

Abstract
The myelodysplastic syndromes (MDS) are a group of clonal hematopoietic stem cell disorders characterized by ineffective hematopoiesis, peripheral blood cytopenias, dysplasia and a propensity for transformation to acute myeloid leukemia (AML). A wide spectrum of genetic aberrations has been associated with MDS, including chromosomal translocations involving the NUP98 gene, most commonly leading to fusions of NUP98 with abd-b group HOX genes, including HOXD13. We used vav regulatory elements to direct expression of a NUP98-HOXD13 (NHD13) fusion gene in hematopoietic tissues. NHD13 transgenic mice faithfully recapitulate all of the key features of MDS, including peripheral blood cytopenias, bone marrow dysplasia and apoptosis, and transformation to acute leukemia. The MDS that develops in NHD13 transgenic mice is highly lethal; within 14 months, 90% of the mice died of either leukemic transformation or severe anemia and leukopenia due to progressive MDS. These mice provide a pre-clinical model that can be used for the evaluation of MDS therapy and biology.
Keywords: NUP98, HOXD13, Myelodysplastic syndrome, Leukemia, Transgenic mouse
Introduction
Myelodysplastic syndrome (MDS) is a heterogenous group of diseases characterized by dysplasia, ineffective hematopoiesis, and peripheral blood cytopenias. Clinically, MDS typically has one of three outcomes. Patients may die due to complications of pancytopenia, the disease can transform to an acute leukemia, or patients may survive for an extended period of time with the disease (1). The crude incidence of MDS has been estimated at 3.5–12.6 per 100,000 per year, and there is a suggestion that this incidence may be increasing (2). A large number of chromosomal abnormalities, including deletions, amplifications, inversions, and translocations have been identified in the malignant cells of patients with MDS (1,3,4).
A new class of chromosomal aberration associated with MDS
The most common recurrent genetic abnormalities in patients with MDS are deletions of chromosomes 5q, 7q, and 20q (1,3,4). Despite decades of intensive study, and numerous promising leads, the critical gene(s) located in these regions that are responsible for MDS have not conclusively been identified. Recently, a number of chromosomal translocations involving the NUP98 gene, located on chromosome 11p15.5, have been identified (5) in patients with hematologic malignancy. NUP98 translocations have now been recognized in a wide array of hematologic maliganancies, including MDS, acute myeloid leukemia (AML), chronic myelogenous leukemia (CML), and precursor-T lymphoblastic lymphoma/leukemia (pre-T LBL) (5). With the use of increasingly sophisticated cytogenetic techniques, at least 20 different partner genes for NUP98 have now been identified. Remarkably, half of the partner genes encode homeodomain proteins, primarily those belonging to the abd-b group of HOX genes (Table 1). As indicated in table 1, the NUP98-HOX gene fusions are universally associated with myeloid malignancies, whereas the NUP98 fusions involving non-homeodomain genes are associated with a wider spectrum of hematologic malignancies.
Table 1.
NUP98 translocations associated with hematologic malignancy
| Translocation | Partner Gene | Homeodomain? | Disease |
|---|---|---|---|
| t(7;11)(p15;p15) | HOXA9, 11, 13 | Yes | MDS, AML, CML |
| t(11;12)(p15:q13) | HOXC11,13 | Yes | MDS, AML |
| t(2;11)(q31;p15) | HOXD9, 11, 13 | Yes | MDS, AML, CML |
| t(1;11)(q23;p15) | PMX1(PRRX1) | Yes | MDS, AML |
| t(9;11)(q34;p15) | PRRX2 | Yes | MDS, AML |
| t(4;11)(q21;p15) | RAP1GDS1 | No | pre-T LBL |
| t(11;20)(p15;q11) | TOP1 | No | MDS, AML |
| t(9;11)(p22;p15) | LEDGF | No | AML |
| t(5;11)(q35;p15) | NSD1 | No | AML |
| t(8;11)(p11;p15) | NSD3 | No | AML |
| t(10;11)(q25;p15) | ADD3 | No | pre-T LBL |
| inv11(p15q22) | DDX10 | No | MDS, AML |
| t(6;11)(q24;p15) | C6orf80 | No | AMKL, pre-T LBL |
| t(3;11)(p24;p15) | TOP2B | No | AML |
| t(11;21;12)(p15;p13;p13) | JAR1D1A | No | AMKL |
The NUP98 gene encodes a 98 kD component of the nuclear pore complex (NPC) that mediates nucleo-cytoplasmic transport of RNA and protein (6,7). NUP98 contains N-terminal phenylalanine-glycine (FG) repeats, which are commonly found in NPC proteins. The NUP98 gene fusions associated with hematologic malignancy invariably encode fusion proteins that fuse the amino-terminal portion of NUP98, containing the FG repeats, with the carboxy-terminal portion of the partner gene (5). In the case of NUP98-HOX fusions, the DNA-binding homeodomain is contained in the NUP98-HOX fusion protein.
