

Abstract
Parkinson’s disease (PD) is a neurologic disorder characterized by dopaminergic cell death in the substantia nigra. PD pathogenesis involves mitochondrial dysfunction, proteasome impairment, and α-synuclein aggregation, insults that may be especially toxic to oxidatively stressed cells including dopaminergic neurons. The enzyme methionine sulfoxide reductase A (MsrA) plays a critical role in the antioxidant response by repairing methionine-oxidized proteins and by participating in cycles of methionine oxidation and reduction that have the net effect of consuming reactive oxygen species. Here, we show that MsrA suppresses dopaminergic cell death and protein aggregation induced by the complex I inhibitor rotenone or mutant α-synuclein, but not by the proteasome inhibitor MG132. By comparing the effects of MsrA and the small-molecule antioxidants N-acetyl-cysteine and vitamin E, we provide evidence that MsrA protects against PD-related stresses primarily via methionine sulfoxide repair rather than by scavenging reactive oxygen species. We also demonstrate that MsrA efficiently reduces oxidized methionine residues in recombinant α-synuclein. These findings suggest that enhancing MsrA function may be a reasonable therapeutic strategy in PD.
Keywords: aggresome, dopamine, glutathione, heat shock protein, methionine sulfoxide reductase, neurodegeneration, oxidative stress, Parkinson’s disease, proteasome, protofibril, rotenone, synuclein
Parkinson’s disease (PD) is a neurologic disorder that involves a selective loss of dopaminergic neurons from the substantia nigra [1, 2]. The postmortem brains of PD patients are characterized by reduced activity of mitochondrial complex I, an enzyme of the mitochondrial electron transport chain [3, 4]. In turn, this defect may cause a ‘leakage’ of electrons from mitochondria, leading to the accumulation of reactive oxygen species (ROS) that damage proteins, lipids, and nucleic acids [3, 5]. The brains of PD patients also show evidence of impaired proteasomal function [6], a defect that results in increased oxidative stress and decreased elimination of oxidatively damaged polypeptides [7–9]. Dopaminergic neurons of the substantia nigra contain relatively high basal levels of ROS resulting from dopamine metabolism and auto-oxidation [10]. Therefore, these neurons may be selectively vulnerable to insults that increase oxidative stress in PD, including complex I inhibition and proteasome impairment.
Surviving neurons in the brains of PD patients contain ‘Lewy bodies,’ cytosolic inclusions enriched with aggregated forms of the presynaptic protein α-synuclein [11]. Mutations in the α-synuclein gene have been identified in patients with early-onset, autosomal-dominant PD [1, 2]. Two familial mutations, A30P and A53T, promote the conversion of α-synuclein to potentially toxic oligomers or ‘protofibrils’, suggesting that the enhanced formation of these assemblies contributes to PD pathogenesis [12, 13]. Oxidative stress may play a role in α-synuclein neurotoxicity in two ways. First, oxidative modifications promote the formation of α-synuclein oligomers but not mature fibrils [14–16]. Second, aggregated forms of α-synuclein may cause an accumulation of ROS, thereby triggering a vicious cycle [17].
A number of antioxidant systems are available to prevent oxidative damage leading to protein aggregation. The enzyme methionine sulfoxide reductase A (MsrA) plays an important role in the antioxidant response by reducing the S-stereoisomer of methionine sulfoxide (MetSO) to methionine [18]. MsrA is present throughout the brain, including the substantia nigra [19]. Cells with increased or decreased levels of MsrA are relatively resistant or vulnerable to oxidative insults, respectively [18, 20–23]. MsrA protects cells from oxidative stress not only by repairing proteins damaged by methionine oxidation, but also by engaging in a cycle of methionine oxidation and reduction that ultimately results in ROS scavenging [18, 22]. Two active-site cysteine residues play key roles in MsrA activity. Cysteine 72 carries out a nucleophilic attack at the sulfur atom of the methionine sulfoxide substrate, leading to the formation of a covalent intermediate, whereas cysteine 218 attacks cysteine 72 to trigger breakdown of the covalent complex [24].
The results of studies in MsrA-knockout mice [25] and in flies over-expressing MsrA in neural tissues [26] suggest that down- or up-regulation of MsrA is associated with decreased or increased longevity, respectively. MsrA activity decreases with age in rats [27], and this decrease coupled with the resulting age-related increase in oxidative stress may explain in part why the incidence of PD increases with age [1, 2]. A very recent study revealed that MsrA expression slows age-related motor defects and neurodegeneration in transgenic Drosophila over-expressing α-synuclein [28].
Although evidence suggests that MsrA has a neuroprotective function associated with longevity [25], it is uncertain whether the enzyme specifically inhibits dopaminergic cell death in PD. Because oxidative stress is critically involved in dopaminergic neurodegeneration, we hypothesized that MsrA plays a major role in dopamine neuron survival. To address this hypothesis, we investigated whether cells over-expressing MsrA were resistant to dopaminergic cell death and protein aggregation induced by various PD-related insults. Our rationale was that by examining the effects of MsrA over-expression, we would amplify (and, therefore, reveal) neuroprotective activities associated with the endogenous enzyme. The results of our study indicate that MsrA protects dopaminergic neurons from the toxic effects of complex I inhibition and α-synuclein expression, but not proteasome dysfunction.
Materials and methods
Antibodies
The following antibodies were used in this study: mouse anti-β-actin (Sigma-Aldrich, St. Louis, MO); mouse anti-Hsp70 (Assay Designs, Ann Arbor, MI); mouse anti-iHsp70 (Assay Designs); mouse anti-MAP2 (Chemicon, Temecula, CA); rabbit anti-MsrA (Upstate USA, Charlottesville, VA); mouse anti-α-synuclein Syn-1 (BD PharMingen, San Diego, CA); rabbit anti-TH (Chemicon); mouse anti-TIM23 (BD PharMingen); rabbit anti-ubiquitin (Chemicon); mouse anti-vimentin (Chemicon); anti-mouse IgG-Alexa Fluor 488 and anti-rabbit IgG-Alexa Fluor 594 (Invitrogen, Carlsbad, CA); anti-mouse IgG and anti-rabbit IgG conjugated to alkaline phosphatase (Promega, Madison, WI). The anti-Hsp70 antibody recognizes both the constitutive and inducible forms of Hsp70 (Hsc70 and iHsp70, respectively), whereas the anti-iHsp70 antibody recognizes the inducible form only.
Preparation of DNA constructs
Bovine MsrA and human α-synuclein (A53T) were expressed in primary mesencephalic cultures from lentiviruses generated using the ViraPower Lentivirus Expression System (Invitrogen). A lentiviral construct encoding A53T was generated as described previously [29–31]. The construct pET28b-MsrA, which contains the gene encoding bovine MsrA including the N-terminal mitochondrial targeting sequence [24, 32], was provided by Dr. Todd Lowther (Wake Forest University). The MsrA gene was amplified from this construct by PCR and subcloned as a BamH I-Xho I fragment into pENTR1A to generate the construct, pENTR-MsrA. The insert from pENTR-MsrA was transferred into the pLENTI6/V5 DEST lentiviral expression vector via recombination. Bovine MsrA was expressed in stably transfected MES23.5 cells from a construct derived from the mammalian expression vector, pcDNA3 (Invitrogen). The cDNA encoding bovine MsrA (from pENTR-MsrA) was subcloned as a BamH I-Xho I fragment into pcDNA3 to generate the construct, pcDNA3-MsrA.
In other experiments, A53T α-synuclein, wild-type, bovine MsrA and two catalytically inactive MsrA mutants, C72S and C218S, were expressed in primary midbrain cultures from adenoviruses generated using the ViraPower Adenoviral Expression System (Invitrogen). Constructs encoding C72S and C218S were produced via overlap extension and subcloned as BamH I-Xho I fragments into pENTR1A, yielding pENTR-C72S and pENTR-C218S. The inserts from pENTR-A53T, pENTR-MsrA, pENTR-C72S, and pENTR-C218S were transferred into the pAd/CMV/V5 DEST adenoviral expression vector via recombination.
Each DNA construct was sequenced using an Applied Biosystems DNA sequencer (Purdue University or University of Wisconsin).
Treatment of primary midbrain cultures
Primary midbrain cultures were prepared from day 17 rat embryos as described previously [29–31, 33]. All of the animal handling procedures were reviewed and approved by the Purdue Animal Care and Use Committee. Briefly, whole brains were dissected from day 17 embryos obtained from pregnant Sprague-Dawley rats (Harlan, Indianapolis). The mesencephalic region containing the substantia nigra and ventral tegmental area was isolated stereoscopically and dissociated with trypsin (26 μg/mL in 0.9% [w/v] NaCl). The cells were plated on coverslips pre-treated with poly-L-lysine (5 μg/mL) in media consisting of DMEM, 10% (v/v) FBS, 10% (v/v) HS, penicillin (100 U/mL), and streptomycin (100 μg/mL). Four days later, the cells were treated for 48 h with β-D-arabinofuranoside hydrochloride (AraC) (20 μM) to inhibit the growth of glial cells.
Next, the primary cultures (7 days in vitro) were transduced with adenoviruses (Figures 1D and 6D only) or lentiviruses prepared using the ViraPower II Gateway Expression System (Invitrogen). Unless otherwise specified, the viral transductions were carried out for 72 h at a multiplicity of infection (MOI) of 10. In experiments aimed at determining the effects of MsrA expression on complex I inhibition or proteasome impairment, the cells were incubated with virus for 72 h. The cells were then treated with fresh media containing rotenone (100 nM) or MG132 (10 μM). Control cells were incubated in the absence of virus and then treated with fresh media supplemented with vehicle (DMSO, 0.002–1% [v/v]) for 24, unless otherwise specified. In experiments aimed at determining the effects of MsrA expression on α-synuclein neurotoxicity, the cells were incubated with virus for 72 h and treated with fresh media for an additional 24 h, unless otherwise specified. Control experiments with LacZ virus were carried out to confirm that the toxic effects of A53T and the protective effects of MsrA were specific.
