downstream DNA. Since initial substrate binding (in particular, GTP at position +1) is weak, it makes sense to use the energy of promoter binding (alone) to maintain the melted bubble.
This result is also consistent with our previous fluorescence mapping of an elongation complex transcription bubble and with recently solved crystal structures of elongation complex models (12–14). In these structures, DNA is melted at most one base beyond the last incorporated base. During RNA synthesis, the enzyme melts the DNA no farther downstream than necessary.
Estimation of fractional melting
A potentially important mechanistic step in the initiation of transcription is the transition from an initially closed complex to an open complex, melted near the start site. Studies in the E. coli enzyme suggest that this simple question can be complicated by the existence of multiple forms of open and closed complexes (31, 41). It is tempting to use the fluorescence changes associated with DNA binding in a quantitative sense to determine the thermodynamic distribution between open and closed states. However, as illustrated above, since changes in fluorescence arise primarily from changes in base-base stacking interactions, and only indirectly from melting of the duplex, ambiguities necessarily arise (similar uncertainties exist for other probes of melting, for similar reasons). The results presented here report anywhere from 62–92% melting at equilibrium, depending on the placement of the probe in the bubble and the choice of control for the fluorescence signal from a fully melted complex. Given the range of variation in this value with the location of the probe, measurements of this sort can therefore be considered semi-quantitative at best.
Comparison of FRET efficiencies in the presence and absence of 3′dGTP should also provide information on the distribution between open and closed complexes at equilibrium. If in the absence of the initiating nucleotide, a substantial fraction of the population exists in the closed and therefore, approximately linear, state, then the measured FRET efficiencies should be lower than expected, representing an average of the linear (long distance) and bent (short distance) populations. The distances measured in Figure 2 approximate the model distances, suggesting near-complete open complex formation, but more importantly, the FRET efficiencies (and resulting distances) do not change substantially on binding of 3′dGTP. These results, reproduced with probes placed at a number of positions, argue that at equilibrium in the absence of substrate, the complex is largely open, consistent with our measurements using fluorescent base analogs.
These data are in agreement with our earlier work (21), but are in quantitative disagreement with other published studies, using similar approaches (26). The latter studies have determined most recently that about 20% of the complexes are open at equilibrium, while we find greater than 80% open. The difference in free energy between these two conditions, however, is small. Indeed, these fractions correspond to a barrier to melting in the complex of from 1.0 to 0.1 kcal mol−1, respectively. Considering that melting 8 base pairs of DNA should require substantially more than 1 kcal mol−1 of energy, the primary conclusion from both sets of experiments must be that promoter binding, rather than substrate GTP binding, pays the bulk of the cost of melting. It is likely that the mechanism of melting arises from the obligatory coupling of the intrusion of the intercalating hairpin into the DNA at position −5 and the establishment of tight-binding duplex contacts upstream of this position (42, 43).
Structural support for the initially-bound complex model
Fluorescence energy transfer measurements provide a measure not only of the overall bend of the DNA in the initiation complex, but by mapping distances to points along the helical path of the template strand, can support the details of the model. Although the fluorescent probes are anchored to the bases by a linker with a flexible component, the model-derived and experimental distances are in remarkable agreement (17, 18, 44).
Both the FRET and base analog fluorescence results are fully consistent with the model for the initiation complex derived from alignment of the protein domains in initiation and elongation complexes and the simple transfer of the downstream duplex from the latter to the former. In the modeling presented in Figure 4, only the DNA is shown for clarity. The initiation complex crystal structure resolves template strand DNA downstream through position +5. Although the complementary nontemplate strand did not exist in the crystallized complex, the path of the template strand backbone is largely coincident with the path of the backbone in the corresponding elongation complex containing a complete downstream duplex. The model also predicts a return to duplex pairing by position +5, a prediction confirmed by the fluorescence results presented here.