Although the molecular mechanism(s) that generate chromosomal translocations involving NUP98 remain poorly understood, it is important to note that many of the NUP98 translocations have been recognized in patients with therapy-related AML or MDS (t-AML or t-MDS). Intriguingly, analysis of breakpoints from t-MDS patients with NUP98-TOP1 fusions revealed an almost perfect reciprocal chromosomal translocation, consistent with a topoisomerase II “subunit exchange” model for the translocation (8).
A mouse model for MDS
Our lab has used two approaches in an attempt to generate an in vivo model for MDS using an NHD13 fusion gene. In our first approach, we used homologous recombination to “Knock-In” a HOXD13 cassette at mouse Nup98 exon 12 (9). Although we were able to successfully target the Nup98 locus and produce a NUP98-HOXD13 (hereafter NHD13) mRNA and protein, we were unable to generate chimeric mice, as there was no contribution from the ES cells to adult mouse tissues. The NHD13 ES cells grew as rapidly as the parental cells in vitro, and we were able to detect contributions from the ES cells to chimeric mice up to embryonic day 14.5, consistent with a hypothesis that the NHD13 ES cells were impaired in their ability to differentiate and contribute to adult tissues. We next attempted to differentiate the NHD13 ES cells in vitro, using a cytokine cocktail including IL3, IL6, SCF, and Epo. In this experiment, although the parental ES cells differentiated to CFU-GM, CFU-GEMM, and CFU-E colonies, the NHD13 cells primarily formed dense clusters of cells reminiscent of blast colonies (BL-CFC) (10). Morphologically, these cells had an appearance consistent with undifferentiated blast cells, displaying a high nuclear/cytoplasmic ratio, uncondensed chromatin, and prominent nucleoli (9). These findings again suggest that the NHD13 ES cells are impaired in their ability to differentiate.
In a complementary approach, we generated transgenic mice on an FVB/N background that express an NHD13 fusion gene under the control of vav regulatory elements (11), leading to expression of the NHD13 fusion gene in all hematopoietic tissues (12). These mice did develop MDS, with leukopenia, neutropenia, and anemia (Table 2) despite a hypercellular or normocellular bone marrow (Figure 1). The disease was highly lethal, as over 90% of the NHD13 transgenic mice died by 14 months of age. Similar to human MDS, 20% of the mice died of severe pancytopenia without evidence of transformation to acute leukemia, 20% of the mice died of unknown causes, and 60% of the mice progressed to acute leukemia.
Table 2.
CBC from NHD13 transgenic mice aged 4–7 months
| WBCa | Neutrophila | Lympha | Hgbb | Plta | BM blasts | BM erythroid dysplasia | BM myeloid dysplasia | BM mega dysplasia | |
|---|---|---|---|---|---|---|---|---|---|
| Transgenicc | 1.8 +/− 0.5 | 0.4 +/− 0.2 | 1.2 +/− 0.4 | 11.8+/− 2.1 | 958 +/− 569 | 14.1 ± 1.9% | 12.8 ± 2.1% | 6.0 ± 1.3% | rare |
| Controlc | 6.5 +/− 1.8 | 1.4 +/− 0.9 | 4.8 +/− 0.9 | 14.2 +/− 0.7 | 841 +/− 130 | 5.6 ± 1.8 % | 1.5± 0.9% | 1.8 ± 1.0% | rare |
| p valued | <0.001 | <0.001 | <0.001 | <0.001 | 0.709 | <0.01 | <0.01 | <0.05 |
White blood cell, neutrophil, lymphocyte, and platelet count expressed as 109/L.