Figure 1.

MsrA protects primary dopaminergic neurons against rotenone toxicity. (A) Western blot analysis of MsrA expression levels in primary midbrain cultures. Lane 1: lysate from cells transduced with LacZ lentivirus for 72 h and then incubated in fresh media for 48 h; lanes 2–6: lysates from cells transduced with MsrA lentivirus for 24 h (lane 2), 48 h (lane 3), 72 h (lane 4), 72 h followed by a 24-h incubation in fresh media (lane 5), or 72 h followed by a 48-h incubation in fresh media (lane 6). 12 μg of protein was loaded in each lane. (B) Western blot showing the distribution of MsrA between mitochondrial fractions (lanes 1 and 2) and cytosolic fractions (lanes 3 and 4). MES23.5 cells were transduced with adenovirus encoding bovine MsrA (bMsrA) (lanes 1 and 3) or LacZ (lanes 2 and 4). (C) Wild-type MsrA expressed from a lentiviral construct inhibits dopaminergic cell death induced by rotenone (48 h). (D) Wild-type MsrA (but not the catalytically inactive mutants C72S and C218S) expressed from an adenoviral construct suppresses neurotoxicity elicited by rotenone (24 h). The data in (C) and (D) are presented as the mean +/− SEM, N = 3; *p<0.05.
Figure 6.

MsrA protects primary dopaminergic neurons from toxicity elicited by mutant α-synuclein. (A) Western blot showing α-synuclein and MsrA levels in primary midbrain cultures. Bands corresponding to bovine and rat MsrA (bMsrA and rMsrA) are indicated by arrows to the right of the blot. 20 μg of protein was loaded in each lane. (B) MsrA expressed from a lentiviral construct suppresses A53T-induced dopaminergic cell death, and the protective effect of MsrA exceeds that of NAC. (C) MsrA expressed from a lentiviral construct protects against A53T-induced dopaminergic cell death with greater efficacy than vitamin E. (D) Wild-type MsrA (but not the catalytically inactive mutants C72S and C218S) expressed from an adenoviral construct suppresses A53T neurotoxicity. The data in (B)-(D) are presented as the mean +/− SEM, N = 3; *p<0.05, **p<0.01, ***p<0.001.
To test the effects of N-acetyl-cysteine (NAC) on neurotoxicity elicited by complex I inhibition, primary midbrain cultures were treated with rotenone (100 nM) in the absence or presence of NAC (1 mM). After a 24-h incubation, the cells were analyzed as described below. A similar approach was used to test the effects of vitamin E, except that in this case the cells were pre-cultured in the absence or presence of vitamin E (2 μM, 72 h) and then treated with rotenone in the absence or presence of vitamin E for 24 h.
To test the effects of small-molecule antioxidants on α-synuclein neurotoxicity, the cells were incubated with A53T lentivirus in the absence or presence of NAC (1 mM) or vitamin E (2 μM). After a 72-h transduction period, the csells were treated with fresh media (in the absence or presence of NAC or vitamin E) for an additional 24 h prior to analysis.
Transfection of MES23.5 cells
The MES23.5 mouse-rat hybrid dopaminergic cell line was provided by Dr. Dennis Selkoe (Brigham and Women’s Hospital) with the permission of Dr. Stanley Appel (Baylor School of Medicine). The cells were routinely propagated in Sato’s N1 medium (87.5% (v/v) DMEM, glutamine (4 mM), NCS (2%, v/v), FBS (5%, v/v), penicillin/streptomycin (1%, v/v), 15 mM HEPES (pH 7.4), and 1x SATO (50x SATO: insulin, 0.25 mg/mL; human transferrin, 0.25 mg/mL; pyruvic acid, 2.43 mg/mL; putrescine, 0.2 mg/mL; sodium selenite, 0.25 μg/mL; progesterone, 0.315 μg/mL) as described [34]. Transfections were carried out by treating the cells with pcDNA3 or pcDNA3-MsrA in the presence of Lipofectamine 2000 (Invitrogen). Stable transfectants were selected in media supplemented with G418 (600 μg/mL). Three to four weeks after transfection, single colonies were isolated using the colony ring method and expanded.
Induction of aggresome formation
MES23.5 cells were plated in 12-well plates on coverslips pretreated with poly-L-lysine (5 μg/mL). The cells were plated at a density of 50,000 to 100,000 cells per well in media supplemented with dibutyryl-cAMP (1 mM) [34]. After 24 h, the cells were treated with fresh media with rotenone (100 nM), MG132 (2 μM), or vehicle (0.0006–0.02% [v/v] DMSO, final concentration in media) for 48 h, unless otherwise specified.
Western blot analyses
Protein expression levels in primary midbrain cultures and MES23.5 cells were determined via Western blot analysis. After cell lysis in buffer L (20 mM Tris HCl, pH 7.4, 25 mM KCl, 5 mM MgCl2, 0.25 M sucrose, 1% (v/v) Triton X-100, 0.5 mg/mL benzamidine, 2 μg/mL aprotinin, 3.6 μg/mL leupeptin, 0.75 mM PMSF, 700 units/mL DNase I), equal amounts of protein from the soluble fraction were separated via SDS-PAGE and transferred to nitrocellulose or polyvinylidene difluoride (PVDF) membranes.
In other experiments, MES23.5 cells were cultured until they were 70%–80% confluent. The cells were transduced with adenovirus encoding LacZ or MsrA (MOI = 50) for 24 h and incubated in fresh media (after a 50:50 split) for an additional 24 h. Cytosolic and mitochondrial fractions were then prepared using the Mitochondria Isolation Kit for Cultured Cells (Pierce). Cytosolic fractions were concentrated with an Ultrafree-0.5 PBCC Centrifugal Filter Unit (Millipore). Mitochondrial fractions were lysed using buffer L. Equal amounts of protein from each fraction were separated via SDS-PAGE and transferred to PVDF membranes.
Western blotting membranes were probed with a primary antibody specific for mammalian α-synuclein (Syn-1, 1:1000), MsrA (1:2000), iHsp70 (1:1000), β-actin (1:10 000) or the mitochondrial marker TIM23 (1:1000) and treated with a secondary anti-mouse or anti-rabbit, alkaline phosphatase-conjugated IgG (1:5000). Chemifluorescence images were obtained and analyzed using a Typhoon imaging system (GE Health Sciences, Piscataway, NJ).
Immunocytochemical analysis
Cells were fixed in 4% (w/v) paraformaldehyde in PBS (pH 7.4) for 30 min and subsequently permeabilized for 1 h with buffer P (PBS, pH 7.4, 0.3% [v/v] Triton X-100, 1% [w/v] BSA, and 10% [v/v] FBS). After washing with PBS, the cells were treated for 1 h at 22 °C or overnight at 4 °C with the following primary antibodies: (i) anti-MAP2 (1:500) plus anti-TH (1:500), to monitor relative dopaminergic cell viability; or (ii) anti-vimentin (1:500), anti-Hsp70 (1:200), or anti-ubiquitin (1:1000), to monitor aggresome formation. After washing, the cells were treated for 1 h at room temperature with secondary antibodies conjugated to Alexa Fluor 488 (1:1000) or Alexa Fluor 594 (1:1000). Cells stained for aggresomes were treated with the nuclear stain 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI) (5 min, 22 °C). The cells were examined using a Nikon Eclipse 1000 or Nikon TE2000-U inverted fluorescence microscope. Fluorescent staining was not observed when the cells were treated with secondary antibody in the absence of primary antibody.
Cell counting
Dopaminergic cell viability was assessed by counting MAP2+ and TH+ primary neurons. The data were expressed as the percentage of MAP2+ neurons that were also TH+ (this ratiometric approach was used to correct for variations in cell density). Aggresome formation was monitored by counting stained MES23.5 cells with or without aggresomes, operationally defined as compact, spherical, perinuclear inclusions that stained positive for one of several classic aggresome ‘markers’ (see ‘Results’). The data were expressed as the percentage of cells that contained aggresomes. Primary neurons and MES23.5 cells were counted blindly in 10 randomly chosen observation fields (corresponding to a minimum of 150 cells) for each treatment. Each experiment was repeated 3 to 4 times using different preparations of cells.
Measurement of protein carbonyls and intracellular glutathione
Primary midbrain cells were dislodged from the plate by trypsinization and collected by centrifugation. To assay protein carbonyls, the cells were washed with PBS and lysed in buffer L. Equal amounts of protein (9 μg) from each sample were characterized with respect to protein carbonyl content via Western blotting using the OxyBlot Oxidized Protein Detection Kit (Chemicon). To assay total intracellular glutathione (GSH), the cells were washed with 0.4 M MES, 0.1 M phosphate, 2 mM EDTA, pH 6.0 and lysed in the same buffer via sonication. The cell lysate (5 μg protein) was assayed using the Glutathione Assay Kit (Cayman Chemical). A typical measurement was 20–25 nmol of GSH per mg of cellular protein.
Purification and oxidation of recombinant α-synuclein
Recombinant, human wild-type α-synuclein was produced in E. coli and purified as described previously [35, 36]. The lyophilized protein was dissolved in 10 mM sodium phosphate buffer (pH 7.0) with 100 mM NaCl and filtered through a 0.22 μm nylon spin filter (Costar) to remove high-molecular mass aggregates. To oxidize α-synuclein, an aliquot of the protein (final concentration, 360–430 μM) was treated with 4% (v/v) H2O2 (20 min, 22 °C) as described [37]. Excess H2O2 was removed via buffer exchange using an Amicon Ultra spin filter. The concentration of oxidized α-synuclein or the unoxidized, control protein was measured using the BCA Protein Assay kit (Pierce).