Finally, by measuring distances to multiple positions along the downstream DNA, we can obtain information on the rotational state of the downstream duplex. The analysis presented in Figure 5 starts with the model derived from alignment of the initiation and elongation complex structures (0°). The experimental data (closed circles and squares) is in reasonable agreement with the model distances. We then rotate the downstream DNA by 90°, 180°, and 270°, comparing the data with the new model distances (open circles). The experimental data are clearly not consistent with rotations of 90° and 180° and are most consistent with the original model (0°). Thus the FRET measurements support not only the modeled trajectory, but also the phasing of the downstream duplex.
Figure 5. Phasing analysis supports the model.
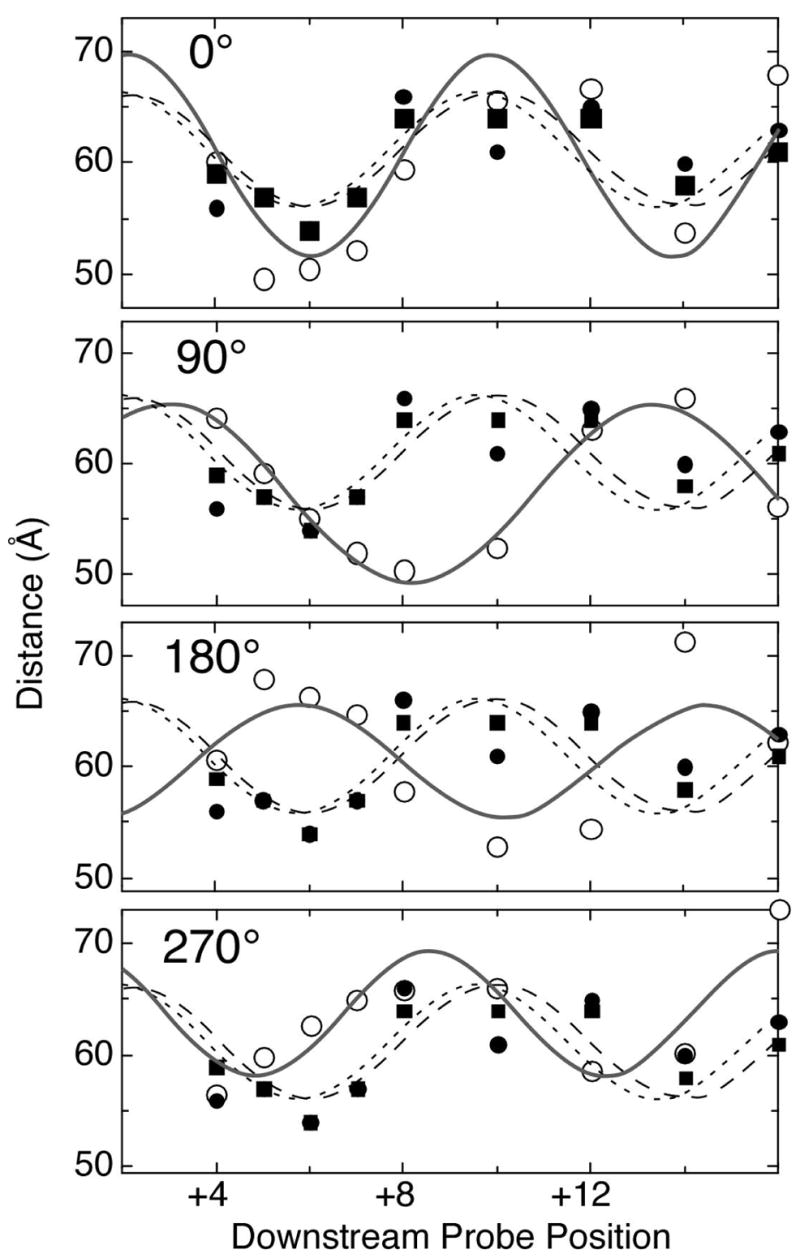
Closed data points are FRET distance measurements from Figure 2 (closed squares: enzyme and DNA only; closed circles: enzyme, DNA, and 3′dGTP). Open data points are distances measured from models in which the downstream duplex has been rotated about its axis. Curves drawn through the various data sets are best fits to a simple sin function, and are not intended to accurately predict distances, as they do not include translocation down the helix axis. Rather, they are presented to aid the viewer in seeing the phasing of the modeled distances, relative to the experimental distances.