Hemoglobin in g/dL
n=22 for transgenic CBC, n=7 for control CBC, n=5 for transgenic bone marrow, n=5 for control bone marrow
p value by Mann-Whitney U-test.
Figure 1. Ineffective hematopoiesis and dysplasia in NHD13 mice.
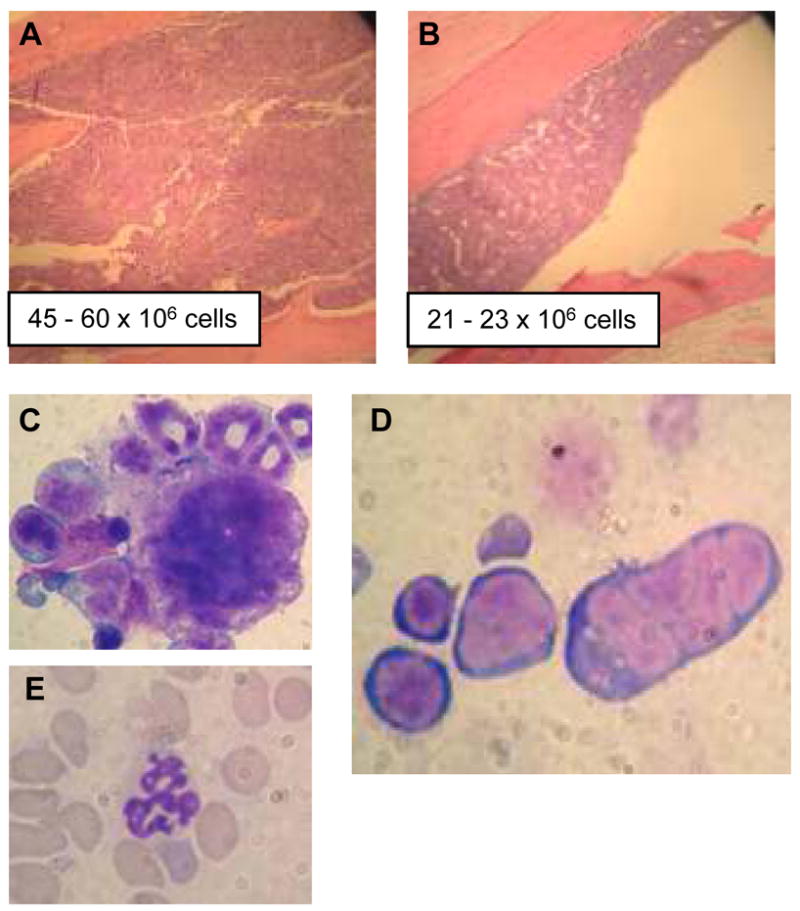
A and B H &E stained bone marrow from NHD13 (A) and wild-type (B) mice; total cell number from 2 femurs are indicated. Note densely cellular bone marrow in (A) (original magnification × 100). Examples of a dysplastic micromegakaryocyte (C) and multinucleate erythroblast (D) and hypersegmented neutrophil (E) from NHD13 mouse are shown (original magnification × 1000).
The leukemic subtype was assessed by FACS, immunohistochemistry, gene expression profiling, and T-cell receptor gene rearrangements. Surprisingly, half of the leukemias we identified in our initial series were pre-T lymphoblastic leukemia/lymphoma (pre-T LBL), with the remainder being non-lymphoid, most commonly myeloid, leukemias. The finding of pre-T LBL in NHD13 mice was unanticipated, since human MDS only rarely transforms into a lymphoid malignancy (13,14), and since NHD13 fusions have not been identified in human patients with T-cell malignancies. However, it should be noted that other NUP98 fusion genes, such as NUP98-RAP1GDS1 and NUP98-ADD3, have been associated with pre-T LBL (15,16), leading to the suggestion that expression of NUP98 fusion genes can result in T-cell malignancies as well as myeloid malignancies. Furthermore, one of the NHD13 mice simultaneously had both an erythroid leukemia and a pre-T LBL (Figure 2), raising the possibility that the erythroid leukemia had evolved from a pre-existing MDS, and the pre-T LBL had originated in the thymus from thymic precursors independent of the MDS clone, and infiltrated the lung (but not liver or spleen). Therefore, we favor the hypothesis that the pre-T LBL in the NHD13 mice arises as an independent malignancy, as opposed to arising from an MDS clone. In either case, these findings indicate that the NHD13 transgene is oncogenic in T-lymphoid precursors as well as myeloid and erythroid cells.