Purification of recombinant bovine MsrA
Recombinant MsrA was prepared using a method adapted from a previous study [24]. Cells of the BL21(DE3) strain of Escherichia coli were transformed with the pET28b-MsrA construct by electroporation. The cells were grown to an OD600 of 0.6 in 2 L of Luria-Bertani broth (LB) plus kanamycin (100 mg/L) at 37 °C, and expression of the MsrA gene was induced by adding isopropyl-β-D-thiogalactopyranoside (1 mM). The cells were grown under inducing conditions for 4 h at 25 °C, harvested by centrifugation, and resuspended in buffer M (50 mM HEPES, pH 7.9, 10% (v/v) glycerol, 0.1% (v/v) Triton X-100, 0.5 M KCl, 1 mM MgCl2, 15 mM methionine, 5 mM imidazole, supplemented with protease inhibitor cocktail (Sigma B8849)). After one round of freeze-thawing, the cells were lysed with a French pressure cell (p.s.i. > 1000) (Thermo Electron Corporation, Waltham, MA). After centrifugation (40,000 g, 30 min), the supernatant was applied to an immobilized metal affinity column (IMAC) charged with NiCl2 and equilibrated with buffer M (IMAC chromatography was carried out at 4 °C with a flow rate of 2 mL/min). After washing with buffer M followed by buffer N (buffer M minus glycerol, Triton X-100, and protease inhibitor cocktail), MsrA was eluted with a linear gradient of imidazole (5 to 250 mM over 150 min) in buffer N. Fractions most highly enriched with MsrA were identified by SDS-PAGE with Coomassie Brilliant Blue staining, pooled, incubated on ice for 1 h, and dialyzed twice against 500 mL of the following buffer: 20 mM MES, pH 6.5, 20 mM NaCl, 0.1 mM EDTA (4 °C). The protein concentration was determined by measuring the absorbance at 280 nm with a theoretical extinction coefficient of 31,390 M−1cm−1. Typical yields were 3.5 mg per L of bacterial culture, and the purity of the final protein sample was estimated to be approximately 95% by SDS-PAGE.
Assay of α-synuclein repair by MsrA
Recombinant, oxidized α-synuclein and MsrA were diluted to a final concentration of 240 μM and 4 μM, respectively, in buffer R (10 mM sodium phosphate, pH 7.0, 100 mM NaCl, 15 mM dithiothreitol (DTT)). A control sample of oxidized α-synuclein was incubated in buffer R in the absence of MsrA. After incubation for 2 h at 37 °C, each sample was injected onto a reversed-phase HPLC (RP-HPLC) column (Vydac C18, 4.6 mm ID X 250 mm, catalog number 218 TP54) equilibrated with mobile phase (5% (v/v) CH3CN in H2O, 0.1% (v/v) trifluoroacetic acid (TFA)). RP-HPLC was carried out at 22 °C with a flow rate of 1 mL/min. The bound proteins were eluted with a linear gradient of CH3CN in mobile phase (5–80% over 180 min). The eluate was monitored at 280 nm, and peak fractions were collected and analyzed by mass spectrometry.
Assay of α-synuclein repair by lysates of PC12 cells over-expressing MsrA
PC12 cells were cultured in 245 mm × 245 mm dishes until they were 50%–60% confluent. The cells were transduced with adenovirus encoding LacZ or MsrA (MOI = 50) for 24 h and incubated in fresh media for an additional 48 h. Cell lysates were prepared in buffer A (20 mM Tris HCl, pH 7.0, 300 mM NaCl, supplemented with protease inhibitor cocktail) via dounce homogenization. A sample of oxidized α-synuclein (300 μg, prepared as described above) was incubated with cell lysate (1.25 mg) in buffer A supplemented with DTT (final concentration, 15 mM) in a total volume of ~300 μL, at 37 °C for 30 min. The mixture was boiled for 5 min, and insoluble proteins were pelleted via centrifugation (13,000 g, 5 min). A control sample was boiled immediately after mixing oxidized α-synuclein with the PC12 cell lysate. The boiling-centrifugation step was used as a method to reisolate α-synuclein, which unlike most proteins remains soluble at elevated temperatures. The supernatant was filtered through a 0.22 μm spin-filter membrane and applied to an RP-HPLC column as described above. The column was pre-equilibrated as described above. For these analyses, the bound proteins were eluted with a stepwise gradient of CH3CN in mobile phase (5–35% over the first 10 min, 35–45% over the next 50 min, and 45–100% over the final 10 min). We verified via control experiments that the mobility of unoxidized and oxidized α-synuclein was unaffected by the boiling step.
Mass spectrometry
Matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) was carried out using a Voyager DE PRO mass spectrometer (Applied Biosystems, Framingham, MA). This instrument is equipped with a nitrogen laser (337 nm) for ionization and a time-of-flight mass analyzer. Positive ion mass spectra were obtained in the linear mode. The accelerating voltage and grid voltage were set at 25 kV and 94%, respectively. The extraction delay time was set at 650 nsec. The acquisition mass range for this study was 1000 to 20,000 Da. There were 100 laser shots per spectrum. The matrix used for these proteins was sinapinic acid. The sample and matrix were mixed in a ratio of 1μL to 1μL on the sample plate. This mixture was allowed to air dry prior to introduction into the mass spectrometer for analysis.
Statistical analyses
Statistical analyses were carried out using the program GraphPad Prism, Version 4.0 (http://www.graphpad.com/prism/Prism.htm). Unless otherwise specified, statistical significance was verified by one-way ANOVA with the Newman-Keuls post-test.
Results
MsrA inhibits dopaminergic cell death induced by rotenone
To determine whether MsrA protected primary dopaminergic neurons from PD-related insults, we first examined its level of expression from a lentiviral construct. Primary midbrain cultures were transduced with lentivirus encoding bovine MsrA, and cell lysates were analyzed via Western blotting (Figure 1A). The results indicated that MsrA was expressed maximally in cells transduced with lentivirus for 72 h, followed by incubation in fresh media for an additional 24–48 h. Endogenous rat MsrA was not detected on our initial Western blot because insufficient amounts of protein were loaded on the SDS-PAGE gel. However, the rat protein was detectable in subsequent analyses in which higher amounts of protein were loaded (Figure 6A). Quantitative analysis of these subsequent blots indicated that the level of bovine MsrA was ~7-fold greater than that of the endogenous rat protein. Additional Western blot analyses revealed that the bovine enzyme was present in cytosolic and mitochondrial fractions (Figure 1B), consistent with previous findings [32].
Next, we examined whether MsrA protected primary dopaminergic neurons from rotenone toxicity. Primary cultures were untransduced or transduced with lentivirus or adenovirus encoding wild-type MsrA or two catalytically inactive mutants, C72S or C218S [24], and subsequently treated with rotenone. The relative number of TH+ neurons was greater in cultures transduced with wild-type MsrA virus compared to untransduced cells or cells infected with virus encoding C72S or C218S (Figure 1C,D). We verified via Western blot analysis that both mutants were expressed at the same level as the wild-type protein (data not shown). Transduction of the cells with control LacZ virus had no significant effect on rotenone-induced TH+ cell loss (data not shown). Together, these data suggested that wild-type but not mutant MsrA rescued dopaminergic neurons from the toxic effects of rotenone in our primary cell-culture model.
MsrA suppresses aggresome formation in dopaminergic cells treated with rotenone
In the next phase of our study, we addressed whether MsrA suppressed rotenone-induced protein aggregation. Previous findings indicate that perinuclear, Lewy-like inclusions termed ‘aggresomes’ are formed in neuronal cells treated with pro-oxidants such as dopamine [38] and rotenone [39]. Characteristic features of aggresomes include (i) a core enriched with ubiquitylated protein aggregates, chaperones, and proteasome subunits, and (ii) a surrounding cage-like structure composed of vimentin [40, 41]. Aggresomes are thought to be generated via an active cellular process involving microtubule-dependent transport of misfolded proteins to the pericentriolar region [40]. We developed a model of aggresome formation by treating MES23.5 cells derived from rat mesencephalon with rotenone (Figure 2) or the proteasome inhibitor, MG132 (see Figure 9, below). Untreated cells exhibited a diffuse pattern of vimentin staining that extended into the neuronal processes (Figure 2Ai). In cells exposed to rotenone (25–50 nM) for only 24 h, vimentin was partially redistributed from the cytosol to the vicinity of the nucleus (Figure 2Aii–iii), whereas it became fully localized to compact, perinuclear structures in cells treated with 50 to 100 nM rotenone for 48 h (Figure 2Aiv). These structures stained positive for Hsp70 and ubiquitin (Figure 2B), and their formation was abrogated in cells treated with the microtubule disruption agent nocodazole (data not shown). These observations indicated that rotenone-treated MES23.5 cells contained perinuclear inclusions with the properties of classic aggresomes [40].
Figure 2.

Rotenone induces the formation of aggresome-like inclusions in the MES23.5 dopaminergic cell line. (A) MES23.5 cells were treated as follows: (i) 0.0006% [v/v] DMSO, 48 h; (ii) 25 nM rotenone, 24 h; (iii) 50 nM rotenone, 24 h; (iv) 50 nM rotenone, 48 h. The cells were stained with a primary antibody specific for vimentin. The arrows in (iv) indicate perinuclear, aggresome-like inclusions. (B) MES23.5 cells were incubated in the presence of DMSO (0.0006%, v/v; top row) or rotenone (50 nM; bottom row) for 48 h. The cells were stained with a primary antibody specific for Hsp70 (left column) or ubiquitin (right column). In (A) and (B), the nuclei were stained with DAPI (blue). The scale bar corresponds to 10 μm.
Figure 9.