MATERIALS and METHODS
T7 RNA Polymerase
T7 RNA polymerase was prepared from E. coli strain BL21 carrying the overproducing plasmid pAR1219 (kindly supplied by F.W. Studier), in which RNA polymerase is expressed under inducible control of the lac UV5 promoter (45). The enzyme was purified and the concentration was determined (ε280 = 1.4 × 105 M−1 cm−1) as described previously (46). Purity of the enzyme (>95%) was verified by SDS–denaturing polyacrylamide gel electrophoresis.
Oligonucleotides
Oligonucleotides were synthesized using phosphoramidite chemistry on an Applied Biosystems (ABI) Expedite 8909 Nucleic Acid Synthesis System. Standard phosphoramidites were purchased from CPG Inc. 2–AP phosphoramidite and pyr–dC phosphoramidite were obtained from Glen Research Corp and coupled normally.
Amino-modified bases dT, dC and dA containing six-carbon linkers (Glen Research) were introduced at select positions during the synthesis. Single-stranded DNA was purified by gel electrophoresis containing 15% polyacrylamide/6M urea followed by ethanol precipitation to ensure that the amine-modified oligonucleotides are free of shorter sequences and of triethylamine and ammonium salts that may interfere with labeling. Oligonucleotides of the desired length were extracted using an electro-separation chamber (EluTrap®, Schleicher and Schuell). Fluorescent labeling of these amine-modified oligonucleotides was done postsynthetically in 1M Na2CO3 reaction buffer (pH adjusted to 9.5 with HCl). The aliquoted fluorescent dyes 5-carboxytetramethylrhodamine succinimidyl ester (Molecular Probes) and fluorescein isothiocyanate (Anaspec, Inc.) were initially dissolved in DMSO. Efficient labeling was achieved by mixing a 25-fold molar excess of dye relative to DNA, in 200mM Na2CO3. The reactions were incubated for 2–3 hours at 37°C. Oligonucleotides were then ethanol precipitated and further purified by gel electrophoresis. Fluorescent bands were visualized using a UV illuminator (Fotodyne Inc.), excised and oligonucleotides from gel slices were again extracted by electroelution. Oligonucleotides were again ethanol precipitated and the labeled pellets were rinsed twice with 70% cold ethanol. The pellets were redissolved in Tris-EDTA (TE) buffer prior to determination of concentration by UV absorbance.
Oligonucleotides containing 2–AP or pyr–dC were synthesized under standard coupling conditions. Single strands were purified using an Amberchrom DG-16C reverse phase resin as previously described (47). 6-MI containing oligonucleotides were synthesized as previously described using an ABI 392-8 synthesizer (coupled normally) ending with the oligonucleotides still attached to the column (manual deprotection selected). 6-MI de-protection was achieved utilizing 1M DBU (1,8-diazabicyclo-[5.4.0]undec-7-ene) in acetonitrile as follows. Set up the column with an empty 3ml syringe on each side. Add 425 μL acetonitrile to one syringe, flush it back and forth then add 75 μL DBU. Flush it back and forth at least three times, then put it in a dark place at room temperature for 5 hours flushing it back and forth about once an hour. After the incubation remove and save the solution. Add 0.5 ml acetonitrile to the column flushing it back and forth several times. Combine this wash with the first eluate and evaporate them using a Speed-Vac. Add 1ml of NH4OH to the column, flush it back and forth and allow it to stand for 1 hour. Add this NH4OH eluate to the corresponding DBU/acetonitryl eluate pellet (from the speed vac) and heat it for 14 hrs at 55°C. Vacuum dry and purify as usual. The pellet will be yellow and oily. It can be put through ethanol precipitation before gel purification or “dissolved” in buffer prior to loading on a gel.
