

Abstract
Cation-mediated RNA folding from extended to compact, biologically active conformations relies on a temporal balance of forces. The Mg2+-mediated folding of the Tetrahymena thermophila ribozyme is characterized by rapid non-specific collapse followed by tertiary contact induced compaction. This paper focuses on an autonomously folding portion of the Tetrahymena ribozyme, its P4–P6 domain, in order to probe one facet of the rapid collapse: chain flexibility. The time evolution of P4–P6 folding was followed by global and local measures as a function of Mg2+ concentration. While all concentrations of Mg2+ studied are sufficient to screen the charge on the helices, the rates of compaction and tertiary contact formation diverge as the concentration of Mg2+ increases; collapse is greatly accelerated by Mg2+ while tertiary contact formation is not. These studies highlight the importance of chain stiffness to RNA folding; at 10 mM Mg2+ a stiff hinge limits the rate of P4–P6 folding. At higher magnesium concentrations the rate limiting step shifts from hinge bending to tertiary contact formation.
Keywords: compaction, persistence length, RNA folding, time-resolved hydroxyl radical foot-printing, time-resolved small angle x-ray scattering
Introduction
RNA folding describes the process by which a biologically active tertiary structure assembles from a single, covalently bonded strand of ribonucleotides. Because secondary structure can form on the microsecond timescale,1 tertiary folding may be viewed as the structuring of a series of short, rigid duplexes, joined by more flexible single stranded regions, loops, hinges or junctions.2,3 Flexibility of the interhelical regions is necessary to accommodate the backbone contortions that accompany RNA folding. From the perspective of this physical model, a structure is folded when the pieces of the molecule assume a unique and stable orientation with each other. These studies highlight the importance of flexible, non-base paired regions to RNA folding. These regions facilitate, but are not directly involved in tertiary contact formation. The approach described herein provides insight into the importance of chain flexibility to RNA folding.
RNA folding in vitro is typically initiated from a low salt condition under which the macro-molecule contains secondary but little or no tertiary structure. In the physical model for this unfolded state, short, rigid helices are arranged in extended structures, reflecting the electrostatic repulsion between their residual negative charges. At monovalent salt concentrations ≤ 8 mM the L-21 Sca1 ribozyme from Tetrahymena thermophila as well as the ribozyme’s P4–P6 domain are rigidly extended. 4,5 Following mixing with additional mono or multivalent cations that screen the negatively charged phosphate oxygens on the polynucleotide backbone, the molecules relax to less conformationally restricted ensembles.4 Electrostatic relaxation occurs within 10 ms in the L-21 Sca1 Tetrahymena ribozyme.6 A second compaction that coincides with tertiary structure formation follows on a 100 ms time scale.7
We have carried out a series of experiments using time resolved SAXS and hydroxyl radical (·OH) footprinting to probe the Mg2+-dependent folding of the Tetrahymena ribozyme’s P4–P6 domain (Figure 1a), with the goal of understanding the interplay of charge neutralization, tertiary contact formation and hinge flexibility. This domain folds in isolation, independent of the rest of the ribozyme.8 The availability of an atomic resolution structure makes P4–P6 an attractive model system for biophysical analysis (Figure 1b).9 P4–P6 is composed of two roughly parallel stacks of helices joined by a non-base paired hinge referred to as J5/5a. The P5abc subdomain of P4–P6, highlighted in red in Figure 1a, is composed of three single stranded regions, the loops L5b and L5c and the ‘A-bulge’, which participate in tertiary interactions and have been suggested to enhance catalytic activity of the full ribozyme.10–12 Mg2+-mediated folding studies of P4–P6 find folding rates that depend on both temperature and cation concentration. 13–15 An analysis of folding that followed the fluorescence of an extrinsic probe attached to P4–P6 showed that the observed folding rate increased with increasing Mg2+ concentration up to 10 – 30 mM after which it reached a plateau; these authors concluded that folding was limited by Mg2+ binding at low cation concentrations.14
Figure 1.
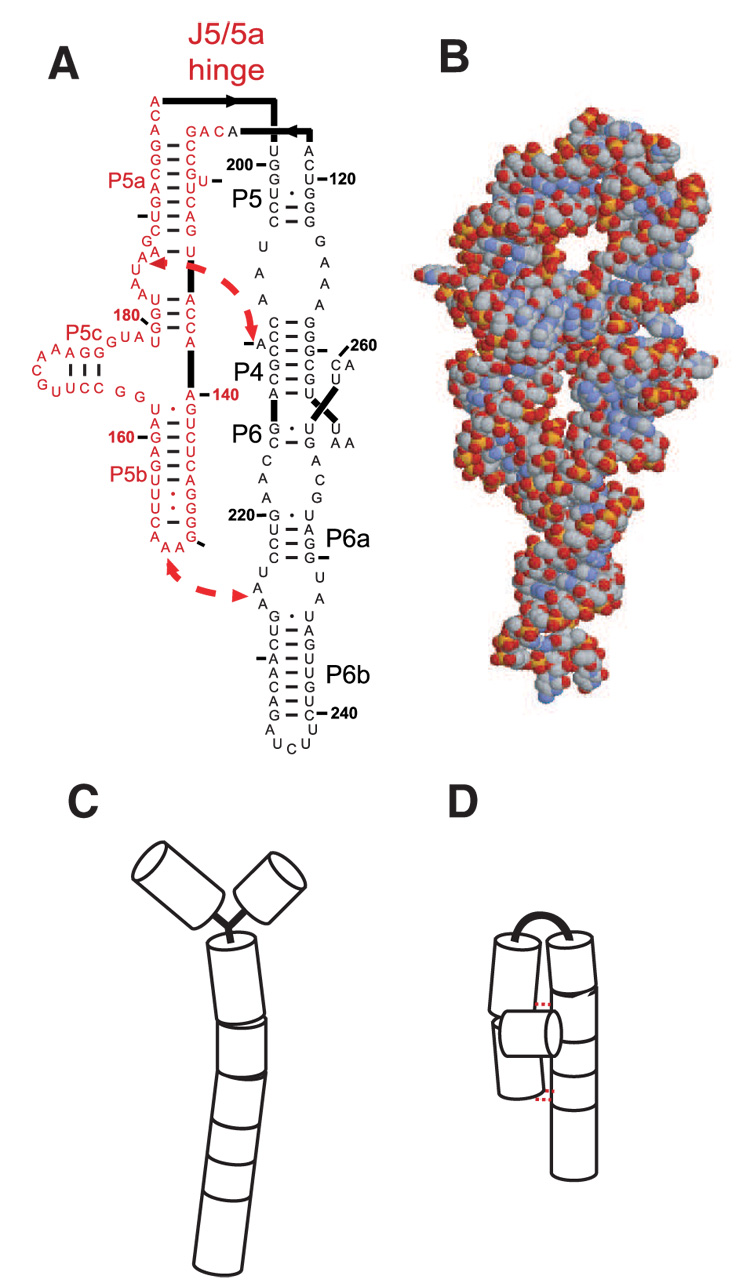
The P4–P6 domain. (A) A two dimensional representation showing secondary structure. The locations of the two tertiary contacts are indicated by the large arrows. In the mutant RNA, the tertiary contacts are disrupted by the following sequence modifications: nucleotides AAUAAG (183–188) are replaced by UUUUUU and GGAAAC (149–154) are replaced by CUUCGG. Bases comprising the P5abc sub-domain are shown in red. (B) Reconstruction of the P4–P6 domain based on a monomeric subunit of 1GID.pdb 9 generated by RasMol. (C & D) Simplified models of the extended, unfolded and compact, folded structures, respectively following the representation of Ref. 4.