Figure 2. Concurrent erythroid and pre-T leukemia in NHD13 mouse 1018.
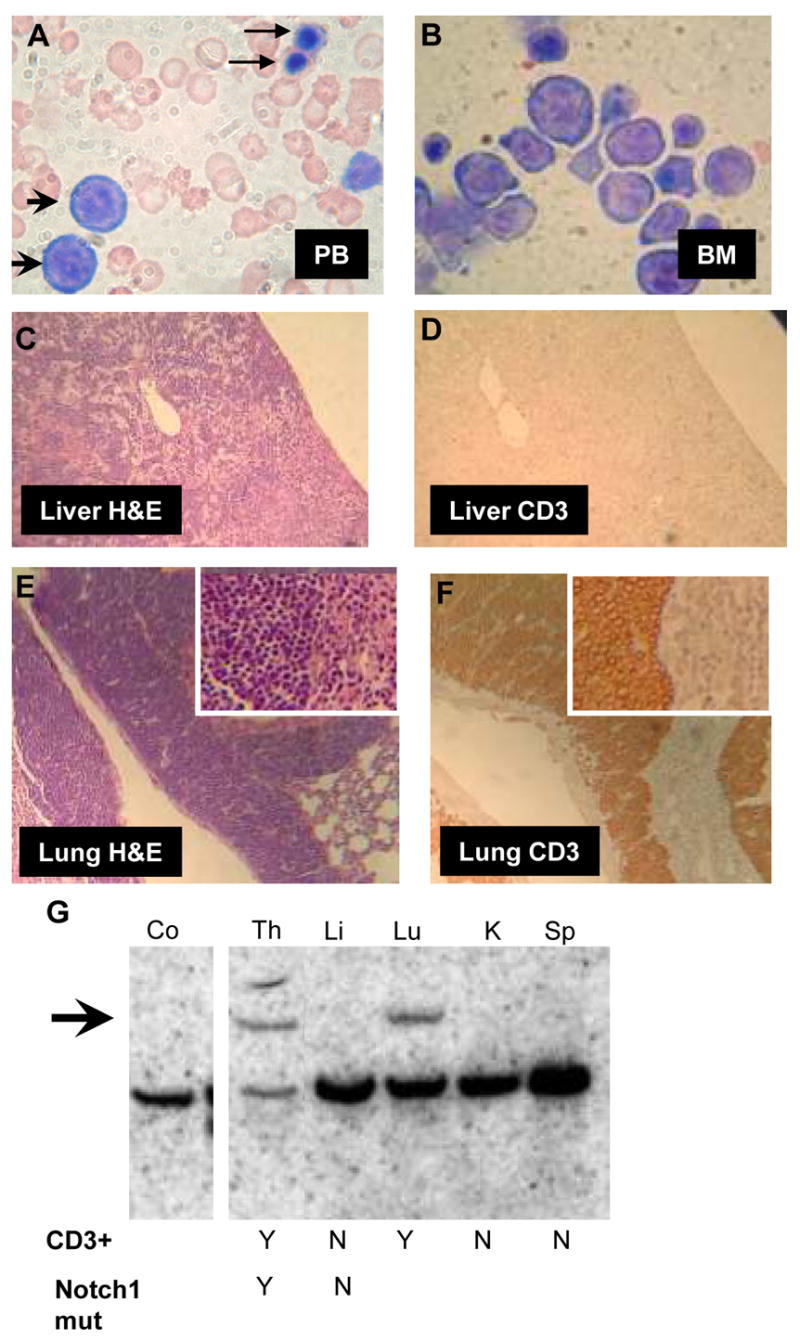
A and B, peripheral blood (A) and bone marrow (B) demonstrating circulating erythroblasts (arrowheads), nucleated red blood cells (arrows), and replacement of bone marrow with erythroblasts (original magnification × 1000). C and D, section of liver infiltrated with leukemic blasts, stained with H&E (C) or anti-CD3 (D) (original magnification × 100). Note lack of CD3 staining in liver. E and F, section of lung infiltrated with leukemic blasts, stained with H&E (E) or anti-CD3 (F). Note strong CD3 staining in lung (original magnification × 100; inset × 400). (G) Southern blot of leukemic tissue hybridized to a TCRB probe. Note germline band in all lanes, and a clonal rearranged band in the Th and Lu (arrow). CD3 staining and presence of Notch1 mutation in infiltrated tissues is indicated as yes (Y) or no (N). Co, non-transgenic control; Th, thymus; Li, liver; Lu, lung; K, kidney; Sp, spleen.