Purity of all oligonucleotides was confirmed by denaturing (urea) gel electrophoresis of 5′ end-labeled single strands. Double-stranded templates for fluorescence and transcription assays were prepared by heating a 1:1 mixture of complementary single strands in TE buffer (10 mM Tris, pH 7.8, 1 mM EDTA) to 75 °C for 5 min. The solutions were then allowed to cool slowly to room temperature and stored frozen until use.
Steady-state fluorescence measurements
Fluorescence measurements were carried out with a PTI (Photon Technology International) L–format fluorimeter with a 75 watt arc lamp and both emission and excitation monochromators, using a 75 μL (light path 3×3 mm, centre 15 mm) ultramicro cell (Hellma). The sample compartment was thermostated at 25°C, and excitation and emission slits were both set to 5 nm. For experiments containing the analog 2–AP, fluorescence was excited at 310 nm and emission was monitored at 375 nm. Samples containing 6–MI were excited at 340 nm and the emission was monitored at 430 nm. In measurements of samples containing pyr–dC, the emission was measured at 460 nm, with excitation at 350 nm. All fluorescence experiments were carried out in fluorescence buffer: 30 mM HEPES, pH 7.8, 15 mM magnesium acetate, 25 mM potassium glutamate, 0.25 mM EDTA and 0.05% TWEEN–20 (Calbiochem, 10% protein grade). The fluorescence changes recorded in all experiments represent an average of three measurements, which were taken after 1 min of incubation, when the reading no longer changed. Background measurements were recorded under identical conditions, but with an unlabeled nucleotide. This allowed correction for changes not only in the fluorescence from the protein, but also for any possible changes of unlabeled DNA during the measurements.
For FRET measurements, a 1 cm path length ultramicro Starna cell (Starna Cells, Inc.) with a nominal volume of 100 μL was used. The slit widths were set to 3 nm and the reactions were carried out at 37 °C. Reported data represent the average of four measurements. Fluorescence buffer consisted of 30 mM HEPES, pH 7.8, 25 mM potassium glutamate, 0.25 mM EDTA, 50 mM NaCl and 0.5% glycerol. Buffer background was subtracted from each measurement.
Determining corrected single stranded DNA concentration
The contribution of fluorescent probes to the measured DNA absorbance was subtracted to correctly determine the concentration of labeled DNA strands. A full spectrum from 220–650 nm was collected for samples containing only free unconjugated dyes and dyes conjugated to DNA. This also ensures the presence of dye in the sample. The labeling efficiencies (~64–90% before purification) were calculated by determining the molar ratio of dye to DNA.
| (1) |
Preparation of duplex DNA molecules
Complementary strands of TAMRA- and FITC- labeled oligonucleotides were hybridized in Tris-EDTA buffer. Samples were again gel-purified using a 12% non-denaturing acrylamide gel to separate labeled double-stranded DNA from excess (labeled) single-stranded DNA. Fluorescent bands were imaged using a Fuji PhosphorImager. These methods were designed to obtain 100% labeling efficiency.
To determine the corrected concentration of the labeled duplexes, the hypochromicity factor, X was first measured using unmodified and unlabeled template and nontemplate strands before and after hybridization. Results showed X = 0.435.
| (2) |
| (3) |
| (4) |
FRET experiment and analysis of data
Labeled duplexes were reacted with enzyme. Final concentration of DNA and T7 RNA polymerase used were 0.026 μM and 0.104 μM, respectively. The reaction was carried out in fluorescence buffer. DNA and enzyme was incubated for 1 min at 37 °C prior to fluorescence measurement. 3′dGTP mix (with concentrations of 3′dGTP, magnesium acetate and Hepes equal to 100 μM,15 mM and 10 mM, respectively in the final reaction mixture) was added to the binary complex and then allowed for 1 min incubation prior to measurement. All steady-state measurements were thermostated at 37 °C.