The present study explores the nature of the rate limiting steps at low and high concentrations of Mg2+ by concordant application of a global probe of conformation and a local probe of tertiary contact formation. Previous thermodynamic folding studies of P4–P6 4 are hereby extended to the time domain. Hydroxyl radical footprinting reports phosphodiester backbone solvent accessibility changes with single nucleotide resolution at times as short a few milliseconds. 16 SAXS distinguishes compact from extended nucleic acid conformations.17 By studying folding under conditions where the residual charge on the helices is fully screened,18 the inter-play between tertiary contact formation and the flexibility of the J5/5a hinge is probed. Wild-type P4–P6 and a mutant in which the domain’s two stabilizing tertiary contacts cannot form were analyzed. Under these experimental conditions, the rate of compaction in the absence of tertiary contact formation reflects the flexibility of the J5/5a hinge. As the concentration of Mg2+ increases, compaction accelerates relative to tertiary contact formation, suggesting that a large ionic-strength dependent energy is associated with the bending of these non-base paired joints to achieve the ~150° overall bend at the center of the domain.13 A simple calculation of this energy is presented. Although joint flexibility has been considered experimentally (e.g. 4,19) and computationally, 20 the present studies find that it can be rate limiting when P4–P6 folding is initiated by the addition of 10 mM Mg2+, a common experimental condition. At higher Mg2+ concentrations where the hinge stiffness decreases, structuring of the P5c subdomain becomes limiting by its minimal dependence on cation concentration. An unexpected interplay between collapse and structuring of P5c is observed.
Results
Global Compaction
Global compaction of the P4–P6 domain accelerates with increases in the concentration of Mg2+ used to initiate folding. Figure 2a shows progress curves for folding initiated by the addition of 10 mM (open circles), 50 mM (open squares) or 100 mM Mg2+ (open triangles). The fraction unfolded (Pu) is computed as the component of the unfolded scattering profile required to best fit each scattering profile with the two-state model (see Materials and Methods). Measurements of Io, the y intercept of the scattering profile (see Materials and Methods) confirm that the P4–P6 domain remains monomeric for ~ 160 ms after the addition of 10 mM Mg2+; increases in Io on longer time scales indicate the onset of dimerization (data not shown). Only data obtained prior to the onset of dimerization are analyzed, for times less than 40 and 15 ms at 50 and 100 mM Mg2+, respectively. All three progress curves are best fit by a single exponential (solidlines). The folding rate of the wild-type P4–P6 domain increases rapidly as the ionic strength is increased, its value is 18 (3) s−1, 56 (18) s−1, and 140 (81) s−1 at 10, 50, and 100 mM Mg2+, respectively.
Figure 2.
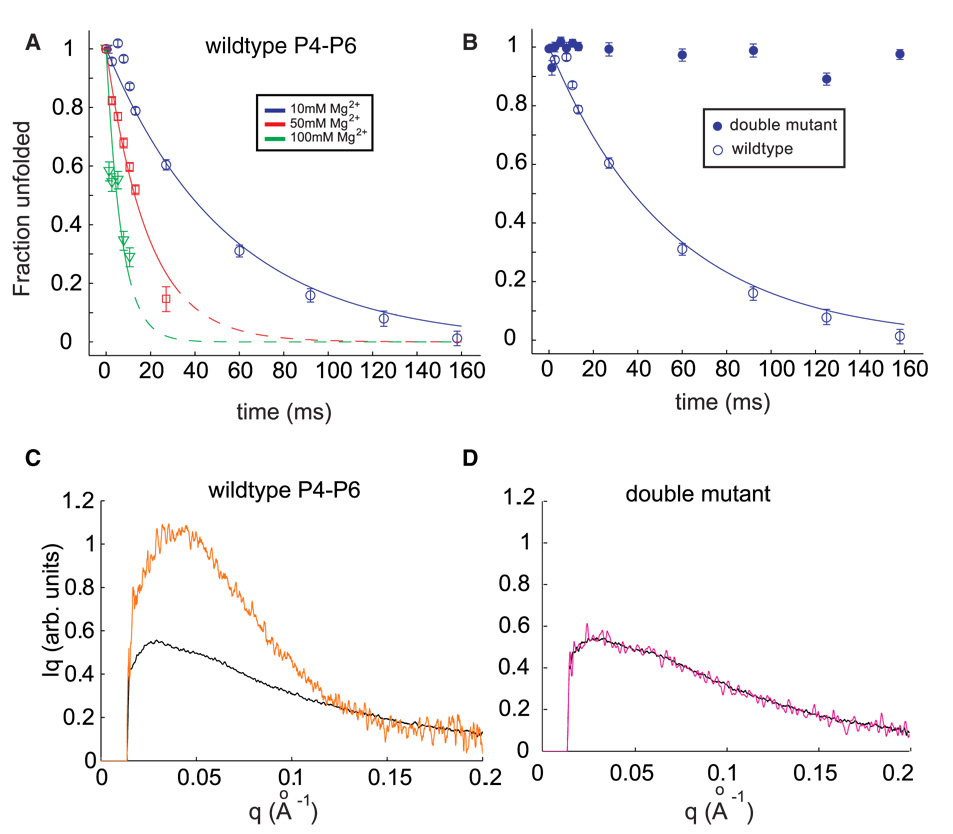
(A) Folding time courses for wild type P4–P6 at different magnesium concentrations. The coefficient Pu, representing the percentage of the unfolded scattering profile required to fit each time-resolved profile (see text) is shown following the addition of 10 mM Mg2+ (circles), 50 mM Mg2+ (squares) and 100 mM Mg2+ (triangles). Changes in the time courses can be explained by a model in which the flexibility of the J5-5a hinge varies with ionic strength. (B) The folding time course following addition of 10 mM Mg2+ for the wild type P4–P6 (open circles) is contrasted with the corresponding time course from the mutant that lacks the ability to form tertiary contacts. (C & D) Comparison of SAXS profiles, displayed as Iq vs. q, for the earliest (unfolded, black) and longest time point (t=160 ms, orange) following the addition of 10 mM Mg2+ to wild type (C) and mutant (D) P4–P6. While wild type P4–P6 folds to a structure compatible with the crystal structure (Figure 1B), no change in the conformation of the mutant is detected.
The Mg2+-mediated compaction of wild-type P4–P6 was compared with the ‘double mutant’ in which both principal tertiary contacts are knocked out. Figure 2b compares progress curves for both RNA molecules following the addition of 10 mM Mg2+ to the unfolded RNA. Whereas the wild type molecule folds to the native form, with PU approaching zero at the longest time (open symbols), there is no change in PU for the mutant on this time scale (closed symbols). Comparison of the full scattering profiles (as I*q vs. q, Figures 2c and 2d) of the wild type and mutant RNA molecules in the initial (unfolded) and longest time-point states (t = 160 ms) shows that only for wild type P4–P6 is a change observed that is consistent with folding. As expected from the PU computation, the scattering profiles of the mutant at both the shortest and longest time points are identical (Figure 2d); the addition of 10 mM Mg2+ does not affect the global conformation of the double mutant RNA. At 50 and 100 mM Mg2+, small changes in the conformation of the mutant are detected that occur within the mixing dead time. This observation is consistent with a small initial decrease in PU. The zero angle scattering, Io begins to increase within tens of milliseconds, indicating dimerization at rates similar to those measured for the wild type RNA (data not shown).
In summary, wild-type P4–P6 compacts to its native dimensions on the millisecond time scale under the present experimental conditions. The observed rate constant is Mg2+ concentration dependent. In contrast, only small changes in the global conformation are measured at high Mg2+ concentrations for the P4–P6 mutant in which the principal tertiary contacts are disrupted. In 10 mM Mg2+ no compaction of the mutant is measured relative to the unfolded state. In order to explore the relation of these changes in global conformation to formation of discrete tertiary interactions, ancillary ·OH footprinting experiments were conducted under consistent experimental conditions and on a comparable time scale. These concomitant studies reveal that the relationship of global compaction and formation of local tertiary contacts is Mg2+-dependent.
Local contact formation
Mg2+ folding isotherms were determined to assess the saturation of tertiary contact formation at the three Mg2+ concentrations analyzed kinetically. These isotherms are shown in Supplementary Figure 2. Both wild-type P4–P6 and the isolated P5abc domain were assayed under the experimental conditions of the kinetics measurements. For P4–P6, values of KMg ranged from 0.22 ± 0.03 mM (nH = 1.29 ± 0.19) at nucleotides 180-181 to 1.64 ± 0.15 mM (nH = 0.95 ± 0.1) at nucleotide 155. For isolated P5abc, values of KMg ranged from 0.66 ± 0.06 mM (nH = 1.12 ± 0.13) to 1.2 ± 0.17 mM (nH = 1.5 ± 0.18) for the same contacts, respectively. Inspection of the P4–P6 isotherms shows tertiary contact saturation greater than 87%, 96% and 99% at 10, 50 and 100 mM Mg2+, respectively. Inspection of the isolated P5abc isotherms shows tertiary contact saturation greater than 95%, 99% and 99% at 10, 50 and 100 mM Mg2+, respectively.