Since the above results were based on offspring from a single founder mouse, we generated additional transgenic lines to eliminate the possibility that the MDS phenotype was secondary to an insertional effect of the transgene; these additional lines were generated on a C57Bl6 background to determine whether the MDS phenotype was strain-specific. Similar to the findings seen on an FVB/N background, these mice showed peripheral blood cytopenias, dysplasia, and transformation to acute leukemia, with a similar high penetrance and similar age of leukemic transformation. In addition to erythroid, myeloid, and pre-T leukemias, some of these mice developed an undifferentiated leukemia that was negative for T-cell (CD3), B-cell (B220), myeloid (myeloperoxidase, MPO), monocyte (F4/80), and megakaryocytic markers (CD41). A single mouse developed a biphenotypic leukemia, characterized by thymic enlargement and hepatosplenomegaly, with infiltration of blasts that were positive for both CD3 and MPO.
Out of 51 transgene positive mice that were followed for at least 14 months, 7 were euthanized because the mice were sick or moribund, and showed signs of MDS, but had not transformed to acute leukemia. Of these 7 mice, three (#1145, 1899, 1196) had severe anemia (hgb < 5.0 g/dl), one (#2747) had a pulmonary hemorrhage, and one (#1903) had a retro-orbital infection, all signs consistent with severe pancytopenia.
The findings we describe here are distinct from results obtained with transduction of bone marrow mobilized with 5-fluorouracil and transduced with an NHD13 retrovirus (17). In those experiments, NHD13 both promoted growth and inhibited differentiation of hematopoietic progenitors in vitro; the inhibition of differentiation was characterized by a marked decrease in ter119+ erythroid cells in a colony forming unit-spleen (CFU-S) assay. Mice reconstituted with bone marrow that expressed the NHD13 retrovirus initially displayed a diminished engraftment of cells that expressed NHD13, and leukopenia, principally due to lymphopenia. A minority of these mice developed anemia and leukocytosis, consistent with a myeloproliferative disease (MPD), whereas other mice from this cohort were markedly anemic but had normal WBC levels. Therefore, both the retroviral transduction and transgenic models lead to anemia and lymphopenia in mice, but the retroviral transduction model leads to MPD with increased neutrophil counts in a minority of mice, whereas the transgenic model leads to decreased neutrophil counts and frequent transformation to acute leukemia. The reasons for the differences in these two models are not clear, but could be due to mouse strain differences, the nature of the cells targeted for retroviral infection, in vitro expansion of infected cells, activation of host genes located at the retroviral insertion sites, and/or relative levels of NHD13 expression.
It has been suggested that mutations in at least two pathways, one leading to impaired differentiation, and one leading to increased proliferation and/or decreased apoptosis are required to produce AML (18). We strongly suspect that the NHD13 fusion protein exerts an oncogenic effect through impaired differentiation for several reasons. NHD13 ES cells do not contribute to adult chimeric mice, and fail to differentiate in vitro (discussed above). In addition, simultaneous expression of a NUP98-HOXA9 fusion and BCR-ABL fusion leads to a fulminant AML in a mouse transduction/transplantation model, whereas expression of BCR-ABL (a “proliferative class” mutation) alone leads to a non-fatal myeloproliferative disease (19). Furthermore, as opposed to cells transfected with an empty vector, K562 cells expressing an NHD13 fusion fail to differentiate to megakaryocytes following treatment with phorbol esters (12). Finally, some human CML patients, who have a “proliferative” mutation (BCR-ABL), develop a NUP98-HOX translocation at the time of blast crisis and transformation to AML (20).
