

Abstract
The scope of heterocycle ortho-alkylation has been dramatically expanded to include pharmaceutically important pyridines and quinolines, which contain only a single nitrogen. The reactions, which are conducted at a high concentration (0.8 M), can be performed with catalyst loadings as low as 1% Rh. Substitution ortho to the heterocycle ring nitrogen is required for efficient alkylation and is consistent with the intermediacy of a Rh-carbene intermediate similar to those proposed in our earlier work.
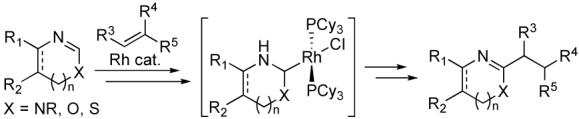
Elaboration of heterocycles through the application of carbon-carbon bond forming reactions to simple heterocyclic cores via C-H bond activation constitutes a powerful approach for the preparation of functional molecules ranging from ligands for biomolecular targets to electroactive materials.1 Our group developed a general method for the ortho-alkylation of nitrogen-containing heterocycles with olefins using a Rh(I)-phosphine catalyst2 and gathered extensive evidence supporting the intermediacy of substrate-based N-heterocyclic carbene (NHC) complexes (eq 1).3
 |
(1) |
Consistent with this mechanism, the catalytic alkylation reaction to date has been reported only for heterocycles with two heteroatoms adjacent to and thereby capable of stabilizing the carbene center of the proposed intermediates.4 Herein, we report dramatic expansion of substrate scope for this reaction by demonstrating the catalytic alkylation of heterocycles containing a single nitrogen, specifically pyridines and quinolines, which are extensively used in the pharmaceutical industry.5 The alkylation of these electron deficient heterocycles marks a significant departure from other direct functionalization methods,6 which typically require directing groups7 or electron rich (hetero)arenes and proceed via electrophilic metallation.8
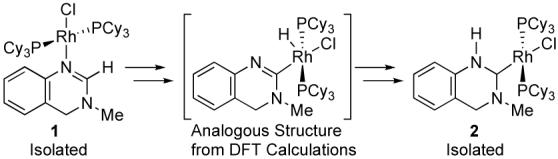
We recently conducted a detailed kinetic analysis of the conversion of heterocycle complex 1 to NHC complex 2 and utilized the acquired data, along with DFT calculations, to propose a reaction coordinate (eq 2).9 This work provided strong evidence that coordination of the heterocycle to the catalytically active RhCl(PCy3)2 fragment precedes an intramolecular C-H activation step, which provides a Rh-H intermediate that ultimately tautomerizes to the observed carbene complex.
 |
(2) |
The Carmona and Esteruelas groups have since reported the synthesis of (2-substituted)-pyridine- and quinoline-based Os, Ru, and Ir-NHC complexes directly from the corresponding heterocycles and a late transition metal complex.10 The authors propose mechanisms analogous to that shown in eq 2, with substitution ortho to the heterocycle ring nitrogen being necessary to drive the equilibrium from an N-bound to the desired NHC complexes. We therefore sought to determine whether our Rh/PCy3 catalyst system could be used to not only activate but also alkylate these heterocycles, presumably via an NHC intermediate.11
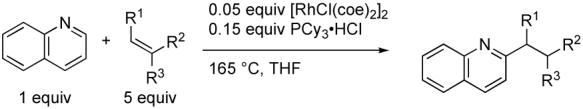
Our investigation commenced with an examination of catalysts and additives to affect the coupling of 2-methylpyridine and 3,3-dimethylbutene (eq 3, Table 1).12 No conversion was observed when no additive or Lewis acids such as MgBr2 were used. However, we were pleased to find that the use of the Rh/PCy3 catalyst system in combination with a Brϕnsted acid provided the desired alkylated product, 3. PCy3·HCl was found to be the optimal acid additive as was previously observed for azoles.2b Notably, increasing the substrate concentration to 0.8 M greatly improved the yield of 3 to 64%.
 |
(3) |
Table 1.
Alkylation of 2-methylpyridinea
| Entry | Phosphine | Additive | Conc. (M)b | 1 (%)c |
|---|---|---|---|---|
| 1 | PCy3 | None | 0.1 | 0 |
| 2 | PCy3 | MgBr2 | 0.1 | 0 |
| 3 | PCy3 | LutBr | 0.1 | 16 |
| 4 | PCy3 | LutCl | 0.1 | 20 |
| 5 | PCy3·HCl | None | 0.1 | 17 |
| 6 | PCy3·HCl | None | 0.4 | 42 |
| 7 | PCy3·HCl | None | 0.8 | 64 |
coe = cis-cyclooctene; Lut = lutidinium.