Time-resolved ·OH footprinting studies were conducted at the three concentrations of Mg2+ analyzed by SAXS. Every nucleotide of the RNA was individually analyzed; time progress curves were generated for each nucleotide that displayed a significant change in sensitivity to ·OH cleavage (see supplementary figures). Sequential sets of nucleotides that display an identical time-dependence are grouped together into a ‘protection’. This procedure simplifies the analysis and facilitates structural interpretation. We interpret these reactivity changes to reflect changes in the solvent accessibility of the backbone sugars. 10
The formation of discrete tertiary contacts were followed by footprinting for wild type P4–P6, its ‘double mutant’ (L5b/UUCG and A-bulge/U), and the isolated P5abc domain (Figure 1). While the footprinting analysis yields a cornucopia of local measures, only those that bear directly upon the hinge bending hypothesis are discussed in this article; these results are summarized in Figure 3 (top).a Structuring of the J5/5a hinge of P4–P6 was followed at three discrete sites; nucleotides 126 and 200–201 are protected against ·OH cleavage during Mg2+-mediated folding whereas nucleotide 122 becomes more solvent accessible. The tertiary contact formed between the tetraloop and its receptor is monitored by protection progress of nucleotides 153–155 and 224–225, respectively. Nucleotides 180–187 within the A-bulge interact with nucleotides 212–213 of P4 to form the second tertiary contact; these nucleotides become protected against ·OH radicals upon Mg2+ addition. Formation of the P5c subdomain is reported by nucleotides 163–164, 166–169 and 175–176 (Figure 3, top).
Figure 3.
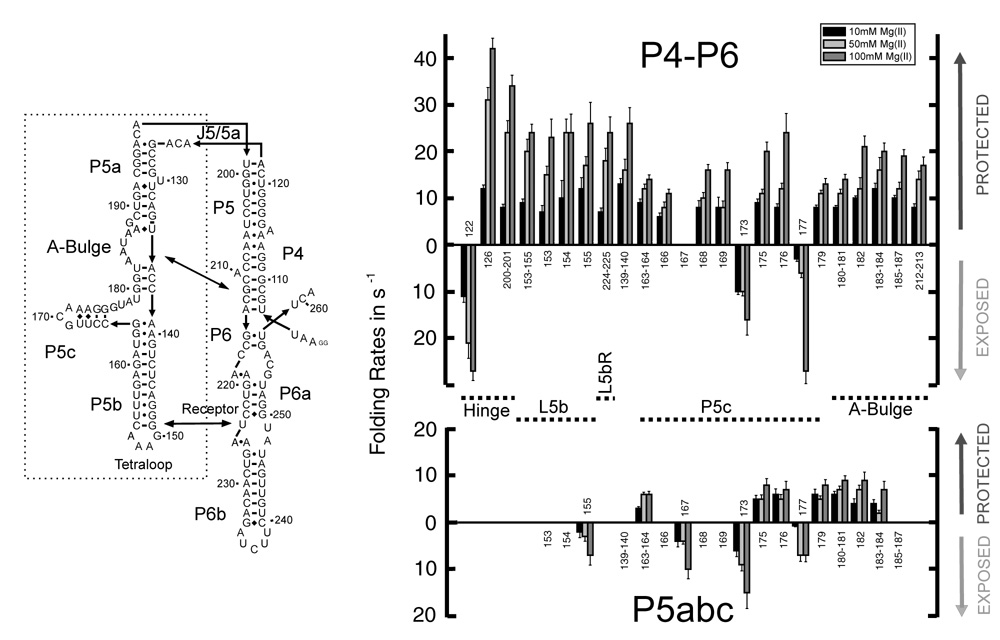
Folding rates of P4–P6 (top) and P5abc (bottom) at 10 mM (black), 50 mM (light grey) and 100 mM Mg2+ (dark grey). The numbering corresponds to the nucleotide location shown in the secondary structure representation. Protections are indicated by columns above, exposures by columns below the base line.
No significant change in ·OH reactivity is observed for the P4–P6 domain double mutant at any of the Mg2+ concentrations analyzed. The local changes in solvent accessibility observed for wild type P4–P6 folding at 10 mM Mg2+ range from 7 to 12 s−1 (Figure 3, top). Although the differences between fastest and slowest footprinting rates under this condition exceed the confidence limits of the measurement, these differences do not group to the different regions of the domain (Hinge, Tetraloop and Tetraloop Receptor, A-bulge and Receptor and P5c).
Differences among the discrete regions of the P4–P6 domain become accentuated when folding is initiated at higher Mg2+ concentrations and in general increase with increasing [Mg2+]. Nonetheless, the protection rates measured by footprinting increase much less than compaction as a function of increasing Mg2+ concentration (Figure 4). For example, the rates measured for hinge nucleotide 122 are 11, 21 and 27 s−1 at 10, 50 and 100 mM Mg2+, respectively. Nucleotides in the hinge showed the same overall trends but the rates were not identical. For example, hinge nucleotide 126 displays rates of 12, 31 and 42 s−1. In contrast, the tetraloop and tetraloop receptor protections increase more modestly and in lockstep. Thus, portions of the hinge become structured at twice the rate of the tetraloop – tetraloop receptor interaction at 100 mM Mg2+. Formation of contacts that comprise the interaction between the A-bulge and its receptor displays a relatively uniform, even more moderate response to Mg2+ concentration. Structuring of P5c is the least sensitive to Mg2+ concentration; at 100 mM Mg2+ folding of this subdomain is rate limiting. Thus, the rough ordering of folding events at high concentrations of Mg2+ is rapid collapse, structuring of the hinge, formation of the tetraloop – tetraloop receptor interaction closely followed by formation of the A-bulge – receptor interaction and lastly, structuring of P5c.
Figure 4.
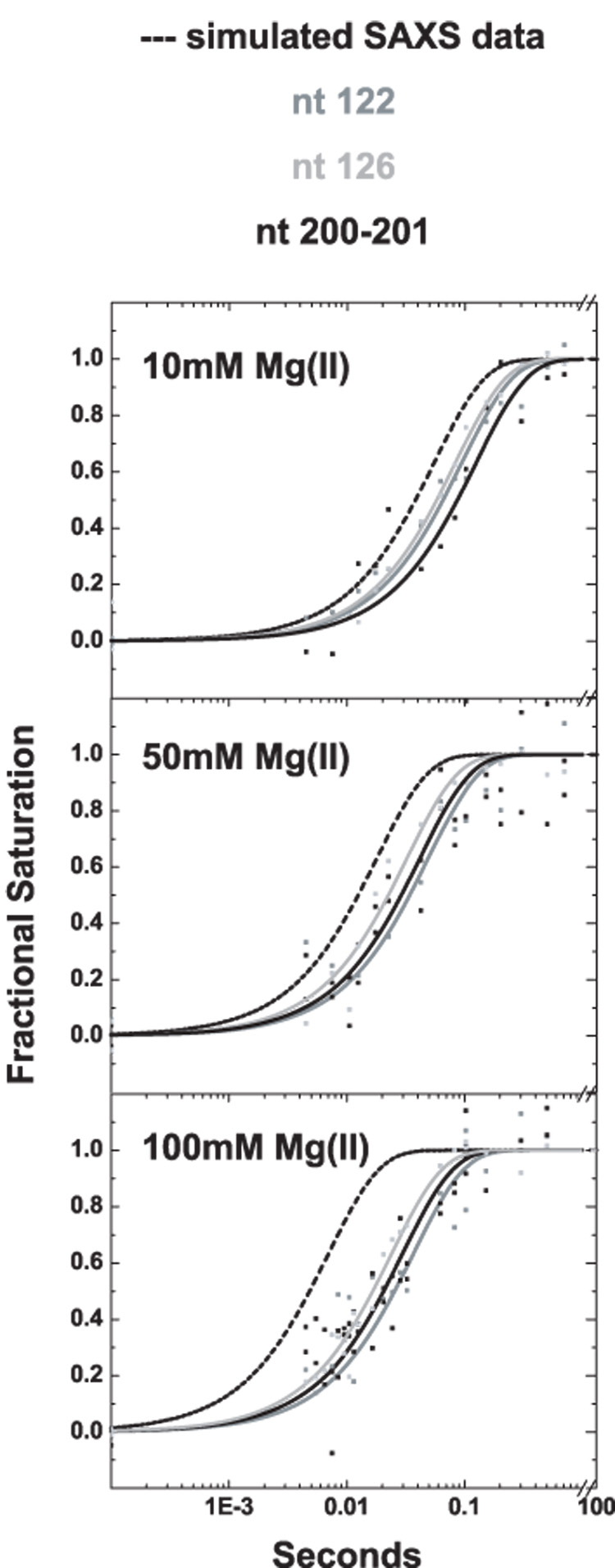
Comparison of time constants for molecular compaction measured by SAXS (dashed line) with time constant for structuring of three residues in the hinge region of P4–P6, nucleotides 122 (grey), 126 (light grey) , and 201–202 (black). At 10 mM Mg2+ (top panel) the rate of compaction is slightly faster than hinge structuring, however, as the ionic strength in the folding buffer increases to 50 mM Mg2+ (middle panel) and 100 mM Mg2+ (bottom panel), the rate of compaction far exceed the structuring of the hinge measured by ·OH footprinting.