MsrA fails to suppress dopaminergic cell death and aggresome formation elicited by proteasome dysfunction. (A) Primary midbrain cultures were treated with MG132 without lentiviral infection or after transduction with MsrA lentivirus. (B) MES23.5 cells were cultured in the presence of DMSO (0.02%, v/v; top row) or MG132 (bottom row). The cells were stained with a primary antibody specific for vimentin (left column), Hsp70 (middle column), or ubiquitin (right column), and the nuclei were stained with DAPI (blue). The scale bar corresponds to 10 μm. (C) MES23.5 cells stably transfected with pcDNA3 or pcDNA3-MsrA were treated with MG132, and the cells were stained for vimentin (‘vim’), Hsp70, or ubiquitin (‘ub’). The data are expressed as the percentage of total cells with aggresomes (mean +/− SD, N = 3, p<0.001, Student’s t test for each stain).
Given that aggresome formation involves the self-assembly of oxidatively damaged proteins [38, 42], we predicted that aggresomes should occur less frequently in neuronal cells over-expressing MsrA. To address this hypothesis, we established a monoclonal MES23.5 cell line stably transfected with a construct encoding bovine MsrA (pcDNA3-MsrA) or with the control vector. Western blotting data revealed the presence of a band corresponding to bovine MsrA in neuronal cells transfected with pcDNA3-MsrA, whereas the endogenous rat protein was not detectable in the vector-control cells (Figure 3A). Vector-control and MsrA-expressing cells were treated with rotenone and analyzed immunocytochemically using a primary antibody specific for vimentin, Hsp70, or ubiquitin. Aggresomes that stained positive for each marker protein were observed less frequently in rotenone-treated cells expressing MsrA than in control cells (Figure 3B,C). From these data, we inferred that MsrA inhibited rotenone-induced aggresome formation.
Figure 3.

MsrA suppresses aggresome formation induced by rotenone in MES23.5 cells. (A) Western blot analysis of MsrA expression levels in MES23.5 cells stably transfected with pcDNA3 (lane 1) or pcDNA3-MsrA (lane 2). (B) Immunocytochemical analysis of aggresome formation. MES23.5 cells stably transfected with pcDNA3 (top row) or pcDNA3-MsrA (bottom row) were treated with rotenone. The cells were stained with a primary antibody specific for vimentin (left column), Hsp70 (middle column), or ubiquitin (right column). Nuclei were stained with DAPI (blue). The scale bar corresponds to 10 μm. (C) Quantification of rotenone-induced aggresome formation in cells transfected with pcDNA3 or pcDNA3-MsrA and stained for vimentin (‘vim’), Hsp70, or ubiquitin (‘ub’). The data are expressed as the percentage of total cells with aggresomes (mean +/− SD, N = 3, p<0.001, Student’s t test for each stain).
MsrA suppresses rotenone toxicity with little effect on protein carbonyl or glutathione levels
Because rotenone triggers dopaminergic cell death by causing a buildup of ROS [43], MsrA could protect against rotenone-induced neurodegeneration by (i) suppressing intracellular oxidative stress via a ROS scavenging mechanism, and/or (ii) repairing methionine-oxidized proteins. To address these mechanisms, we tested the effect of MsrA on indices of oxidative stress in rotenone-treated, primary midbrain cultures. MsrA was compared to NAC, a membrane-permeable antioxidant that suppresses oxidative stress by quenching ROS directly and by promoting the synthesis of glutathione (GSH) [44]. We found that cultures exposed to rotenone had increased levels of protein carbonyls (Figure 4A). Rotenone-treated cells also had decreased levels of total, intracellular GSH compared to untreated cells (Figure 4B), presumably due to increased protein glutathionylation and/or export of oxidized GSH (GSSG) from the cell [44]. MsrA over-expression had little impact on protein carbonyl formation or GSH depletion, whereas both effects were mitigated in cells treated with NAC (Figure 4A,B). Rotenone neurotoxicity was alleviated both by MsrA over-expression and by treatment of the cells with NAC (Figure 4C). Similar results were obtained when these experiments were repeated using vitamin E, a lipid-soluble antioxidant, instead of NAC: vitamin E suppressed rotenone-induced protein carbonyl formation and GSH depletion with greater efficacy than MsrA (Figure 5A,B), and both MsrA and vitamin E protected primary dopaminergic neurons from rotenone toxicity (Figure 5C). From these results, we inferred that MsrA inhibited rotenone-induced dopaminergic cell death via a mechanism distinct from that of the ROS scavengers, NAC and vitamin E.
Figure 4.

MsrA protects against rotenone toxicity without affecting protein-carbonyl or glutathione levels. (A) Western blot showing that MsrA only marginally suppresses protein carbonyl formation in rotenone-treated primary cultures. The bracket indicates bands corresponding to the most abundant carbonylated species. Dashed arrows indicate a small number of bands that are less intense in the ‘rotenone + MsrA’ lane compared to the ‘rotenone’ lane. (B) MsrA fails to reverse GSH depletion in rotenone-treated primary cultures, in contrast to NAC. (C) MsrA and NAC suppress rotenone-induced dopaminergic cell death. The data in (B) and (C) are presented as the mean +/− SEM, N = 3; *p<0.05, **p<0.01.
Figure 5.

MsrA and vitamin E protect against rotenone neurotoxicity via apparently different mechanisms. (A) Western blot showing that vitamin E suppresses rotenone-induced protein carbonyl formation in primary midbrain cultures with greater efficacy than MsrA. The bracket indicates bands corresponding to the most abundant carbonylated species. (B) MsrA fails to reverse GSH depletion in rotenone-treated primary cultures, whereas vitamin E partially restores GSH levels. (C) MsrA and vitamin E suppress rotenone-induced dopaminergic cell death. The data in (B) and (C) are presented as the mean +/− SEM, N = 3; *p<0.05, **p<0.01, ***p<0.001.
MsrA protects dopaminergic neurons from α-synuclein toxicity and aggregation
Next, we examined whether MsrA inhibits α-synuclein-induced dopaminergic cell death. These experiments were carried out using A53T mutant α-synuclein because we and others have found that the wild-type protein is only weakly neurotoxic [33, 45, 46]. The cells were transduced with lentivirus encoding A53T or A53T plus MsrA for 72 h, followed by incubation in fresh media for an additional 24 h. Data from Western blot analyses of cell lysates indicated that these conditions were suitable to achieve robust expression of α-synuclein and MsrA (Figure 6A) [29–31]. The relative number of TH+ neurons was lower in primary cultures transduced with A53T lentivirus compared to uninfected control cells (Figure 6B,C). Dopaminergic cell viability was partially rescued in primary cultures that co-expressed A53T and MsrA compared to cultures that expressed only A53T or A53T and LacZ (Figure 6B,C and data not shown). In other experiments, we found that the relative number of TH+ neurons was greater in cultures cotransduced with adenoviruses encoding A53T and wild-type MsrA compared to cells infected with A53T virus alone, or cells co-transduced with virus encoding A53T and LacZ, C72S, or C218S (Figure 6D). These results indicated that wild-type but not mutant MsrA protected dopaminergic neurons from α-synuclein neurotoxicity.
Next, we explored the mechanism by which MsrA suppressed A53T-induced dopaminergic cell death. Because we and others have reported that α-synuclein neurotoxicity correlates with the formation of aggregates [33, 47, 48], we hypothesized that MsrA-mediated neuroprotection against A53T involves inhibition of α-synuclein self-assembly. To address this hypothesis, we analyzed primary cell lysates via Western blotting with a primary antibody specific for mammalian α-synuclein. Soluble, SDS-resistant α-synuclein oligomers were more abundant in cells expressing A53T than in untransduced control cells (Figure 7A). Oligomer formation was markedly suppressed in cells co-expressing MsrA, but not in cells treated with NAC (Figure 7A). SDS-resistant α-synuclein oligomers were also more abundant in the insoluble fraction of cells expressing A53T compared to untransduced control cells, and the formation of these insoluble aggregates was inhibited by MsrA expression but not by treatment with NAC (data not shown). MsrA suppressed A53T-induced dopaminergic cell death with greater efficacy than NAC or vitamin E (Figure 6B,C), suggesting that the ROS-scavenging mechanism of MsrA did not play a significant role in alleviating α-synuclein neurotoxicity. Previous studies have shown that DJ-1, another protein involved in the oxidative stress response, protects against α-synuclein toxicity and aggregation by upregulating Hsp70 [33, 47]. However, in contrast to DJ-1, MsrA had no effect on Hsp70 levels in cells expressing A53T (Figure 7B).
Figure 7.