FRET efficiency was measured using enhanced fluorescence of TAMRA upon excitation at FITC λmax = 490nm. Emission scans from 500 to 650 nm were measured. Standard FITC-only labeled duplexes were constructed to subtract FITC contribution to the total emission to give pure TAMRA emission. In this study, we normalized the total acceptor emission by dividing the resulting acceptor emission by the fluorescence value of the same doubly-labeled duplex sample that is excited at 560 nm, the wavelength where only acceptor TAMRA absorbs. By so doing, we also normalize the obtained FRET signal for the concentration, for the quantum yield and for possible error in percent labeling of the acceptor TAMRA (18, 44).
| (5) |
Parameters ε and E above are the extinction coefficients and efficiency of transfer, respectively, and labeling efficiency, after purification, was near 100%. Experimental values for extinction ratios were measured and found equal to 0.734 for εfluorescein490nm/εTAMRA560nm and 0.05769 for εTAMRA490nm/εTAMRA560nm. For normalized ratio values to calculate for FRET efficiency, ratios of signals taken at 580 nm (the experimental λmax emission for TAMRA) were used. The FRET efficiencies measured are then transformed into distance values from E= R06/(R6 + R06), where R0 is equal to 45 Å (18).
Supplementary Material
A table with FRET efficiencies, corresponding to the data in Figure 2. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by National Institutes of Health grant 1R01GM55002 to C. T. M.
Footnotes
Abbreviations: 2–AP, 2–aminopurine; 6–MI, 6–methylisoxanthopterin; DBU, 1,8-diazabicyclo-[5.4.0]undec-7-ene; FITC, fluorescein-5-isothiocyanate; FRET, fluorescence resonance energy transfer; pyr-dC, pyrrolo-dC; SDS, sodium dodecyl sulfate; TAMRA, 5-carboxytetramethylrhodamine.
References
- 1.McAllister WT. Structure and function of the bacteriophage T7 RNA polymerase (or, the virtues of simplicity) Cell Mol Biol Res. 1993;39:385–91. [PubMed] [Google Scholar]
- 2.Sousa R. Structural and mechanistic relationships between nucleic acid polymerases. Trends Biochem Sci. 1996;21:186–90. [PubMed] [Google Scholar]
- 3.deHaseth PL, Zupancic ML, Record MT., Jr RNA polymerase-promoter interactions: the comings and goings of RNA polymerase. J Bacteriol. 1998;180:3019–25. doi: 10.1128/jb.180.12.3019-3025.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.von Hippel PH, Rees WA, Rippe K, Wilson KS. Specificity mechanisms in the control of transcription. Biophys Chem. 1996;59:231–46. doi: 10.1016/0301-4622(96)00006-3. [DOI] [PubMed] [Google Scholar]
- 5.von Hippel PH, Bear DG, Morgan WD, McSwiggen JA. Protein-nucleic acid interactions in transcription: a molecular analysis. Annu Rev Biochem. 1984;53:389–446. doi: 10.1146/annurev.bi.53.070184.002133. [DOI] [PubMed] [Google Scholar]
- 6.Weston BF, Kuzmine I, Martin CT. Positioning of the start site in the initiation of transcription by bacteriophage T7 RNA polymerase. J Mol Biol. 1997;272:21–30. doi: 10.1006/jmbi.1997.1199. [DOI] [PubMed] [Google Scholar]