Whether collapse of the P4–P6 domain influences folding of the P5c subdomain was investigated by analysis of isolated P5abc. Its expected tertiary structure (and the absence of contacts with the P4 and P6 helices present in the intact domain) was observed at all the Mg2+ concentrations analyzed (Figure 3, bottom). Overall, folding of P5c is slower within P5abc relative to P4–P6 under these experimental conditions with minimal Mg2+ concentration dependence (Figure 3). For example, the time progress curves measured for nucleotides 163–164, 175, 176 and 179 are characterized by rates of 3 – 6 s−1 at 10 mM Mg2+ and 6 – 8 s−1 at 100 mM Mg2+. It is striking that collapse of P4–P6 facilitates rather than impedes folding of P5c (compare top and bottom of Figure 3). The observed slow folding of P5c within isolated P5abc is the opposite of a previous Mg2+-mediated folding analysis conducted in a background of very low ionic strength.13 In contrast to the other corresponding A-bulge nucleotides in P4–P6, no significant change in solvent accessibility of nucleotides 185–187 of the isolated P5abc domain could be monitored as a function of Mg2+ concentration; the detectable A-bulge protections fold at rates of 4 – 6 s−1 and 7 – 9 s−1 at 10 and 100 mM Mg2+, respectively.
The single nucleotide analysis shows that several nucleotides (167, 173 and 177) within P5c become more solvent exposed upon folding. The time progress curves for these nucleotides show a large dependence on Mg2+ concentration. An unexpected observation is that nucleotide 155, within the tetraloop, also exhibits an increase in solvent exposure upon the addition of Mg2+. Its slow rate of formation, relative to the tetraloop – tetraloop receptor interaction in P4–P6, is only minimally Mg2+ concentration dependent. Since the tetraloop receptor is not present in P5abc, this transition cannot be related to formation of a native tertiary contact.
Finally, we note that the Mg2+-mediated folding kinetics of P4–P6 tertiary contact formation conducted with a background salt concentration of ~ 20 mM presented herein are significantly faster compared with published studies conducted at ≤ 8 mM.13 SAXS studies at the ≤ 8 mM salt condition of the previous footprinting kinetics studies are complicated by strong interparticle interference effects and thus not reliable. The relative rates of P4–P6 and isolated P5abc folding differ significantly between ~ 20 and ≤ 8 mM salt. At ≤ 8 mM salt, P5c within isolated P5abc folded > 50 fold faster than P4–P6. In the present study at 20 mM salt, folding of P5c is slower within isolated P5abc compared with the P4–P6 domain. The characteristic(s) of the initial conformational ensemble present at ≤ 8 mM salt that impede P4–P6 but not P5abc folding are unknown and were not explored in the present study.
Discussion
P4–P6 is in an extended but not rigid conformation under the initial condition of these studies. 4 At the ~ 20 mM free K+ concentration present in the initial folding solution, strong electrostatic repulsion between the helices is expected: 18 a repulsive energy of about 10 kT is computed from studies of a model system of two DNA helices coupled by a completely flexible tether.17 Reconstructions of the unfolded state from its scattering profile are consistent with an extended structure (J. Lamb, manuscript in preparation). Additional evidence supporting the extended unfolded state comes from measurements of the J5/5a BP mutant of P4–P6, within which the J5/5a hinge is locked into an extended conformation by complementary base pairing.21 The physical properties of the unfolded J5/5a BP mutant and the wild type P4–P6 are indistinguishable.13,21 Upon the addition of Mg2+, the isolated P4–P6 domain folds to its well documented compact structure. The measured SAXS profile of P4–P6 at 10 mM Mg2+ is in good agreement with the scattering curve predicted by CRYSOL 22 derived from the crystal structure. Agreement was also observed between the solvent accessible surfaces reported by ·OH footprinting at the three Mg2+ concentrations investigated and the crystallographically defined structure.b
That significant compaction of P4–P6 requires formation of stabilizing tertiary contacts is shown by the indistinguishable scattering profiles of the P4–P6 double mutant before and after the addition of 10 mM Mg2+ (Figure 2d). This conclusion is confirmed by the absence of discernible changes in the ·OH reactivity of the double mutant upon the addition of the tested concentrations of Mg2+. Charge screening due to ionic strength alone is insufficient to overcome the energetic folding barrier to stable compaction of the P4–P6 domain.
Thus, the wild type P4–P6 folding reactions discussed herein progress from an initial distribution of extended but flexible conformations to a common unique folded structure (Figure 1 C&D). At 10, 50, and 100 mM Mg2+ P4–P6 shows fully formed tertiary contacts (Supplementary Figure 2). The different dependencies of global compaction and tertiary contact formation on the Mg2+ concentration used to initiate folding was exploited in order to separate compaction and tertiary contact formation and thereby clearly define their relationship. The concordant SAXS and footprinting analysis of wild type P4–P6 delineates an emergent hierarchy to tertiary contact formation.
A model for ionic-strength dependent hinge stiffness
The global compaction of the P4–P6 domain to a state consistent in size and shape with the native structure occurs in an apparent single phase (Figure 2a) nearly two orders of magnitude slower than the initial compaction measured for the full-length Tetrahymena ribozyme at 10 mM Mg2+ under comparable experimental conditions. 6,23 The latter compaction reflects electrostatic relaxation from extended structures due to the complete screening of residual backbone charge by the added Mg2+; tertiary contact formation is not observed to overlap with the fast transition.7
The large discrepancy between the initial compaction rates of the P4–P6 domain and the full-length ribozyme suggests that the isolated domain does not undergo relaxation upon initiation of Mg2+-mediated folding. The increase in P4–P6 compaction rate with Mg2+ concentration occurs at concentrations far in excess of those required to neutralize the charge on the RNA. Thus, another process limits molecular compaction of P4–P6. We propose that this process is the inflexibility of the J5/5a hinge. This hypothesis is consistent with the absence of detectable relaxation of the double mutant upon the addition of 10 mM Mg2+ (Figure 2b) and its minimal compaction at the higher concentrations of Mg2+.
The folding barrier due to electrostatic repulsion drops to values below kT even with modest increases in ionic strength.17 Here, we consider an additional barrier to P4–P6 folding, the energy expended in bending the flexible non-base paired strands present in the J5/5a hinge that subtend an angle of ~ 150° in the folded structure (Figure 1). The J5/5a internal loop consists of 4 unpaired bases along one strand and 5 along the other.21 Our model depicts this region as parallel single strands of RNA that act as entropic springs. We assume that the springs are initially unbent and that the junction is unstructured in the unfolded state (e.g. there is no base stacking). 21 These assumptions are consistent with the models of the initial state discussed above. We also assume that the bending energy of both strands together is at least the sum of the energies required to bend each individual strand. This model strives to capture the importance of chain flexibility to folding. Thus, the strands are treated in the most general terms as homopolymers without regard for effects due to base specific stacking or exclusion, or for sequence optimization through evolution.21 This simple model also does not account for ion specific effects, such as stabilization of the junction by Mg2+ or displacement of K+ ions.