Outstanding questions and future directions
MDS is a heterogeneous group of diseases, that displays a wide spectrum of chromosomal abnormalities. Although rare, at least seven different NUP98 translocations, including the NUP98-HOXD13 translocation, have been recognized in patients with therapy-related MDS. NHD13 mice recapitulate all of the key findings of human MDS, including blood cell dysplasia, peripheral blood cytopenias, ineffective hematopoiesis, and transformation to acute leukemia, and thus provide an excellent model for the human disease. Our future and ongoing studies are designed to address the following questions. First, we suspect that additional mutations, especially those in proliferative and/or apoptotic pathways, are required to convert the MDS to an acute leukemia. What are those mutations? We have begun experiments using retroviral insertional mutagenesis to identify collaborating genes; based on historical precedent, we anticipate that this approach will primarily identify activation or gain of function events. Complementary experiments that are more likely to identify loss of function events include screens using restriction landmark genome scanning (RLGS) (21) and array-based comparative genomic hybridization (aCGH) (22). We also have begun experiments designed to determine the utility of these mice as a pre-clinical model for drug development. We think that this type of model is particularly important for assessing a disease such as MDS, since available MDS cell lines typically have been established from MDS patients that have converted to AML, or have acquired additional chromosomal abnormalities, suggesting that these MDS cell lines are indistinguishable from AML cell lines (23). Finally, we are interested in determining if MDS, which can be viewed as a “pre-malignant” condition, at least in some cases, can be transplanted prior to conversion to AML.
Acknowledgments
We would like to acknowledge Du H. Lam and Linda Lowe for technical assistance, and W. Michael Kuehl, R. Keith Humphries, J.P. Issa, Eli Estey, as well as past and present members of our lab group for helpful discussions and encouragement. This research was supported by the Intramural Research Program of the NIH, NCI.
References
- 1.Mufti G, List AF, Gore SD, Ho AY. Myelodysplastic syndrome. Hematology (Am Soc Hematol Educ Program) 2003:176–99. doi: 10.1182/asheducation-2003.1.176. [DOI] [PubMed] [Google Scholar]
- 2.Aul C, Germing U, Gattermann N, Minning H. Increasing incidence of myelodysplastic syndromes: real or fictitious? Leuk Res. 1998;22:93–100. doi: 10.1016/s0145-2126(97)00089-1. [DOI] [PubMed] [Google Scholar]
- 3.Kurzrock R. Myelodysplastic syndrome overview. Semin Hematol. 2002;39:18–25. doi: 10.1053/shem.2002.35981. [DOI] [PubMed] [Google Scholar]
- 4.Steensma DP, Tefferi A. The myelodysplastic syndrome(s): a perspective and review highlighting current controversies. Leuk Res. 2003;27:95–120. doi: 10.1016/s0145-2126(02)00098-x. [DOI] [PubMed] [Google Scholar]
- 5.Slape C, Aplan PD. The role of NUP98 gene fusions in hematologic malignancy. Leuk Lymphoma. 2004;45:1341–50. doi: 10.1080/10428190310001659325. [DOI] [PubMed] [Google Scholar]
- 6.Radu A, Moore MS, Blobel G. The peptide repeat domain of nucleoporin Nup98 functions as a docking site in transport across the nuclear pore complex. Cell. 1995;81:215–22. doi: 10.1016/0092-8674(95)90331-3. [DOI] [PubMed] [Google Scholar]
- 7.Powers MA, Forbes DJ, Dahlberg JE, Lund E. The vertebrate GLFG nucleoporin, Nup98, is an essential component of multiple RNA export pathways. Journal of Cell Biology. 1997;136:241–50. doi: 10.1083/jcb.136.2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahuja HG, Felix CA, Aplan PD. Potential role for DNA topoisomerase II poisons in the generation of t(11;20)(p15;q11) translocations. Genes Chromosomes Cancer. 2000;29:96–105. doi: 10.1002/1098-2264(2000)9999:9999<::aid-gcc1013>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 9.Slape CI, Tessarollo L, Aplan PD. Generation of immortal, IL3-dependent cell lines from embryonic stem (ES) cells expressing a NUP98-HOXD13 fusion gene. AACR Meeting Abstracts. 2006;2006:61. [Google Scholar]