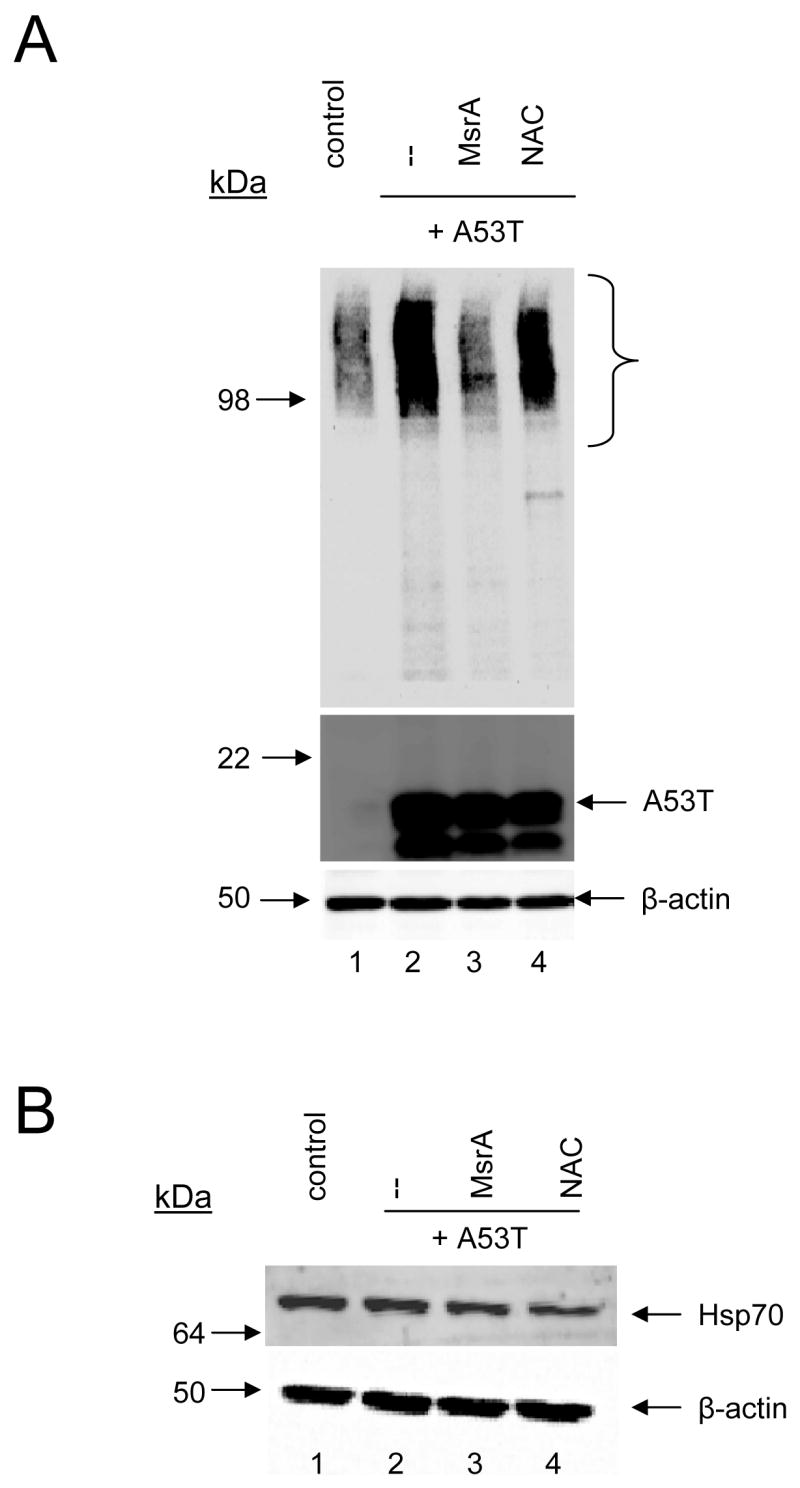
MsrA suppresses α-synuclein self-assembly without inducing Hsp70 expression. (A) Western blot showing that MsrA but not NAC inhibits the formation of soluble α-synuclein oligomers in primary midbrain cultures. The bracket indicates bands or ‘smears’ corresponding to oligomeric α-synuclein. (B) Western blot showing the absence of iHsp70 upregulation by MsrA.
MsrA repairs oxidized α-synuclein in cell-free systems
The results of our primary cell-culture analyses implied that MsrA alleviates A53T neurotoxicity primarily by repairing oxidized methionine residues, presumably in α-synuclein itself. To address this hypothesis, we examined whether oxidized α-synuclein is a substrate of MsrA. A sample of oxidized, wild-type α-synuclein was generated by incubating the recombinant protein with 4% (v/v) H2O2, corresponding to a ~7 00-fold molar excess relative to the number of methionine residues. The oxidized protein eluted from a RP-HPLC column with a markedly shorter retention time than untreated α-synuclein (Figure 8A,B), suggesting that the polarity of the protein was increased by oxidation with H2O2. Analysis of the eluted fractions by MALDI-MS revealed that the untreated protein was fully unoxidized, whereas the treated protein consisted of a major species with all four methionine residues oxidized to MetSO (Table 1; Supplementary Figure S1A,B), consistent with previous results [37].
Figure 8.
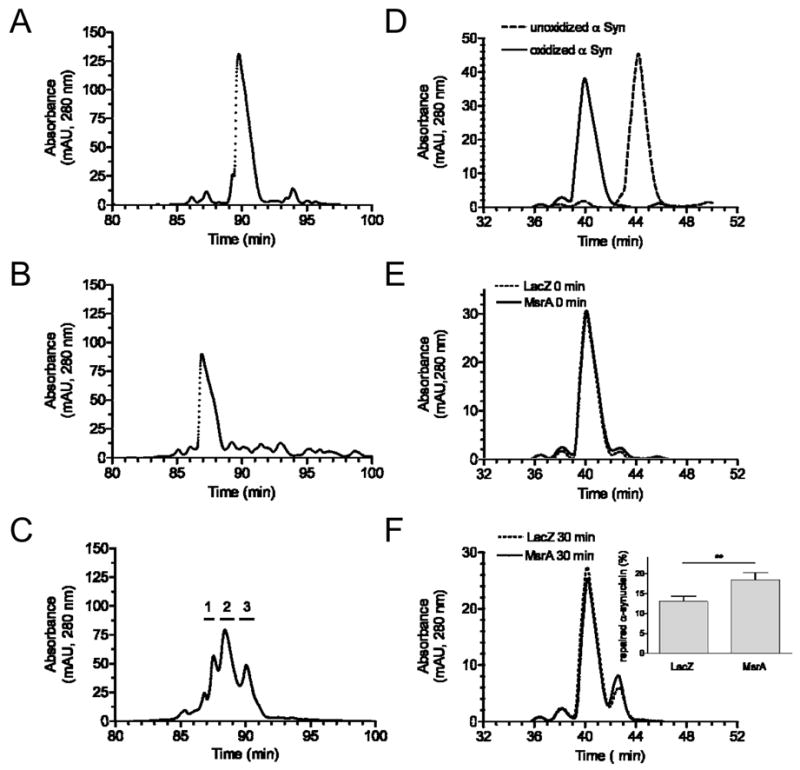
RP-HPLC chromatograms showing MsrA-mediated repair of recombinant α-synuclein. (A) Unoxidized α-synuclein. (B) α-Synuclein oxidized by incubation with H2O2. (C) Oxidized α-synuclein after incubation with recombinant MsrA (2 h, 37 °C). Horizontal lines above the chromatogram correspond to the collection times of fractions 1, 2, and 3. (D) Mixture of unoxidized and oxidized α-synuclein. (E) Oxidized α-synuclein after mixing with lysate of PC12 cells transduced with MsrA or LacZ adenovirus, followed by immediate termination of the reaction (i.e. ‘0 min’ incubation). (F) Oxidized α-synuclein after a 30-min incubation with lysate of PC12 cells transduced with MsrA or LacZ adenovirus. Inset: Quantification of the species eluting at ~42.5 min (corresponding to 3-MetSO α-synuclein) via peak area measurements. The data are expressed as the percentage of total α-synuclein converted to the 3-MetSO species (mean +/− SEM, N = 3, *p<0.05, Student’s t test). Note that different elution gradients were used in (A)-(C) versus (D)-(F).
Next, we investigated whether methionine-oxidized α-synuclein is repaired by MsrA. A sample of α-synuclein pretreated with H2O2 was incubated for 2 h at 37 °C with recombinant, bovine MsrA in the presence of DTT, which served as an electron donor for MetSO repair [49]. Following the incubation, α-synuclein eluted from the RP-HPLC column as three major peaks, referred to as peaks 1, 2, and 3 in order of their retention times (Figure 8C). Analysis of the fractions by MALDI-MS indicated that these peaks corresponded to the following mixtures of α-synuclein species: peak 1, α-synuclein with 3 or 4 oxidized methionine residues; peak 2, α-synuclein with 2 or 3 oxidized methionine residues; peak 3, α-synuclein with 1 or 2 oxidized methionine residues (Table 1; Supplementary Figure S1C-E). A similar conversion to partially reduced species was observed upon incubating oxidized α-synuclein in the presence of MsrA and DTT for 10 min instead of 2 h (data not shown). In contrast, incubation in the presence of DTT but in the absence of MsrA had no effect on the retention time or mass of oxidized α-synuclein. From these results, we inferred that MsrA efficiently reduces oxidized methionine residues in α-synuclein.
Finally, we used the methods described above to determine whether the α-synuclein repair activity is increased in cells over-expressing MsrA. Oxidized α-synuclein was incubated for 30 min at 37 °C in the presence of DTT and lysate from rat PC12 cells transduced with MsrA or LacZ virus. α-Synuclein was then repurified from the mixture and analyzed via RP-HPLC and MALDI-MS. Incubation with either lysate led to the time-dependent appearance of a species that eluted from the RP-HPLC column later than fully oxidized α-synuclein but earlier than the unoxidized protein (Figure 8D-F). Analysis by MALDI-MS revealed that this species had a molecular mass of 14,508 Da, corresponding to α-synuclein with 3 oxidized methionine residues. The fact that this species had one fewer MetSO than the original oxidized protein strongly suggests that it was generated via MsrA-mediated repair. Presumably, significant amounts of the 3-MetSO isoform were observed in the control reaction due to α-synuclein repair by endogenous MsrA and MsrB. Nevertheless, the reduced species was ~1.5-fold more abundant in the mixture containing lysate from MsrA-expressing cells compared to the control reaction, based on measurements of the corresponding peak areas (Figure 8F, inset). From these data, we infer that cells transduced with our MsrA-encoding viruses had enhanced α-synuclein repair activity.