Intuitively, the energy required to bend a polymer strand should depend on its stiffness, or resistance to bending, quantified as persistence length (lp).24 Measurement of lp for single strands of RNA have been made at only a few sodium concentrations and/or as a function of force;25 however, the lp of DNA single strands has been measured as a function of monovalent ionic strength 26,27 at concentrations close to the initial state of these experiments. Thus, we estimate lp ≈ 3 nm for single strands in the unfolded state.26 Given the length of the J5/5a single strands and an upper limit to the base-to-base separation, b = 0.7 nm (consistent with the assumption of non-interacting bases in the unfolded state21) the persistence length is greater than the distance between neighboring monomers. Hence we model the RNA strands as wormlike chains (WLC). The WLC model accurately represents the behavior of short strands (10 bases) of ss DNA, only a factor of 2 longer than the strands associated with J5/5a.26 The bending energy of a WLC is Ubend = (k·T·lp / 4b) Σθi 2, where θi is the bend angle of each bond (assumed to be distributed uniformly) and kT is thermal energy.28 This simple, back-of-the-envelope calculation predicts that ~ 3 kT is required to bend J5/5a to its native conformation in the absence of Mg2+.
Like electrostatic repulsion, lp decreases as ionic strength increases, though its dependence is weak at the concentrations studied herein. Empirical descriptions are consistent with variation as the inverse one half power of ionic strength.27 Assuming this functional dependence, lp drops to ≈ 2 nm upon the addition of 10 mM Mg2+ to our initial solution. The bending energy Ubend is linearly proportional to persistence length in this model, and is therefore reduced from ~ 3 to ~ 2 kT. Figure 5 illustrates the proposed free energy barrier to P4–P6 folding resulting from hinge stiffness for 10 and 100 mM Mg2+. This barrier is shown in blue in Figure 5b. At 10 mM Mg2+, wild type P4–P6 folds (albeit slowly) because of the significant drop in free energy due to native contact formation. Since the double mutant lacks the tertiary contacts required to stabilize the compact state, the extended state is favored, though occasional excursions to the fully bent position can occur.
Figure 5.
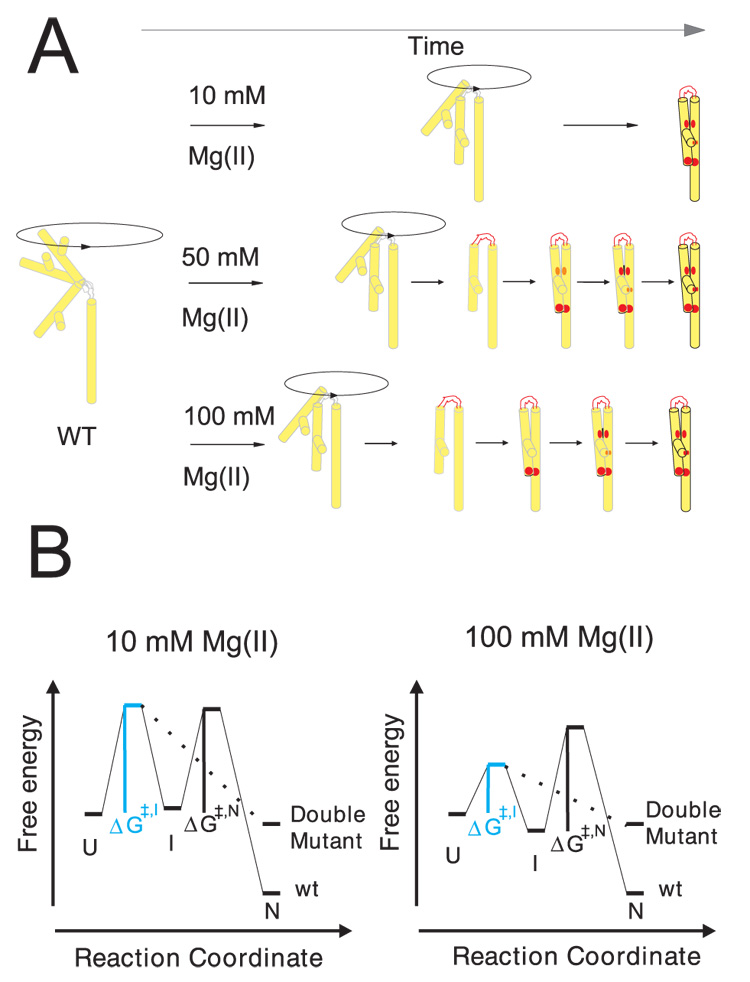
Folding of P4–P6 RNA at the low and high ionic strength conditions of this study. A) The Mg2+ dependent folding pathway of wild type P4–P6 is represented schematically. The outlined transition from light grey to black indicates the time dependent local structure formation within P4–P6 upon Mg2+ addition. The red color highlights the folding sequence of key elements within the RNA. On the left, P4–P6 is depicted in the initial, extended and flexible position. Upon addition of 10 mM Mg2+ (top) the extended conformation of P4–P6 is preferred but bending of the hinge results in a trapped structure by two stabilizing, long range interactions simultaneously (red dots). At 50 mM Mg2+ (middle) the structure formation occurs in sequence: 1) hinge, 2) tetraloop/tetraloop receptor, and 3) A-bulge/P4 helix. The relative difference in the formation of the structural elements in sequence is more developed at 100 mM Mg2+ (bottom). B) The physical barriers to RNA folding are shown by a tri-state free energy diagram. The unfolded, extended state (U state) is pictured and represents the reference state for this diagram. The intermediate state (I) refers to the bent RNA structure whereas the native molecule (N) includes tertiary structure. The rate limiting step in folding shifts from hinge bending to tertiary structure formation at high ionic strength. At 10 mM Mg2+ the activation energy ΔG≠,I is substantial (left) whereas the second barrier, ΔG≠,N, is limiting as ion concentration increases (right). The lower free energy of the intermediate state at 100 mM Mg2+ reflects the observed accumulation of a relaxed molecule. This structure may be stabilized by base stacking in the bent conformation. Because we do not have detailed information about the contacts in the various states, these diagrams are schematic representations, and the energy scales are arbitrary.
At high salt concentrations lp approaches a limiting value of 1 nm for single stranded DNA; the behavior of RNA is assumed to be comparable.27 Thus, the energy required to bend J5/5a drops to kT as the ionic strength increases above 0.1 M. The predicted decrease of barrier height with increasing ionic strength is consistent with the measured increase in compaction rate (Figure 2a). The SAXS rate increases almost ten fold when folding is initiated by 10 and 100 mM Mg2+, respectively, consistent with theoretical treatments of RNA loop stiffness computed as function of ionic strength.20 Because J5/5a bending presents a decreasing barrier to folding as ionic strength increases, we have the interesting scenario of the rate limiting step of folding ‘trading off’ from compaction to tertiary contact formation. The importance of persistence length to RNA folding has also been reported by other authors.29
To summarize the predictions of this model, the initial compaction of the RNA that results from the addition of Mg2+ to a low salt solution requires that two independent conditions be satisfied. The first involves screening of the charge on the helices through counterion condensation, which decreases electrostatic repulsion. The second requires flexibility of the regions that connect the helices, which is modeled here as a function of the (ionic strength dependent) persistence length of the chain. Although both effects are electrostatic, they are distinct. For example, even if the charge on the helices is completely screened, compaction would not occur if the hinge remained rigid. A limiting case is that of the J5/5a BP mutant, which does not collapse, even if the charge on the helices is screened, because the joint is ‘locked open’ by mutation. We also note that, at the highest ionic strength considered, it is a challenge to compute the exact number of excess cations due to the presence of RNA. Exclusion of anions occurs when they are present in large quantities, e.g. in a solution of 100 mM MgCl2.30 However, a rapid collapse consistent with electrostatic relaxation has been observed in the full length ribozyme at all Mg2+ concentrations reported here (L.W.K. and L.P., unpublished), suggesting that the RNA charge is fully screened under these conditions.