- 7.Chapman KA, Gunderson SI, Anello M, Wells RD, Burgess RR. Bacteriophage T7 late promoters with point mutations: quantitative footprinting and in vivo expression. Nucleic Acids Res. 1988;16:4511–24. doi: 10.1093/nar/16.10.4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Újvári A, Martin CT. Identification of a minimal binding element within the T7 RNA polymerase promoter. J Mol Biol. 1997;273:775–81. doi: 10.1006/jmbi.1997.1350. [DOI] [PubMed] [Google Scholar]
- 9.Cheetham GM, Jeruzalmi D, Steitz TA. Structural basis for initiation of transcription from an RNA polymerase- promoter complex. Nature. 1999;399:80–3. doi: 10.1038/19999. [DOI] [PubMed] [Google Scholar]
- 10.Cheetham GM, Steitz TA. Structure of a transcribing T7 RNA polymerase initiation complex. Science. 1999;286:2305–9. doi: 10.1126/science.286.5448.2305. [DOI] [PubMed] [Google Scholar]
- 11.Jiang M, Rong M, Martin C, McAllister WT. Interrupting the template strand of the T7 promoter facilitates translocation of the DNA during initiation, reducing transcript slippage and the release of abortive products. J Mol Biol. 2001;310:509–22. doi: 10.1006/jmbi.2001.4793. [DOI] [PubMed] [Google Scholar]
- 12.Liu C, Martin CT. Fluorescence characterization of the transcription bubble in elongation complexes of T7 RNA polymerase. J Mol Biol. 2001;308:465–75. doi: 10.1006/jmbi.2001.4601. [DOI] [PubMed] [Google Scholar]
- 13.Yin YW, Steitz TA. Structural basis for the transition from initiation to elongation transcription in T7 RNA polymerase. Science. 2002;298:1387–95. doi: 10.1126/science.1077464. [DOI] [PubMed] [Google Scholar]
- 14.Tahirov TH, Temiakov D, Anikin M, Patlan V, McAllister WT, Vassylyev DG, Yokoyama S. Structure of a T7 RNA polymerase elongation complex at 2.9 A resolution. Nature. 2002;420:43–50. doi: 10.1038/nature01129. [DOI] [PubMed] [Google Scholar]
- 15.Újvári A, Martin CT. Evidence for DNA bending at the T7 RNA polymerase promoter. J Mol Biol. 2000;295:1173–84. doi: 10.1006/jmbi.1999.3418. [DOI] [PubMed] [Google Scholar]
- 16.Tang GQ, Patel SS. T7 RNA polymerase-induced bending of promoter DNA is coupled to DNA opening. Biochemistry. 2006;45:4936–46. doi: 10.1021/bi0522910. [DOI] [PubMed] [Google Scholar]
- 17.Dietrich A, Buschmann V, Muller C, Sauer M. Fluorescence resonance energy transfer (FRET) and competing processes in donor-acceptor substituted DNA strands: a comparative study of ensemble and single-molecule data. J Biotechnol. 2002;82:211–31. doi: 10.1016/s1389-0352(01)00039-3. [DOI] [PubMed] [Google Scholar]
- 18.Clegg RM, Murchie AI, Zechel A, Lilley DM. Observing the helical geometry of double-stranded DNA in solution by fluorescence resonance energy transfer. Proc Natl Acad Sci U S A. 1993;90:2994–8. doi: 10.1073/pnas.90.7.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin CT, Muller DK, Coleman JE. Processivity in early stages of transcription by T7 RNA polymerase. Biochemistry. 1988;27:3966–74. doi: 10.1021/bi00411a012. [DOI] [PubMed] [Google Scholar]
- 20.Stano NM, Levin MK, Patel SS. The +2 NTP binding drives open complex formation in T7 RNA polymerase. J Biol Chem. 2002;277:37292–300. doi: 10.1074/jbc.M201600200. [DOI] [PubMed] [Google Scholar]
- 21.Újvári A, Martin CT. Thermodynamic and kinetic measurements of promoter binding by T7 RNA polymerase. Biochemistry. 1996;35:14574–82. doi: 10.1021/bi961165g. [DOI] [PubMed] [Google Scholar]