Concentration of heterocycle in total reaction volume.
Determined by 1H NMR spectroscopy relative to internal standard.
The scope of heterocycles compatible with the optimized alkylation conditions was next investigated (eq 4, Table 2). Increasing the bulk of the group located ortho to the pyridine ring nitrogen from methyl to isopropyl led to an increase in both alkylation rate and isolated yield of alkylated product 4. o-Triisopropylsilyl (TIPS) substituted pyridine was also an effective substrate (entry 3). This has significant synthetic utility because the silyl group can be removed, enabling additional transformations (vide infra). Consistent with the findings of Carmona and Esteruelas for carbene formation,10 pyridine itself was alkylated in less than 5% yield when heated in the presence of excess olefin and catalyst.
Table 2.
Investigation of Heterocycle Scope
| Entry | Heterocycle | Time (h) | # | Yield (%)a |
|---|---|---|---|---|
| 1 | 14 | 3 | 59 | |
| 2 | 14 | 4 | 83 | |
| 3 | 24 | 5 | 64 | |
| 4 | 14 | 6 | 96 | |
| 5 | 9.5 | 7 | 98 | |
| 6 | 7 | 8 | 96 |
Isolated yield of pure product.
A variety of quinolines were also alkylated under the reaction conditions. Parent quinoline provided nearly quantitative conversion to the corresponding alkylated quinoline (entry 5). Both ether and ester substitution were tolerated in the quinoline 6-position (entries 4 and 6). On the other hand, isoquinoline was not alkylated, which again supports the fact that ortho substitution, not simply the differing electronics of the benzo-fused heterocycle, is responsible for alkylation.
 |
(4) |
We next investigated the scope of olefins compatible with the reaction conditions (eq 5, Table 3). The isomerizable olefin, n-hexene, coupled to quinoline to provide quantitative conversion to the alkylated quinoline (entry 2). An 80:14 mixture of linear to branched isomers was observed which, in addition to providing a synthetically useful yield of the linear isomer, also indicated the feasibility of using disubstituted olefins as coupling partners. Indeed, cyclohexene could be used to alkylate quinoline in extremely high yield (entry 3). 1,1-Disubstituted olefins, including 2-methylpropene and camphene, were also effective coupling partners (entries 4 and 5). In a very preliminary investigation of functional group tolerance, both esters and phthalimides were found to be compatible with the reaction conditions (entries 6-8); however, styrene was not.
 |
(5) |
Table 3.
Investigation of Olefin Scopea
| Entry | Olefin | Time (h) | # | Yield (%)b |
|---|---|---|---|---|
| 1 | 9.5 | 7 | 98 | |
| 2 | 9.5 |
9 10 |
80 (linear) 14 (branched) |
|
| 3 |  |
9.5 | 11 | 96 |
| 4 | 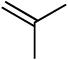 |
19 | 12 | 91 |
| 5 | 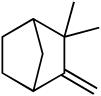 |
19 | 13 | 90c |
| 6 |  |
3.5 | 14 | 53 |
| 7 | 16 | 15 | 53d | |
| 8 | 14 | 16 | 57 |
Unless specified, only the linear isomer was observed in cases where linear and branched products were possible.
Isolated yield of pure product.
c.a. 2:1 mixture of diastereomers.
0.1 equiv [RhCl(coe)2]2, 0.3 equiv PCy3·1HCl used.
While substitution ortho to the pyridine nitrogen was required to obtain high yields of alkylated products, an orthosilyl group serves as a suitable blocking group that can readily be removed to provide mono-alkylated pyridines. For example, treatment of 5 with aqueous HF in refluxing THF provided the mono-alkylated pyridine product 17 in good yield (eq 6).
 |
(6) |
We were also able to substantially reduce the catalyst loading required to affect the alkylation reaction. Specifically, quinoline was alkylated with 3,3-dimethylbutene in 91% yield using only 1% of the Rh catalyst (eq 7).
 |
(7) |
In summary, we have developed a method for the Rh(I)-catalyzed alkylation of pyridines and quinolines. Consistent with the work of Carmona and Esteruelas, steric interactions provided by the ortho-substituent presumably increase the equilibrium from an N-bound to a C-bound Rh complex. We are currently investigating this hypothesis by undertaking efforts to isolate intermediate complexes and by performing DFT calculations on model structures. Continued expansion of the catalytic alkylation process to new classes of heterocycle and alkene inputs are also in progress.
Supplementary Material
Acknowledgement
This work was supported by the NIH GM069559 to J.A.E. and by the Director and Office of Energy Research, Office of Basic Energy Sciences, Chemical Sciences Division, U.S. Department of Energy, under Contract DEAC03-76SF00098 to R.G.B.