The rate limiting step of folding is different at low and high Mg2+ concentrations
The increase in folding rate observed by SAXS due to the flexibility of the J5/5a hinge as a function of increasing Mg2+ concentration occurs at concentrations of Mg2+ over which folding rate changes little when assayed by the anisotropy change of pyrene-labeled P4–P6.14 Thus, this fluorescence probe appears to more closely reflect the formation of stable tertiary contacts reported by ·OH footprinting (Figure 3) than compaction reported by SAXS. The Φ – value analysis of the folding of wild type and mutant P4–P6 derived from the pyrene anisotropy data indicates a folding transition state in which most of the native-state tertiary contacts are not yet formed. Our results are consistent with this interpretation at low but not high Mg2+. Therefore, a simple two-state energy diagram is an incomplete description of Mg2+- induced P4–P6 tertiary folding.
Figure 5A illustrates the folding scenarios for the wild type RNA as a function of Mg2+ concentration. Panel B represents the proposed energetics of folding at low and high Mg2+ concentration. At 10 mM Mg2+ the fully extended state is favored although the hinge samples other conformations. In the wild type molecule these excursions may be trapped by formation of the stabilizing, tertiary contacts. This scheme is supported by the substantial overlap of the compaction and footprinting progress curves (Figure 4). Near-coincident formation of the two primary tertiary contacts within P4–P6, the tetraloop-receptor and A-rich bulge interactions is observed at rates that are only one and a half times slower than the compaction rate reported by SAXS. These data are consistent with a stiff hinge that limits molecular compaction of P4–P6 under these folding conditions. Thus, folding at 10 mM Mg2+ is rationalized by a simple model in which the stiffness of the hinge separating the two halves of P4–P6 allows only an occasional excursion to the ‘fully bent’ position that brings the two sides of each contact into proximity (Figure 5A, top). The rate limiting step is reflected by a substantial ΔG≠,I towards a bent folding intermediate (Figure 5B, left). Tertiary structure formation occurs subsequently by passing the smaller energy barrier ΔG≠,N whose existence was concluded by experiments at high divalent ion concentrations.
If tertiary contact formation was limited only by hinge stiffness, the rate of contact formation should track the increasing compaction rate. It does not. At 100 mM Mg2+, the compaction rate is almost seven fold faster than the average rate of tertiary contact formation. Compaction precedes tertiary contact formation at high ionic strength with the two processes occurring sequentially (Figure 5A bottom). Therefore, a tri-state energy-diagram is proposed. At high Mg2+ concentration the hinge folds first, followed by formation of the tetraloop – tetraloop receptor interaction and then the A-bulge-P4 contact. The disparity between the time-dependence of structuring of these regions is more pronounced as the Mg2+ concentration further increases. Fast formation of the tetraloop-receptor contact is readily rationalized if compaction is fast; a more flexible hinge brings the two sides of this contact into more frequent proximity. However, the weaker Mg2+ concentration dependence demonstrated by the latter two contacts clearly represents another process. The free energy diagram for this folding reaction illustrates the small ΔG≠,I whereas the transition from the bent intermediate (possibly stabilized by base stacking within the junction) to the native conformation is still affiliated with a substantial ΔG≠,N (Figure 5B, right). The formation of tertiary contacts is rate limiting.
Interstrand contact facilitates local structure formation
The near-absence of Mg2+ dependence displayed by some of the tertiary contacts merited further examination. Notably, P5c is least sensitive to changes in the Mg2+ concentration during folding of P4–P6. Structuring of this region within the P4–P6 domain is independent of hinge flexibility and becomes rate-limiting when collapse is rapid. The folded P5abc structure is notable for the five bound Mg2+ visualized crystallographically.31 Three nucleotides from the P5c stem and the A-bulge (166, 168, & 182) coordinate a single Mg2+ in a conformation stabilized by stacking between A183 and U168. The independence of P5c folding with Mg2+ concentration was unexpected in light of the structural intimacy of Mg2+ with the RNA in this region. NMR analysis of P5abc has revealed a secondary structure rearrangement upon Mg2+-mediated folding.32
Mg2+ equilibrium experiments with isolated P5abc suggested that at all Mg2+ concentrations tertiary structure is fully formed (Supplementary Figure 2). The time-resolved footprinting studies of P5abc in isolation were conducted to determine whether contacts of P5c with P4 were responsible for the observed folding behavior. The equally unexpected retardation of P5abc formation in the isolated subdomain relative to within P4–P6, suggests that contact between the P4 strand and P5abc initiates and facilitates folding in the intact domain. This conclusion is supported by the increase in the relative difference between P5c folding in isolation and within P4–P6 with increasing Mg2+ concentration (Figure 3); fast bending of the hinge brings the A-bulge and P4 quickly together. The A-bulge / P4 contact could ‘activate’ nucleotides 182 and 183 for metal ion and stacking interactions, respectively. In this model, the rate limiting step is associated with secondary structure rearrangement rather than Mg2+ binding. The absence of P5c structuring in the P4–P6 double mutant highlights the necessity of the native A-bulge for structuring of this sub-domain.
The tetraloop – tetraloop receptor tertiary contact first observed in P4–P6 is a ubiquitous RNA structural motif.33 The changes in ·OH reactivity of the P5b tetraloop (nucleotides 153 – 155) and the P6a receptor (nucleotides 212 – 213) observed upon Mg2+-mediated contact formation are consistent with the change in solvent accessible surface calculated from the structure of the domain.10 However, the behavior of the tetraloop in isolated P5abc suggests that formation of the ubiquitous contact may not be a simple association of rigid bodies. The unique ·OH reactivity enhancement of nucleotide 155 in P5abc upon the addition of Mg2+ (obscured by tetraloop docking in P4–P6) reflects a conformational change due either to the Mg2+ binding at the tetraloop or global restructuring. That the rate of this transition is comparable to those of the P5c protections argues for the latter explanation. These data are also consistent with propagation of the secondary structure shift initiated by the A-bulge/P4 contact and fuel the speculation about conditions which force fast tertiary contact formation prior to the development of the native secondary structure.
Conclusions
The coupled application of global and local probes to study folding of a small RNA domain has identified two barriers to its folding. At low ionic strength, the barrier is the stiffness of the connecting hinge of the P4–P6 domain. A simplified model of the bending energy for non-base paired chains dependent on the persistence length of single stranded RNA rationalizes the barrier. The persistence length in turn depends on ion concentration and valence. Since the P4–P6 domain is the first to form in the full-length ribozyme under several experimental conditions,7 any barrier to P4–P6 folding becomes a barrier for all subsequent folding events. At higher ionic strength, where hinge bending is no longer limiting, P4–P6 folding is limited by tertiary contact formation; the P4–P6 folding pathway includes at least one populated folding intermediate. These results suggest that an ionic-strength-dependent combination of electrostatic effects, tertiary contact formation and flexibility of non base-paired regions sculpts the RNA folding landscape.
Materials and Methods
RNA preparation
RNA molecules were generated by T7 RNA polymerase in vitro transcription of PCR-generated DNA templates containing wild type or mutant P4–P6 domain of the ribozyme (nucleotides 104 to 261). The mutant has the two tertiary contacts of the P4–P6 domain destabilized, the A-rich bulge (AAUAAG (183 – 188) to UUUUUU) and the tetraloop receptor (UAAG (224 – 227) to AUA). All transcribed RNA was gel purified prior to use in SAXS or footprinting experiments.
Small angle X-ray scattering
A continuous flow, diffusive mixer similar to one previously described,23 was used to trigger and monitor folding on time scales from 1–160 ms. The longer measurement times achieved here, relative to the previous use of this device 23 results from decreasing the flow speed from its nominal value of 20 cm/s to 1.5 cm/s for several data points. The mixing devices were fabricated at the Cornell Nanoscale Science Facility (CNF) from 1 mm thick silicon wafers. RNA at 2 mg/mL (16µM) in 50 mM potassium MOPS [3-(N-morpholino)propanesulfonic acid], pH 7.0 was mixed with equivalently buffered solutions containing 10, 50 or 100 mM MgCl2 at 25 °C.