- 22.Hawkins ME, Pfleiderer W, Balis FM, Porter D, Knutson JR. Fluorescence properties of pteridine nucleoside analogs as monomers and incorporated into oligonucleotides. Anal Biochem. 1997;244:86–95. doi: 10.1006/abio.1996.9879. [DOI] [PubMed] [Google Scholar]
- 23.Martin CT, Ujvari A, Liu C. Evaluation of fluorescence spectroscopy methods for mapping melted regions of DNA along the transcription pathway. Methods Enzymol. 2003;371:13–33. doi: 10.1016/S0076-6879(03)71002-7. [DOI] [PubMed] [Google Scholar]
- 24.Jia Y, Kumar A, Patel S. Equilibrium and stopped-flow kinetic studies of interaction between T7 RNA polymerase and its promoters measured by protein and 2-aminopurine fluorescence changes. J Biol Chem. 1996;271:30451–8. doi: 10.1074/jbc.271.48.30451. [DOI] [PubMed] [Google Scholar]
- 25.Sastry S, Ross B. A direct real-time spectroscopic investigation of the mechanism of open complex formation by T7 RNA polymerase. Biochemistry. 1996;35:15715–25. doi: 10.1021/bi960729d. [DOI] [PubMed] [Google Scholar]
- 26.Tang GQ, Patel SS. Rapid binding of T7 RNA polymerase is followed by simultaneous bending and opening of the promoter DNA. Biochemistry. 2006;45:4947–56. doi: 10.1021/bi052292s. [DOI] [PubMed] [Google Scholar]
- 27.Tu AH, Turnbough CL., Jr Regulation of upp expression in Escherichia coli by UTP-sensitive selection of transcriptional start sites coupled with UTP-dependent reiterative transcription [published errata appear in J Bacteriol 1997 Dec;179(24):7887 and 1998 Sep;180(18):4991] J Bacteriol. 1997;179:6665–73. doi: 10.1128/jb.179.21.6665-6673.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Millar DP. Fluorescence studies of DNA and RNA structure and dynamics. Curr Opin Struct Biol. 1996;6:322–6. doi: 10.1016/s0959-440x(96)80050-9. [DOI] [PubMed] [Google Scholar]
- 29.Walter NG, Harris DA, Pereira MJ, Rueda D. In the fluorescent spotlight: global and local conformational changes of small catalytic RNAs. Biopolymers. 2001;61:224–42. doi: 10.1002/bip.10144. [DOI] [PubMed] [Google Scholar]
- 30.Menger M, Tuschl T, Eckstein F, Porschke D. Mg(2+)-dependent conformational changes in the hammerhead ribozyme. Biochemistry. 1996;35:14710–6. doi: 10.1021/bi960440w. [DOI] [PubMed] [Google Scholar]
- 31.Craig ML, Suh WC, Record MT., Jr HO• and DNAse I Probing of Eσ70 RNA Polymerase - λPR Promoter Open Complexes: Mg2+ Binding and Its Structural Consequences at the Transcription Start Site. Biochemistry. 1995;34:15624–32. doi: 10.1021/bi00048a004. [DOI] [PubMed] [Google Scholar]
- 32.Murakami KS, Masuda S, Campbell EA, Muzzin O, Darst SA. Structural basis of transcription initiation: an RNA polymerase holoenzyme-DNA complex. Science. 2002;296:1285–90. doi: 10.1126/science.1069595. [DOI] [PubMed] [Google Scholar]
- 33.Murakami KS, Masuda S, Darst SA. Structural basis of transcription initiation: RNA polymerase holoenzyme at 4 A resolution. Science. 2002;296:1280–4. doi: 10.1126/science.1069594. [DOI] [PubMed] [Google Scholar]
- 34.Zhang G, Campbell EA, Minakhin L, Richter C, Severinov K, Darst SA. Crystal structure of Thermus aquaticus core RNA polymerase at 3.3 A resolution. Cell. 1999;98:811–24. doi: 10.1016/s0092-8674(00)81515-9. [DOI] [PubMed] [Google Scholar]