References
- (1)(a).Dyker G. Angew. Chem. Int. Ed. 1999;38:1698. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1698::AID-ANIE1698>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]; (b) Kakiuchi F, Murai S. Top. Curr. Chem. 1999;3:47. [Google Scholar]; (c) Miura M, Nomura M. Top. Curr. Chem. 2002;219:212. [Google Scholar]; (d) Kakiuchi F, Chatani N. Adv. Synth. Catal. 2003;345:1077. [Google Scholar]; (e) Campeau L-C, Fagnou K. Chem. Commun. 2006:1253. doi: 10.1039/b515481m. [DOI] [PubMed] [Google Scholar]; (f) Daugulis O, Zaitsev VG, Shabashov D, Pham Q-N, Lazareva A. Syn. Lett. 2006;20:3382. [Google Scholar]
- (2)(a).Tan KL, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2001;123:2685. doi: 10.1021/ja0058738. [DOI] [PubMed] [Google Scholar]; (b) Tan KL, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2002;124:13964. doi: 10.1021/ja0281129. [DOI] [PubMed] [Google Scholar]; (c) Tan KL, Park S, Ellman JA, Bergman RG. J. Org. Chem. 2004;69:7329. doi: 10.1021/jo048666p. [DOI] [PubMed] [Google Scholar]; (d) Wiedemann SH, Bergman RG, Ellman JA. Org. Lett. 2004;6:1685. doi: 10.1021/ol049417q. [DOI] [PubMed] [Google Scholar]; (e) Wiedemann SH, Ellman JA, Bergman RG. J. Org. Chem. 2006;71:1969. doi: 10.1021/jo052345b. [DOI] [PubMed] [Google Scholar]
- (3)(a).Tan KL, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2002;124:3202. doi: 10.1021/ja017351d. [DOI] [PubMed] [Google Scholar]; (b) Lewis JC, Wiedemann SH, Bergman RG, Ellman JA. Org. Lett. 2004;6:35. doi: 10.1021/ol035985e. [DOI] [PubMed] [Google Scholar]
- (4).Herrmann WA, Köcher C. Angew. Chem. Int. Ed. Engl. 1997;36:1047. [Google Scholar]
- (5).Carey JS, Laffan D, Thomson C, Williams MT. Org. Biomol. Chem. 2006;4:2337. doi: 10.1039/b602413k. [DOI] [PubMed] [Google Scholar]
- (6)(a).Ru-catalyzed heterocycle acylation is an exception.Moore EJ, Pretzer WR, O’Connel TJ, Harris J, LaBounty L, Cou L, Grimmer SS. J. Am. Chem. Soc. 1992;114:5888.Chatani N, Fukuyama T, Kakiuchi F, Murai S. J. Am. Chem. Soc. 1996;118:493.
- (7).Kakiuchi F, Murai S. Acc. Chem. Res. 2002;35:826. doi: 10.1021/ar960318p. [DOI] [PubMed] [Google Scholar]
- (8)(a).Jia C, Kitamura T, Fujiwara Y. Acc. Chem. Res. 2001;34:633. doi: 10.1021/ar000209h. [DOI] [PubMed] [Google Scholar]; (b) Tunge JA, Foresee LN. Organometallics. 2005;24:6440. [Google Scholar]; (c) Lane BS, Brown MA, Sames D. J. Am. Chem. Soc. 2005;127:8050. doi: 10.1021/ja043273t. [DOI] [PubMed] [Google Scholar]
- (9).Wiedemann SH, Lewis JC, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2006;128:2452. doi: 10.1021/ja0576684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10)(a).Alvarez E, Conejero S, Paneque M, Petronilho A, Poveda ML, Serrano O, Carmona E. J. Am. Chem. Soc. 2006;128:13060. doi: 10.1021/ja0646592. [DOI] [PubMed] [Google Scholar]; (b) Esteruelas MA, Fernandez-Alvarez FJ, Onate E. J. Am. Chem. Soc. 2006;128:13044. doi: 10.1021/ja064979l. [DOI] [PubMed] [Google Scholar]
- (11).Jordan reported the Zr-catalyzed alkylation of 2-picoline using simple olefins. In all cases, the branched isomer of the product was favored.Jordan RF, Taylor DF. J. Am. Chem. Soc. 1989;111:778.Rodewald S, Jordan RF. J. Am. Chem. Soc. 1994;116:4491.Murakami reported the Ru-catalyzed alkenylation of pyridine using alkynyl silanes.Murakami M, Hori S. J. Am. Chem. Soc. 2003;125:4720. doi: 10.1021/ja029829z.
- (12).Reactions were conducted in sealed tubes fitted with Kontes stoppers and heated in an oil bath.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.