Molecular conformations were assessed using small angle x-ray scattering from molecules at different positions along the outlet channel of the flow cell. Several positions were sampled, corresponding to different times after the initiation of folding. High intensity, pink beam available at the Advanced Photon Source beamlime 8-ID I 34 was spatially defined with slits set to 13 µm in the vertical direction and 50 µm in the direction of flow. The beam flux into this small rectangle was 3 × 1010 photons/s. Scattering intensity is measured as a function of q=4πsinθ/λ, where 2θ is the scattering angle and λ is the x-ray wavelength. Scattering profiles were collected with a Princeton Instruments x-ray detector placed at the end of an evacuated, 50-cm long beampipe. The images were analyzed using MATLAB software.
Compaction of the P4–P6 domain is accurately described by projecting each time dependent scattering profile onto a linear combination of the initial (unfolded, U) and final (native, N) states, Iu(q) and In(q) respectively. 23 Thus, each time dependent scattering profile, I(q,t) can be expressed as I(q,t) = Pu(t)Iu(q) + Pn(t) In(q). The scattering profile of the U state is measured for each construct. The scattering profile of the N state is calculated with CRYSOL 22 from the monomeric form of the 1GID crystal structure. This computed scattering profile agrees well with the measured profile of the wild-type domain acquired in 10 mM Mg2+, 160 ms after mixing. Although similar time courses are obtained from analysis of the time-dependent radius of gyration, the projections use data at larger scattering angles, where the effect of interparticle interference is negligible. 4 Because the Mg2+-folded P4–P6 domain dimerizes rapidly at the relatively high concentrations required for the SAXS experiments, the scattering intensity in the forward direction Io, was also monitored for each data point. This potential for rapid aggregation highlights the importance of carrying out time-resolved measurements where folding can be monitored prior to oligomerization. Further details of the molecular conformations were assessed by examining the product of I(q,t) and q as a function of q (as in Ref. 17). This formalism, emphasizing the cylindrical nature of the unfolded state, effectively distinguishes extended from relaxed conformations of the molecule. Finally, Kratky plots, displaying the product of I(q,t) and q2 as a function of q, were used to qualitatively assess molecular compaction.35 Supplementary Figure 3 shows time-dependent Kratky plots for both constructs following the addition of 10 mM Mg2+.
Time-resolved hydroxyl radical footprinting
P4–P6 RNA was radiolabeled at the 5’ end with [γ-32P]ATP and purified as described.36 The 32P-RNA was dissolved in 18 mM potassium cacodylate (pH 7.0) whose ionic strength corresponds to the 50 mM potassium MOPS (pH 7.0) used in the SAXS studies. The 1 µM solutions of RNA were denatured by heating to 95°C for 1 min, then cooled slowly and incubated at 25°C prior to initiating the kinetics experiment. The time-resolved ·OH footprinting studies were carried out by Fast Fenton Footprinting at 25°C using a KinTek® RQF-3 three-syringe mixer.16. RNA folding was initiated by the addition of 10, 50, or 100 mM MgCl2, respectively. All experiments were repeated.
The RNA samples were expelled into ethanol to quench the footprinting reaction and initiate precipitation. The footprinting reaction products were separated by denaturing polyacrylamide gel electrophoresis and imaged as described. 16 The change in the density of each band was quantified using SAFA. 37 Either single bands or groups of bands were scaled to fractional saturation by fi = L + (U − L) · Y̅ where f denotes the integrated density of the band(s) being analyzed, L and U represent the lower and upper limits to the transition and Y̅ is fractional saturation. L and U were determined from samples lacking MgCl2 and samples equilibrated in 10 mM MgCl2. Scaling of each data set to these limits allowed multiple data sets, including replicates, rapid-mix and hand-mix experiments to be globally analyzed. The data scaled to fractional saturation were fit globally using non-linear, least-squares analysis in Origin® v6.1 (OriginLab®) to:
| <1> |
where αi and ki are the amplitude and rate constant, respectively, of the ith kinetic phase. The 65% confidence intervals are reported.
Mg2+ -folding isotherms of wild-type P4–P6 and isolated P5abc RNA
5’-32P –labeled RNA samples were denatured by heating to 95°C for 1 min, then cooled slowly in 18 mM potassium cacodylate (pH 7.0). After the addition of various concentrations of Mg2+, the solution was incubated at 25°C for 1 hour. The cleavage reaction was initiated by addition of 0.01 mM Fe(NH4)2(SO4)2/0.02 mM EDTA solution, following addition of 0.03% H2O2 and 1mM ascorbic acid. The reaction was allowed to proceed at 25°C for 2 min and was quenched by the addition of thiourea to a final concentration of 15 mM. RNA fragment and data analysis followed standard procedure. 38
Supplementary Material
Acknowledgements
We thank Alec Sandy and Suresh Narayanan for their assistance at beamline 8-IDI at the APS and Inna Shcherbakova and Somdeb Mitra for their assistance in initiating the time-resolved footprinting studies. This work was supported by grant P01-GM066275 from the National Institute of General Medical Sciences. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. We acknowledge additional support from the National Science Foundation through grant MCB-0347220 (to L.P.) and the Cornell Nanobiotechnology Center. Fabrication of the SAXS mixing devices was conducted in the Cornell Nanoscale Science and Technology Facility that is supported by the NSF, Cornell University and industrial affiliates.
Footnotes
Supplementary Table 1 summarizes the rate constants obtained for all of the nucleotides that were analyzed.
The RNA concentrations employed for ·OH footprinting are much lower than those required for SAXS. Dimerization was not detected by native gel shift assays at the RNA concentrations employed for footprinting (data not shown).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Crothers DM, Cole PE, Hilbers CW, Shulman RG. Molecular Mechanism of Thermal Unfolding of Escherichia-Coli Formylmethionine Transfer-RNA. J. Mol. Biol. 1974;87:63–72. doi: 10.1016/0022-2836(74)90560-9. [DOI] [PubMed] [Google Scholar]
- 2.Duckett DR, Murchie AIH, Clegg RM, Bassi GS, Giraud-Panis MJE, Lilley DMJ. Nucleic acid structure and recognition. Biophys. Chem. 1997;68:53–62. doi: 10.1016/s0301-4622(97)00007-0. [DOI] [PubMed] [Google Scholar]
- 3.Hagerman PJ. Flexibility of RNA. Annual Review of Biophysics and Biomolecular Structure. 1997;26:139–156. doi: 10.1146/annurev.biophys.26.1.139. [DOI] [PubMed] [Google Scholar]
- 4.Takamoto K, Das R, He Q, Doniach S, Brenowitz M, Herschlag D, Chance MR. Principles of RNA compaction: Insights from the equilibrium folding pathway of the P4–P6 RNA domain in monovalent cations. J. Mol. Biol. 2004;343:1195–1206. doi: 10.1016/j.jmb.2004.08.080. [DOI] [PubMed] [Google Scholar]
- 5.Takamoto K, He Q, Morris S, Chance MR, Brenowitz M. Monovalent cations mediate formation of native tertiary structure of the Tetrahymena thermophila ribozyme. Nat. Struct. Biol. 2002;9:928–933. doi: 10.1038/nsb871. [DOI] [PubMed] [Google Scholar]