MsrA fails to inhibit dopaminergic cell death elicited by MG132
In a final set of experiments, we examined whether MsrA protected primary dopaminergic neurons from the toxic effects of proteasome dysfunction. Primary cultures were untransduced or transduced with lentivirus encoding MsrA for 72 h and subsequently treated with the proteasome inhibitor MG132 for 24 h. The relative number of TH+ neurons was similar in cultures expressing MsrA and in untransduced cells exposed to MG132 (Figure 9A). In parallel, we investigated whether MsrA inhibited MG132-induced protein aggregation in the MES23.5 aggresome model. MES23.5 cells were treated with MG132 (2 μM, 48 h) and analyzed immunocytochemically using a primary antibody specific for vimentin, Hsp70, or ubiquitin. Similar to what was observed with rotenone-treated cells, MG132 elicited the formation of perinuclear inclusions that stained positive for all three aggresome markers (Figure 9B). The inclusions occurred at similar frequencies in cells expressing MsrA and in vector-control cells (Figure 9C). From these data, we inferred that MsrA failed to inhibit dopaminergic cell death or aggresome formation induced by MG132.
Discussion
In this study, we characterized the neuroprotective effects of MsrA against PD-related insults. We focused on MsrA because it has a critical function as part of the oxidative stress response, and, therefore, we hypothesized that it might play an important role in suppressing oxidative damage associated with PD pathogenesis. Consistent with this hypothesis, we found that wild-type MsrA (but not two catalytically inactive mutants, C72S and C218S) protected dopaminergic neurons from two PD-related stresses: complex I inhibition and α-synuclein over-expression. Other groups have reported that MsrA is neuroprotective in vitro [21, 50] and in vivo [25], and data from one study indicate that MsrA levels are decreased in the brains of Alzheimer’s disease patients compared to age-matched controls [51]. A very recent study revealed that MsrA alleviates motor dysfunction and neurodegeneration in transgenic flies over-expressing α-synuclein [28]. Our data are the first to show in a mammalian system that MsrA suppresses dopaminergic cell death and protein aggregation elicited by toxic phenomena involved in PD.
The suppression of rotenone-induced neurodegeneration and aggresome formation by MsrA suggests that the enzyme interferes with oxidative stress triggered by complex I inhibition [43]. This result agrees with previous data showing that MsrA protects against cell death elicited by other oxidative insults [20–23]. Evidence suggests that aggresome formation (coupled with autophagy) is a protective mechanism initiated by the cell to eliminate misfolded proteins with a high propensity to form toxic aggregates [16, 40]. Accordingly, we infer that MsrA inhibits rotenone-induced neurodegeneration by preventing the accumulation of methionine-oxidized polypeptides that would normally be directed into the aggresome pathway.
Previous findings suggest that MsrA protects against oxidative stress via two mechanisms: (i) the direct repair of oxidized methionine residues, or (ii) the depletion of ROS via acceleration of a redox cycle (Figure 10A) [18, 22]. In our study, NAC and vitamin E restored normal (basal) levels of protein carbonyls and GSH in rotenone-treated cells, suggesting that these small-molecule antioxidants protect against mitochondrial dysfunction via free-radical quenching and, in the case of NAC, stimulation of GSH biosynthesis [44]. In contrast, MsrA had a substantially diminished ability to suppress GSH depletion and protein carbonyl formation elicited by rotenone. If MsrA protected dopaminergic neurons via global ROS scavenging, then it would be expected to inhibit protein carbonyl formation and rescue GSH levels to a similar extent as NAC and vitamin E. Accordingly, we infer that ROS quenching does not play a major role in the MsrA-dependent suppression of rotenone neurotoxicity. Instead, MsrA apparently mitigates oxidative protein damage primarily via its other protective mechanism, the repair of methionine-oxidized proteins, although ROS scavenging may also be involved to some extent. Our findings suggest that there are at least two antioxidant defense mechanisms in dopaminergic neurons (Figure 10A). The first mechanism involves upregulation of GSH, a response modulated by the PD-related protein DJ-1 [33, 47], whereas the second mechanism involves the repair of methionine-oxidized proteins by MsrA.
Figure 10.

Models illustrating the neuroprotective effects of MsrA against PD-related insults. (A) Neuroprotection against complex I impairment. Complex I inhibitors such as rotenone trigger a buildup of ROS, which in turn cause oxidative damage to cellular proteins. MsrA prevents the accumulation of oxidatively damaged proteins by repairing methionine-oxidized proteins. This mechanism is complementary to other antioxidant responses, including the DJ-1-mediated upregulation of GSH [33, 47], modeled in this study by treating the cells with NAC. (B) Neuroprotection against α-synuclein toxicity. α-Synuclein readily undergoes oxidative modification in dopaminergic neurons, which have high levels of cytosolic ROS even under basal conditions due to the auto-oxidation of dopamine. In turn, oxidized α-synuclein has a high propensity to form potentially neurotoxic oligomers or protofibrils [14, 53], which can then undergo further assembly to yield mature fibrils and Lewy bodies. Evidence from this study suggests that MsrA interferes with protofibril formation by repairing methionine-oxidized α-synuclein. This mechanism is complementary to other protective responses, including the DJ-1-mediated upregulation of Hsp70 [33, 47].
Our study also revealed that MsrA inhibits α-synuclein neurotoxicity in primary midbrain cultures, and this protective effect correlates with a decrease in the levels of soluble, SDS-resistant α-synuclein oligomers. These oligomers may be similar to protofibrillar forms of α-synuclein that are suggested to play a significant role in PD pathogenesis [14, 52]. Accordingly, we infer that MsrA protects dopaminergic neurons from A53T-induced cell death by inhibiting the accumulation of methionine-oxidized α-synuclein isoforms with a high propensity to form toxic protofibrils. The expression of A53T does not cause a decrease in GSH levels, suggesting that a buildup of ROS does not play a major role in A53T-induced dopaminergic cell death. Moreover, NAC and vitamin E have a substantially lower ability to protect against A53T neurotoxicity than MsrA. Together, these results argue that MsrA inhibits A53T-induced cell death primarily by repairing oxidized methionine residues, presumably in α-synuclein itself, rather than by globally suppressing ROS.
We propose that MsrA-mediated α-synuclein repair is neuroprotective because it prevents a buildup of toxic protofibrils (Figure 10B). In support of this model, α-synuclein protofibrils have been found to accumulate in mixtures of the methionine-oxidized and unoxidized protein [14, 53]. Moreover, here we show for the first time that α-synuclein is a substrate of MsrA. Assuming that methionine residues exposed to H2O2 have an equal probability of becoming oxidized to the R- and S-stereoisomers of MetSO, we predict that the peroxide treatment yields fully oxidized α-synuclein isoforms with different combinations of R- and S-MetSO in a 1:4:6:4:1 molar ratio, as follows: 1 mol 4R; 4 mol 3R, 1S; 6 mol 2R, 2S; 4 mol 3S, 1R; 1 mol 4S. If all of the S-MetSO in these α-synuclein isoforms were completely reduced by MsrA, then we should observe a 1:4:6:4:1 ratio of α-synuclein species with different numbers of oxidized methionine residues (i.e. 1, 4, 6, 4, or 1 mol with 4, 3, 2, 1, or 0 MetSO, respectively). Because we observed a roughly symmetric distribution of α-synuclein species after 2 h of repair by recombinant MsrA (Figure 8C; Table 1), we infer that (i) the MsrA-mediated reduction of S-MetSO was nearly quantitative under these conditions, and (ii) all four oxidized methionine residues in α-synuclein were repaired with equal efficiency, consistent with the fact that α-synuclein is a natively unfolded polypeptide. Importantly, α-synuclein repair was enhanced in lysates from cells transduced with MsrA virus, supporting our hypothesis that this activity plays a major role in protecting dopaminergic neurons against α-synuclein toxicity.
We note that MsrA alleviated oxidative damage to a subset of proteins in our primary midbrain cultures (Figures 4A and 5A), and α-synuclein neurotoxicity and aggregation were partially relieved by NAC. Moreover, the methionine analog S-methyl-L-cysteine was recently found to delay the onset of motor deficits in α-synuclein transgenic flies, possibly by activating an MsrA-driven redox cycle [28]. These observations imply that the MsrA repair function is not the only activity involved in mitigating α-synuclein-induced cell death – rather, other functions such as MsrA-mediated ROS scavenging may also contribute to neuroprotection.
Our findings suggest an apparent paradox: if the MsrA repair function plays a major role in preventing α-synuclein toxicity, then why do NAC and vitamin E not have a similar protective effect, given that each should suppress oxidative stress upstream of MsrA-catalyzed reduction? As one possibility, the antioxidant activity of these small molecules may be insufficient to completely eliminate α-synuclein methionine oxidation. Consistent with this idea, NAC fails to induce the upregulation of total GSH above normal, basal levels (Figure 4B and data not shown), most likely because GSH biosynthesis is subject to feedback inhibition [44]. In addition, α-synuclein readily undergoes methionine oxidation in dopaminergic cells even in the absence of oxidative insults [54], presumably because these cells have elevated basal levels of oxidative stress and because the natively unfolded structure of α-synuclein renders the protein highly vulnerable to oxidative modification. Accordingly, we propose that MsrA activity is essential to keep the methionine residues of α-synuclein in a reduced state in dopaminergic neurons (Figure 10B) [53, 54]. The physical interaction of MsrA with α-synuclein during enzymatic repair may also result in decreased protofibril formation by interfering with early steps on the α-synuclein self-assembly pathway. The reduction of methionine-oxidized α-synuclein by MsrA complements other mechanisms used by the cell to protect against α-synuclein toxicity, including upregulation of the molecular chaperones Hsp27 and Hsp70 (Figure 10B) [16, 30, 33, 47].
In contrast to its protection against rotenone and α-synuclein, MsrA failed to alleviate dopaminergic cell death and aggresome formation elicited by MG132. This finding indicates that the repair of MetSO has little impact on neurotoxicity associated with proteasome dysfunction. Previous studies have revealed that proteasomal dysfunction triggers increased oxidative stress [9], perhaps via the accumulation of enzymes (e.g. nitric oxide synthase [7]) or protein aggregates [55] that promote oxidative stress, thereby creating a vicious cycle. Accordingly, we would predict that proteasome dysfunction leads to increased levels of protein-bound MetSO. The results of our study do not refute this hypothesis – rather, our data suggest that the reduction of MetSO is not sufficient to protect against the toxic effects of proteasome dysfunction. Presumably, MsrA is ineffective in this case because proteasome impairment is expected to trigger a buildup of potentially toxic polypeptides even in the absence of methionine oxidation.