- 35.Gnatt AL, Cramer P, Fu J, Bushnell DA, Kornberg RD. Structural basis of transcription: an rna polymerase ii elongation complex at 3.3 a resolution. Science. 2001;292:1876–82. doi: 10.1126/science.1059495. [DOI] [PubMed] [Google Scholar]
- 36.Cramer P, Bushnell DA, Kornberg RD. Structural basis of transcription: rna polymerase ii at 2.8 angstrom resolution. Science. 2001;292:1863–76. doi: 10.1126/science.1059493. [DOI] [PubMed] [Google Scholar]
- 37.Bushnell DA, Kornberg RD. Complete, 12-subunit RNA polymerase II at 4.1-A resolution: implications for the initiation of transcription. Proc Natl Acad Sci U S A. 2003;100:6969–73. doi: 10.1073/pnas.1130601100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murakami KS, Darst SA. Bacterial RNA polymerases: the wholo story. Curr Opin Struct Biol. 2003;13:31–9. doi: 10.1016/s0959-440x(02)00005-2. [DOI] [PubMed] [Google Scholar]
- 39.Sousa R, Chung YJ, Rose JP, Wang BC. Crystal structure of bacteriophage T7 RNA polymerase at 3.3 Å resolution. Nature. 1993;364:593–9. doi: 10.1038/364593a0. [DOI] [PubMed] [Google Scholar]
- 40.Place C, Oddos J, Buc H, McAllister WT, Buckle M. Studies of contacts between T7 RNA polymerase and its promoter reveal features in common with multisubunit RNA polymerases. Biochemistry. 1999;38:4948–57. doi: 10.1021/bi982689e. [DOI] [PubMed] [Google Scholar]
- 41.Suh WC, Leirmo S, Record MT., Jr Roles of Mg2+ in the mechanism of formation and dissociation of open complexes between Escherichia coli RNA polymerase and the λPR promoter: kinetic evidence for a second open complex requiring Mg2+ Biochemistry. 1992;31:7815–25. doi: 10.1021/bi00149a011. [DOI] [PubMed] [Google Scholar]
- 42.Brieba LG, Sousa R. The T7 RNA polymerase intercalating hairpin is important for promoter opening during initiation but not for RNA displacement or transcription bubble stability during elongation. Biochemistry. 2001;40:3882–90. doi: 10.1021/bi002716c. [DOI] [PubMed] [Google Scholar]
- 43.Stano NM, Patel SS. The intercalating beta-hairpin of T7 RNA polymerase plays a role in promoter DNA melting and in stabilizing the melted DNA for efficient RNA synthesis. J Mol Biol. 2002;315:1009–25. doi: 10.1006/jmbi.2001.5313. [DOI] [PubMed] [Google Scholar]
- 44.Lorenz M, Hillisch A, Diekmann S. Fluorescence resonance energy transfer studies of U-shaped DNA molecules. J Biotechnol. 2002;82:197–209. doi: 10.1016/s1389-0352(01)00038-1. [DOI] [PubMed] [Google Scholar]
- 45.Davanloo P, Rosenberg AH, Dunn JJ, Studier FW. Cloning and expression of the gene for bacteriophage T7 RNA polymerase. Proc Natl Acad Sci U S A. 1984;81:2035–9. doi: 10.1073/pnas.81.7.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.King GC, Martin CT, Pham TT, Coleman JE. Transcription by T7 RNA polymerase is not zinc-dependent and is abolished on amidomethylation of cysteine-347. Biochemistry. 1986;25:36–40. doi: 10.1021/bi00349a006. [DOI] [PubMed] [Google Scholar]
- 47.Schick C, Martin CT. Identification of specific contacts in T3 RNA polymerase-promoter interactions: kinetic analysis using small synthetic promoters. Biochemistry. 1993;32:4275–80. doi: 10.1021/bi00067a016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A table with FRET efficiencies, corresponding to the data in Figure 2. This material is available free of charge via the Internet at http://pubs.acs.org.