- 6.Das R, Kwok LW, Millett IS, Bai Y, Mills TT, Jacob J, Maskel GS, Seifert S, Mochrie SGJ, Thiyagarajan P, Doniach S, Pollack L, Herschlag D. The fastest global events in RNA folding: Electrostatic relaxation and tertiary collapse of the Tetrahymena ribozyme. J. Mol. Biol. 2003;332:311–319. doi: 10.1016/s0022-2836(03)00854-4. [DOI] [PubMed] [Google Scholar]
- 7.Kwok LW, Shcherbakova I, Lamb JS, Park HY, Andresen K, Smith H, Brenowitz M, Pollack L. Concordont exploration of the kinetics of RNA folding from global and local perspectives. J. Mol. Biol. 2006;355:282–293. doi: 10.1016/j.jmb.2005.10.070. [DOI] [PubMed] [Google Scholar]
- 8.Murphy FL, Cech TR. An Independently Folding Domain of RNA Tertiary Structure within the Tetrahymena Ribozyme. Biochemistry. 1993;32:5291–5300. doi: 10.1021/bi00071a003. [DOI] [PubMed] [Google Scholar]
- 9.Cate JH, Gooding AR, Podell E, Zhou KH, Golden BL, Kundrot CE, Cech TR, Doudna JA. Crystal structure of a group I ribozyme domain: Principles of RNA packing. Science. 1996;273:1678–1685. doi: 10.1126/science.273.5282.1678. [DOI] [PubMed] [Google Scholar]
- 10.Cate JH, Gooding AR, Podell E, Zhou K, Golden BL, Szewczak AA, Kundrot CE, Cech TR, Doudna JA. RNA tertiary structure mediation by adenosine platforms. Science. 1996;273:1696–1699. doi: 10.1126/science.273.5282.1696. [DOI] [PubMed] [Google Scholar]
- 11.Ikawa Y, Okada A, Imahori H, Shiraishi H, Inoue T. Identification of the nucleotides in the A-rich bulge of the Tetrahymena ribozyme responsible for an efficient self-splicing reaction. J Biochem (Tokyo) 1997;122:878–882. doi: 10.1093/oxfordjournals.jbchem.a021836. [DOI] [PubMed] [Google Scholar]
- 12.Murphy FL, Cech TR. GAAA tetraloop and conserved bulge stabilize tertiary structure of a group I intron domain. J Mol Biol. 1994;236:49–63. doi: 10.1006/jmbi.1994.1117. [DOI] [PubMed] [Google Scholar]
- 13.Deras ML, Brenowitz M, Ralston CY, Chance MR, Woodson SA. Folding mechanism of the Tetrahymena ribozyme P4–P6 domain. Biochemistry. 2000;39:10975–10985. doi: 10.1021/bi0010118. [DOI] [PubMed] [Google Scholar]
- 14.Silverman SK, Cech TR. An early transition state for folding of the P4–P6 RNA domain. RNA. 2001;7:161–166. doi: 10.1017/s1355838201001716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silverman SK, Deras ML, Woodson SA, Scaringe SA, Cech TR. Multiple folding pathways for the P4–P6 RNA domain. Biochemistry. 2000;39:12465–12475. doi: 10.1021/bi000828y. [DOI] [PubMed] [Google Scholar]
- 16.Shcherbakova I, Mitra S, Beer RH, Brenowitz M. Fast Fenton footprinting: a laboratory-based method for the time-resolved analysis of DNA, RNA and proteins. Nucleic Acids Res. 2006;34:e48. doi: 10.1093/nar/gkl055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bai Y, Das R, Millett IS, Herschlag D, Doniach S. Probing counterion modulated repulsion and attraction between nucleic acid duplexes in solution. Proc. Natl. Acad. Sci. U S A. 2005;102:1035–1040. doi: 10.1073/pnas.0404448102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qiu XY, Kwok LW, Park HY, Lamb JS, Andresen K, Pollack L. Measuring inter-DNA potentials in solution. Phys. Rev. Lett. 2006;96 doi: 10.1103/PhysRevLett.96.138101. [DOI] [PubMed] [Google Scholar]
- 19.Penedo JC, Wilson TJ, Jayasena SD, Khvorova A, Lilley DMJ. Folding of the natural hammerhead ribozyme is enhanced by interaction of auxiliary elements. RNA. 2004;10:880–888. doi: 10.1261/rna.5268404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan ZJ, Chen SJ. Electrostatic free energy landscapes for nucleic acid helix assembly. Nucleic Acids Res. 2006;34:6629–6639. doi: 10.1093/nar/gkl810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szewczak AA, Cech TR. An RNA internal loop acts as a hinge to facilitate ribozyme folding and catalysis. RNA. 1997;3:838–849. [PMC free article] [PubMed] [Google Scholar]
- 22.Svergun D, Barberato C, Koch MHJ. CRYSOL - A program to evaluate xray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Cryst. 1995;28:768–773. [Google Scholar]
- 23.Russell R, Millettt IS, Tate MW, Kwok LW, Nakatani B, Gruner SM, Mochrie SGJ, Pande V, Doniach S, Herschlag D, Pollack L. Rapid compaction during RNA folding. Proc. Natl. Acad. Sci. U S A. 2002;99:4266–4271. doi: 10.1073/pnas.072589599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doi M, Edwards SF. Theory of Polymer Dynamics. New York: Oxford University Press; 1986. [Google Scholar]
- 25.Seol Y, Skinner GM, Visscher K. Elastic properties of a single-stranded charged homopolymeric ribonucleotide. Phys. Rev. Lett. 2004;93 doi: 10.1103/PhysRevLett.93.118102. [DOI] [PubMed] [Google Scholar]
- 26.Murphy MC, Rasnik I, Cheng W, Lohman TM, Ha TJ. Probing single-stranded DNA conformational flexibility using fluorescence spectroscopy. Biophys. J. 2004;86:2530–2537. doi: 10.1016/S0006-3495(04)74308-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tinland B, Pluen A, Sturm J, Weill G. Persistence length of single-stranded DNA. Macromolecules. 1997;30:5763–5765. [Google Scholar]
- 28.Schnurr B, MacKintosh FC, Williams DRM. Dynamical intermediates in the collapse of semiflexible polymers in poor solvents. Europhysics Letters. 2000;51:279–285. [Google Scholar]
- 29.Caliskan G, Hyeon C, Perez-Salas U, Briber RM, Woodson SA, Thirumalai D. Persistence length changes dramatically as RNA folds. Phys. Rev. Lett. 2005;95:268303. doi: 10.1103/PhysRevLett.95.268303. [DOI] [PubMed] [Google Scholar]
- 30.Misra VK, Draper DE. The interpretation of Mg2+ binding isotherms for nucleic acids using Poisson-Boltzmann theory. J. Mol. Biol. 1999;294:1135–1147. doi: 10.1006/jmbi.1999.3334. [DOI] [PubMed] [Google Scholar]
- 31.Cate JH, Hanna RL, Doudna JA. A magnesium ion core at the heart of a ribozyme domain. Nat. Struct. Biol. 1997;4:553–558. doi: 10.1038/nsb0797-553. [DOI] [PubMed] [Google Scholar]
- 32.Wu M, Tinoco I. RNA folding causes secondary structure rearrangement. Proc. Natl. Acad. Sci. U S A. 1998;95:11555–11560. doi: 10.1073/pnas.95.20.11555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moore PB. Structural motifs in RNA. Annu. Rev. Biochem. 1999;68:287–300. doi: 10.1146/annurev.biochem.68.1.287. [DOI] [PubMed] [Google Scholar]
- 34.Sandy AR, Lurio LB, Mochrie SGJ, Malik A, Stephenson GB, Pelletier JF, Sutton M. Design and characterization of an undulator beamline optimized for small-angle coherent X-ray scattering at the Advanced Photon Source. J. Synchrotron Rad. 1999;6:1174–1184. [Google Scholar]
- 35.Doniach S. Changes in biomolecular conformation seen by small angle X-ray scattering. Chemical Reviews. 2001;101:1763–1778. doi: 10.1021/cr990071k. [DOI] [PubMed] [Google Scholar]
- 36.Zaug AJ, Grosshans CA, Cech TR. Sequence-specific endoribonuclease activity of the Tetrahymena ribozyme: enhanced cleavage of certain oligonucleotide substrates that form mismatched ribozyme-substrate complexes. Biochemistry. 1988;27:8924–8931. doi: 10.1021/bi00425a008. [DOI] [PubMed] [Google Scholar]
- 37.Das R, Laederach A, Pearlman SM, Herschlag D, Altman RB. SAFA: semi-automated footprinting analysis software for high-throughput quantification of nucleic acid footprinting experiments. RNA. 2005;11:344–354. doi: 10.1261/rna.7214405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uchida T, He Q, Ralston CY, Brenowitz M, Chance MR. Linkage of monovalent and divalent ion binding in the folding of the P4–P6 domain of the Tetrahymena ribozyme. Biochemistry. 2002;41:5799–5806. doi: 10.1021/bi020042v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.