- 10.Huber TL, Kouskoff V, Fehling HJ, Palis J, Keller G. Haemangioblast commitment is initiated in the primitive streak of the mouse embryo. Nature. 2004;432:625–30. doi: 10.1038/nature03122. [DOI] [PubMed] [Google Scholar]
- 11.Ogilvy S, Metcalf D, Gibson L, Bath ML, Harris AW, Adams JM. Promoter elements of vav drive transgene expression in vivo throughout the hematopoietic compartment. Blood. 1999;94:1855–63. [PubMed] [Google Scholar]
- 12.Lin YW, Slape C, Zhang Z, Aplan PD. NUP98-HOXD13 transgenic mice develop a highly penetrant, severe myelodysplastic syndrome that progresses to acute leukemia. Blood. 2005;106:287–95. doi: 10.1182/blood-2004-12-4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sato N, Nakazato T, Kizaki M, Ikeda Y, Okamoto S. Transformation of myelodysplastic syndrome to acute lymphoblastic leukemia: a case report and review of the literature. Int J Hematol. 2004;79:147–51. doi: 10.1532/ijh97.03137. [DOI] [PubMed] [Google Scholar]
- 14.San Miguel JF, Hernandez JM, Gonzalez-Sarmiento R, Gonzalez M, Sanchez I, Orfao A, et al. Acute leukemia after a primary myelodysplastic syndrome: immunophenotypic, genotypic, and clinical characteristics. Blood. 1991;78:768–74. [PubMed] [Google Scholar]
- 15.Hussey DJ, Nicola M, Moore S, Peters GB, Dobrovic A. The (4;11)(q21;p15) translocation fuses the NUP98 and RAP1GDS1 genes and is recurrent in T-cell acute lymphocytic leukemia. Blood. 1999;94:2072–9. [PubMed] [Google Scholar]
- 16.Lahortiga I, Vizmanos JL, Agirre X, Vazquez I, Cigudosa JC, Larrayoz MJ, et al. NUP98 is fused to adducin 3 in a patient with T-cell acute lymphoblastic leukemia and myeloid markers, with a new translocation t(10;11)(q25;p15) Cancer Res. 2003;63:3079–83. [PubMed] [Google Scholar]
- 17.Pineault N, Buske C, Feuring-Buske M, Abramovich C, Rosten P, Hogge DE, et al. Induction of acute myeloid leukemia in mice by the human leukemia-specific fusion gene NUP98-HOXD13 in concert with Meis1. Blood. 2003;101:4529–38. doi: 10.1182/blood-2002-08-2484. [DOI] [PubMed] [Google Scholar]
- 18.Gilliland DG, Tallman MS. Focus on acute leukemias. Cancer Cell. 2002;1:417–20. doi: 10.1016/s1535-6108(02)00081-8. [DOI] [PubMed] [Google Scholar]
- 19.Dash AB, Williams IR, Kutok JL, Tomasson MH, Anastasiadou E, Lindahl K, et al. A murine model of CML blast crisis induced by cooperation between BCR/ABL and NUP98/HOXA9. Proc Natl Acad Sci U S A. 2002;99:7622–7. doi: 10.1073/pnas.102583199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahuja HG, Popplewell L, Tcheurekdjian L, Slovak ML. NUP98 gene rearrangements and the clonal evolution of chronic myelogenous leukemia. Genes Chromosomes Cancer. 2001;30:410–5. doi: 10.1002/1098-2264(2001)9999:9999<::aid-gcc1108>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 21.Plass C, Smiraglia DJ. Genome-wide analysis of DNA methylation changes in human malignancies. Curr Top Microbiol Immunol. 2006;310:179–98. doi: 10.1007/3-540-31181-5_9. [DOI] [PubMed] [Google Scholar]
- 22.Barrett MT, Scheffer A, Ben-Dor A, Sampas N, Lipson D, Kincaid R, et al. Comparative genomic hybridization using oligonucleotide microarrays and total genomic DNA. Proc Natl Acad Sci U S A. 2004;101:17765–70. doi: 10.1073/pnas.0407979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steube KG, Gignac SM, Hu ZB, Teepe D, Harms D, Kabisch H, et al. In vitro culture studies of childhood myelodysplastic syndrome: establishment of the cell line MUTZ-1. Leuk Lymphoma. 1997;25:345–63. doi: 10.3109/10428199709114174. [DOI] [PubMed] [Google Scholar]