It is unclear whether the neuroprotective activity of MsrA against rotenone and mutant α-synuclein occurs in the cytosol or mitochondria, given that the enzyme is present in both subcellular compartments in our cell-culture models. Although α-synuclein is normally considered a cytosolic protein, a recent study revealed that the protein is also imported into mitochondria [56], suggesting that MsrA-mediated α-synuclein repair may occur in either compartment. Another unresolved question is whether MsrA carries out its neuroprotective activity preferentially in neurons or glia in the mixed midbrain cultures, given that the lentiviruses used in this study infect both cell types. Because our data suggest that MsrA inhibits dopaminergic cell death by repairing methionine-oxidized proteins, we infer that this protective activity occurs primarily in the neurons, although we cannot rule out that MsrA carries out part of its neuroprotective function in glia (e.g. by modulating neuron-glia interactions). Finally, it is unknown whether MsrB, the enzyme responsible for reducing the R-stereoisomer of MetSO, also protects dopaminergic neurons from PD-related insults. If this is indeed the case, then we would expect to observe more dramatic neuroprotective effects in cells that co-express MsrA and MsrB.
In conclusion, we report that MsrA inhibits dopaminergic cell death and protein aggregation elicited by complex I inhibition and α-synuclein over-expression. We also provide evidence that MsrA protects against these stresses primarily by catalyzing the repair of methionine-oxidized proteins rather than via ROS scavenging. A loss of this MsrA ‘surveillance’ function may lead to the inactivation of pro-survival proteins via methionine oxidation. Alternatively, a decrease in MsrA activity may trigger a buildup of methionine-oxidized proteins (most notably α-synuclein) with a high propensity to form neurotoxic oligomers. Our findings imply a possible connection between the reported inactivation of MsrA during aging [27] and the age-dependence of sporadic PD (this idea can be tested by examining the effects of MsrA downregulation via RNA silencing). Conversely, therapeutic strategies aimed at enhancing MsrA function may prove beneficial to PD patients.
Supplementary Material
Acknowledgments
We thank Dr. Todd Lowther for sharing the construct pET28b-MsrA, Dr. Dennis Selkoe and Dr. Stanley Appel for providing the MES23.5 cells, and Jun Zhang for assistance with experiments aimed at determining cytosolic and mitochondrial levels of MsrA.
This work was supported by grants RO1 NS049221 and RO3 AG027123 from the National Institutes of Health (J.-C.R.) and awards from the Showalter Trust (J.-C.R., V.J.D.) and the Purdue Research Foundation (J.-C.R.). The research described herein was conducted in a facility constructed with support from Research Facilities Improvement Program Grants Number C06-14499 and C06-15480 from the National Center for Research Resources of the National Institutes of Health.
List of Abbreviations
- AraC
β-D-arabinofuranoside hydrochloride
- BCA
bicinchoninic acid
- BSA
bovine serum albumin
- DAPI
4′,6-diamidino-2-phenylindole, dihydrochloride
- DMEM
Dulbecco’s Minimal Essential Media
- DMSO
dimethyl sulfoxide
- DTT
dithiothreitol
- FBS
fetal bovine serum
- GSH
glutathione
- HS
horse serum
- HSC70
constitutive heat-shock protein 70
- iHsp70
inducible heat-shock protein 70
- LB
Luria-Bertani broth
- MALDI-MS
matrix-assisted laser desorption ionization mass spectrometry
- MAP2
microtubule-associated protein 2
- MetSO
methionine sulfoxide
- MOI
multiplicity of infection
- MsrA
methionine sulfoxide reductase A
- MsrB
methionine sulfoxide reductase B
- NAC
N-acetyl-cysteine
- PBS
phosphate-buffered saline
- PD
Parkinson’s disease
- PVDF
polyvinylidene difluoride
- ROS
reactive oxygen species
- RP-HPLC
reversed-phase HPLC
- TH
tyrosine hydroxylase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson’s disease. Science. 2003;302:819–22. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- 2.Cookson MR. The biochemistry of Parkinson’s disease. Annu Rev Biochem. 2005;74:29–52. doi: 10.1146/annurev.biochem.74.082803.133400. [DOI] [PubMed] [Google Scholar]
- 3.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3:1301–6. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 4.Orth M, Schapira AH. Mitochondrial involvement in Parkinson’s disease. Neurochem Int. 2002;40:533–41. doi: 10.1016/s0197-0186(01)00124-3. [DOI] [PubMed] [Google Scholar]
- 5.Jenner P. Oxidative stress in Parkinson’s disease. Ann Neurol. 2003;53 (Suppl 3):S26–38. doi: 10.1002/ana.10483. [DOI] [PubMed] [Google Scholar]
- 6.McNaught KS, Olanow CW. Proteolytic stress: a unifying concept for the etiopathogenesis of Parkinson’s disease. Ann Neurol. 2003;53 (Suppl 3):S73–84. S84–6. doi: 10.1002/ana.10512. [DOI] [PubMed] [Google Scholar]
- 7.Halliwell B. Hypothesis: proteasomal dysfunction: a primary event in neurogeneration that leads to nitrative and oxidative stress and subsequent cell death. Ann N Y Acad Sci. 2002;962:182–194. doi: 10.1111/j.1749-6632.2002.tb04067.x. [DOI] [PubMed] [Google Scholar]
- 8.Farout L, Friguet B. Proteasome function in aging and oxidative stress: implications in protein maintenance failure. Antioxid Redox Signal. 2006;8:205–16. doi: 10.1089/ars.2006.8.205. [DOI] [PubMed] [Google Scholar]
- 9.Papa L, Gomes E, Rockwell P. Reactive oxygen species induced by proteasome inhibition in neuronal cells mediate mitochondrial dysfunction and a caspase-independent cell death. Apoptosis. 2007;12:1389–405. doi: 10.1007/s10495-007-0069-5. [DOI] [PubMed] [Google Scholar]
- 10.Graham DG. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol Pharmacol. 1978;14:633–43. [PubMed] [Google Scholar]
- 11.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-Synuclein in Lewy bodies. Nature. 1997;388:839–40. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 12.Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT., Jr Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A. 2000;97:571–6. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, Uversky VN, Fink AL. Effect of familial Parkinson’s disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human α-synuclein. Biochemistry. 2001;40:11604–13. doi: 10.1021/bi010616g. [DOI] [PubMed] [Google Scholar]
- 14.Conway KA, Rochet JC, Bieganski RM, Lansbury PT., Jr Kinetic stabilization of the α-synuclein protofibril by a dopamine-α-synuclein adduct. Science. 2001;294:1346–9. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- 15.Cole NB, Murphy DD, Lebowitz J, Di Noto L, Levine RL, Nussbaum RL. Metal-catalyzed oxidation of α-synuclein: helping to define the relationship between oligomers, protofilaments and filaments. J Biol Chem. 2005;280:9678–9690. doi: 10.1074/jbc.M409946200. [DOI] [PubMed] [Google Scholar]
- 16.Rochet JC. Novel therapeutic strategies for the treatment of protein-misfolding diseases. Expert Rev Mol Med. 2007;9:1–34. doi: 10.1017/S1462399407000385. [DOI] [PubMed] [Google Scholar]
- 17.Tabner BJ, Turnbull S, El-Agnaf OM, Allsop D. Formation of hydrogen peroxide and hydroxyl radicals from A(beta) and alpha-synuclein as a possible mechanism of cell death in Alzheimer’s disease and Parkinson’s disease. Free Radic Biol Med. 2002;32:1076–83. doi: 10.1016/s0891-5849(02)00801-8. [DOI] [PubMed] [Google Scholar]
- 18.Levine RL, Moskovitz J, Stadtman ER. Oxidation of methionine in proteins: roles in antioxidant defense and cellular regulation. IUBMB Life. 2000;50:301–7. doi: 10.1080/713803735. [DOI] [PubMed] [Google Scholar]
- 19.Moskovitz J, Jenkins NA, Gilbert DJ, Copeland NG, Jursky F, Weissbach H, Brot N. Chromosomal localization of the mammalian peptide-methionine sulfoxide reductase gene and its differential expression in various tissues. Proc Natl Acad Sci U S A. 1996;93:3205–8. doi: 10.1073/pnas.93.8.3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kantorow M, Hawse JR, Cowell TL, Benhamed S, Pizarro GO, Reddy VN, Hejtmancik JF. Methionine sulfoxide reductase A is important for lens cell viability and resistance to oxidative stress. Proc Natl Acad Sci U S A. 2004;101:9654–9. doi: 10.1073/pnas.0403532101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yermolaieva O, Xu R, Schinstock C, Brot N, Weissbach H, Heinemann SH, Hoshi T. Methionine sulfoxide reductase A protects neuronal cells against brief hypoxia/reoxygenation. Proc Natl Acad Sci U S A. 2004;101:1159–64. doi: 10.1073/pnas.0308215100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Picot CR, Petropoulos I, Perichon M, Moreau M, Nizard C, Friguet B. Overexpression of MsrA protects WI-38 SV40 human fibroblasts against H2O2-mediated oxidative stress. Free Radic Biol Med. 2005;39:1332–41. doi: 10.1016/j.freeradbiomed.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 23.Sreekumar PG, Kannan R, Yaung J, Spee CK, Ryan SJ, Hinton DR. Protection from oxidative stress by methionine sulfoxide reductases in RPE cells. Biochem Biophys Res Commun. 2005;334:245–53. doi: 10.1016/j.bbrc.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 24.Lowther WT, Brot N, Weissbach H, Honek JF, Matthews BW. Thiol-disulfide exchange is involved in the catalytic mechanism of peptide methionine sulfoxide reductase. Proc Natl Acad Sci U S A. 2000;97:6463–8. doi: 10.1073/pnas.97.12.6463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moskovitz J, Bar-Noy S, Williams WM, Requena J, Berlett BS, Stadtman ER. Methionine sulfoxide reductase (MsrA) is a regulator of antioxidant defense and lifespan in mammals. Proc Natl Acad Sci USA. 2001;98:12920–12925. doi: 10.1073/pnas.231472998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruan H, Tang XD, Chen ML, Joiner ML, Sun G, Brot N, Weissbach H, Heinemann SH, Iverson L, Wu CF, Hoshi T, Joiner MA. High-quality life extension by the enzyme peptide methionine sulfoxide reductase. Proc Natl Acad Sci USA. 2002;99:2748–2753. doi: 10.1073/pnas.032671199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petropoulos I, Mary J, Perichon M, Friguet B. Rat peptide methionine sulphoxide reductase: cloning of the cDNA, and down-regulation of gene expression and enzyme activity during aging. Biochem J. 2001;355:819–25. doi: 10.1042/bj3550819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wassef R, Haenold R, Hansel A, Brot N, Heinemann SH, Hoshi T. Methionine sulfoxide reductase A and a dietary supplement S-methyl-L-cysteine prevent Parkinson’s-like symptoms. J Neurosci. 2007;27:12808–16. doi: 10.1523/JNEUROSCI.0322-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science. 2006;313:324–8. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Outeiro TF, Klucken J, Strathearn KE, Liu F, Nguyen P, Rochet JC, Hyman BT, McLean PJ. Small heat shock proteins protect against alpha-synuclein-induced toxicity and aggregation. Biochem Biophys Res Commun. 2006;351:631–8. doi: 10.1016/j.bbrc.2006.10.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Outeiro TF, Kontopoulos E, Altman S, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, Rochet JC, McLean PJ, Young AB, Abagyan R, Feany MB, Hyman BT, Kazantsev A. Sirtuin 2 Inhibitors Rescue alpha-Synuclein-Mediated Toxicity in Models of Parkinson’s Disease. Science. 2007;317:516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- 32.Kim HY, Gladyshev VN. Role of structural and functional elements of mouse methionine-S-sulfoxide reductase in its subcellular distribution. Biochemistry. 2005;44:8059–67. doi: 10.1021/bi0501131. [DOI] [PubMed] [Google Scholar]
- 33.Liu F, Nguyen JL, Hulleman JD, Li L, Rochet J-C. Mechanisms of DJ-1 neuroprotection in a cellular model of Parkinson’s disease. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05333.x. in press. [DOI] [PubMed] [Google Scholar]
- 34.Crawford GD, Jr, Le WD, Smith RG, Xie WJ, Stefani E, Appel SH. A novel N18TG2 × mesencephalon cell hybrid expresses properties that suggest a dopaminergic cell line of substantia nigra origin. J Neurosci. 1992;12:3392–3398. doi: 10.1523/JNEUROSCI.12-09-03392.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Conway KA, Harper JD, Lansbury PT., Jr Fibrils formed in vitro from α-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry. 2000;39:2552–2563. doi: 10.1021/bi991447r. [DOI] [PubMed] [Google Scholar]
- 36.Rochet JC, Conway KA, Lansbury PT., Jr Inhibition of fibrillization and accumulation of prefibrillar oligomers in mixtures of human and mouse alpha-synuclein. Biochemistry. 2000;39:10619–26. doi: 10.1021/bi001315u. [DOI] [PubMed] [Google Scholar]
- 37.Uversky VN, Yamin G, Souillac PO, Goers J, Glaser CB, Fink AL. Methionine oxidation inhibits fibrillation of human alpha-synuclein in vitro. FEBS Lett. 2002;517:239–44. doi: 10.1016/s0014-5793(02)02638-8. [DOI] [PubMed] [Google Scholar]
- 38.Muqit MM, Davidson SM, Payne Smith MD, MacCormac LP, Kahns S, Jensen PH, Wood NW, Latchman DS. Parkin is recruited into aggresomes in a stress-specific manner: over-expression of parkin reduces aggresome formation but can be dissociated from parkin’s effect on neuronal survival. Hum Mol Genet. 2004;13:117–135. doi: 10.1093/hmg/ddh012. Epub 2003 Nov 25. [DOI] [PubMed] [Google Scholar]
- 39.Diaz-Corrales FJ, Asanuma M, Miyazaki I, Miyoshi K, Ogawa N. Rotenone induces aggregation of gamma-tubulin protein and subsequent disorganization of the centrosome: relevance to formation of inclusion bodies and neurodegeneration. Neuroscience. 2005;133:117–35. doi: 10.1016/j.neuroscience.2005.01.044. [DOI] [PubMed] [Google Scholar]
- 40.Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wigley WC, Fabunmi RP, Lee MG, Marino CR, Muallem S, DeMartino GN, Thomas PJ. Dynamic association of proteasomal machinery with the centrosome. J Cell Biol. 1999;145:481–490. doi: 10.1083/jcb.145.3.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee HJ, Shin SY, Choi C, Lee YH, Lee SJ. Formation and removal of alpha-synuclein aggregates in cells exposed to mitochondrial inhibitors. J Biol Chem. 2002;277:5411–7. doi: 10.1074/jbc.M105326200. [DOI] [PubMed] [Google Scholar]
- 43.Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno-Yagi A, Greenamyre JT. Mechanism of toxicity in rotenone models of Parkinson’s disease. J Neurosci. 2003;23:10756–10764. doi: 10.1523/JNEUROSCI.23-34-10756.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maher P. The effects of stress and aging on glutathione metabolism. Ageing Res Rev. 2005;4:288–314. doi: 10.1016/j.arr.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 45.Zhou W, Hurlbert MS, Schaack J, Prasad KN, Freed CR. Overexpression of human α-synuclein causes dopamine neuron death in rat primary culture and immortalized mesencephalon-derived cells. Brain Res. 2000;866:33–43. doi: 10.1016/s0006-8993(00)02215-0. [DOI] [PubMed] [Google Scholar]
- 46.Petrucelli L, O’Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, Choi P, Wolozin B, Farrer M, Hardy J, Cookson MR. Parkin protects against the toxicity associated with mutant α-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron. 2002;36:1007–19. doi: 10.1016/s0896-6273(02)01125-x. [DOI] [PubMed] [Google Scholar]
- 47.Zhou W, Freed CR. DJ-1 up-regulates glutathione synthesis during oxidative stress and inhibits A53T alpha-synuclein toxicity. J Biol Chem. 2005;280:43150–8. doi: 10.1074/jbc.M507124200. [DOI] [PubMed] [Google Scholar]
- 48.Periquet M, Fulga T, Myllykangas L, Schlossmacher MG, Feany MB. Aggregated alpha-synuclein mediates dopaminergic neurotoxicity in vivo. J Neurosci. 2007;27:3338–46. doi: 10.1523/JNEUROSCI.0285-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun H, Gao J, Ferrington DA, Biesiada H, Williams TD, Squier TC. Repair of oxidized calmodulin by methionine sulfoxide reductase restores ability to activate the plasma membrane Ca-ATPase. Biochemistry. 1999;38:105–12. doi: 10.1021/bi981295k. [DOI] [PubMed] [Google Scholar]
- 50.Jung B, Lee EH, Chung WS, Lee SJ, Shin SH, Joo SH, Kim SK, Lee JH. Increased viability of PC12 cells exposed to amyloid-beta peptide by transduction with human TAT-methionine sulfoxide reductase. Neuroreport. 2003;14:2349–53. doi: 10.1097/00001756-200312190-00012. [DOI] [PubMed] [Google Scholar]
- 51.Gabbita SP, Aksenov MY, Lovell MA, Markesbery WR. Decrease in peptide methionine sulfoxide reductase in Alzheimer’s disease brain. J Neurochem. 1999;73:1660–6. doi: 10.1046/j.1471-4159.1999.0731660.x. [DOI] [PubMed] [Google Scholar]
- 52.Rochet JC, Outeiro TF, Conway KA, Ding TT, Volles MJ, Lashuel HA, Bieganski RM, Lindquist SL, Lansbury PT. Interactions among alpha-synuclein, dopamine, and biomembranes: some clues for understanding neurodegeneration in Parkinson’s disease. J Mol Neurosci. 2004;23:23–34. doi: 10.1385/jmn:23:1-2:023. [DOI] [PubMed] [Google Scholar]
- 53.Glaser CB, Yamin G, Uversky VN, Fink AL. Methionine oxidation, alpha-synuclein and Parkinson’s disease. Biochim Biophys Acta. 2005;1703:157–69. doi: 10.1016/j.bbapap.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 54.Mirzaei H, Schieler JL, Rochet J-C, Regnier F. Identification of rotenone-induced modifications in α-synuclein using affinity pull-down and tandem mass spectrometry. Anal Chem. 2006;78:2422–2431. doi: 10.1021/ac051978n. [DOI] [PubMed] [Google Scholar]
- 55.Tabner BJ, El-Agnaf OM, Turnbull S, German MJ, Paleologou KE, Hayashi Y, Cooper LJ, Fullwood NJ, Allsop D. Hydrogen peroxide is generated during the very early stages of aggregation of the amyloid peptides implicated in Alzheimer disease and familial British dementia. J Biol Chem. 2005;280:35789–92. doi: 10.1074/jbc.C500238200. [DOI] [PubMed] [Google Scholar]
- 56.Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of alpha -synuclein impairs complex I in human dopaminergic neuronal cultures and Parkinson’s disease brain. J Biol Chem. 2008 doi: 10.1074/jbc.M710012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.