

Abstract
Small molecules that can be used to specifically control the activity of cellular gene products are key tools in chemical biology. Here, we present a scanning-insertional-mutagenesis approach to identify protein tyrosine phosphatase (PTP) mutants whose activities can be selectively modulated by a small molecule. A six-amino-acid, cysteine-rich ligand-binding peptide, known to specifically bind a biarsenical fluorescein derivative (FlAsH), was inserted site-specifically at twelve locations in the catalytic domain of T-cell PTP (TCPTP). The majority of the resulting insertion mutants expressed efficiently as soluble enzymes and retained catalytic activities comparable to wild-type levels in the absence of the FlAsH ligand. Mutants that contain TetraCys insertions at TCPTP positions 79 and 187 demonstrated novel FlAsH-dependent inhibitor sensitivity: the catalytic activities of these constructs strongly decreased when incubated with FlAsH, compared to a no-FlAsH control. Structural analysis suggests that the inhibitory TetraCys-FlAsH complexes are acting allosterically, as positions 79 and 187 are both located in loops, distal from TCPTP’s active site. These results show that insertion of TetraCys and, potentially, other ligand-biding peptides can be used to readily engineer novel allosteric sites into PTPs, a critical family of cell-signaling enzymes.
Small molecules that can specifically control the activity of cellular gene products are key tools in chemical biology. However, protein-specific small molecules are known for only a small fraction of the proteome. Significant increases in the number of “targetable” proteins will require the systematic development of chemical tools for targeting members of large protein families (e.g., G-protein-coupled receptors, proteases, kinases, phosphatases). We have recently described a method for engineering novel inhibitor sensitivity into protein tyrosine phosphatase (PTP) catalytic sites.1 Active-site-directed PTP inhibitor discovery, however, is somewhat limited by the relatively shallow PTP active site (6–9 Å) and the low cellular permeability of many known competitive PTP inhibitors.2
A potential alternative to such complications lies in the identification, or engineering, of PTP allosteric sites.3 Two naturally occurring allosteric-inhibition sites have been identified in the most thoroughly studied classical PTP, PTP1B.4 However, the small molecules known to target these wild-type allosteric sites, like active-site-directed inhibitors, suffer from a lack of specificity; and unique allosteric sites are not known for other PTPs. Here, we use scanning-insertional mutagenesis to engineer PTP mutants whose activities can be allosterically modulated by the cell-permeable small molecule fluorescein arsenical hairpin binder (FlAsH, Figure 1A), which is known to bind specifically to cysteine-rich peptides.5 While FlAsH was developed as a protein-visualization tool (FlAsH fluorescence increases dramatically upon peptide binding5), we co-opted the ligand as a potential “chemical switch” to control enzyme activity. Specifically, a FlAsH-binding peptide (TetraCys: Cys-Cys-Pro-Gly-Cys-Cys) was inserted at site-directed positions in the catalytic domain of a PTP that is essential for mammalian viability, T-cell PTP (TCPTP).6 Mutants harboring the TetraCys insertion can putatively bind FlAsH, which should have little to no affinity for wild-type TCPTP or other cellular proteins.5 When TetraCys is embedded within the enzyme’s catalytic domain, FlAsH/TetraCys binding can potentially lead to conformational and/or activity changes unique to the mutant/FlAsH pair.
Figure 1.
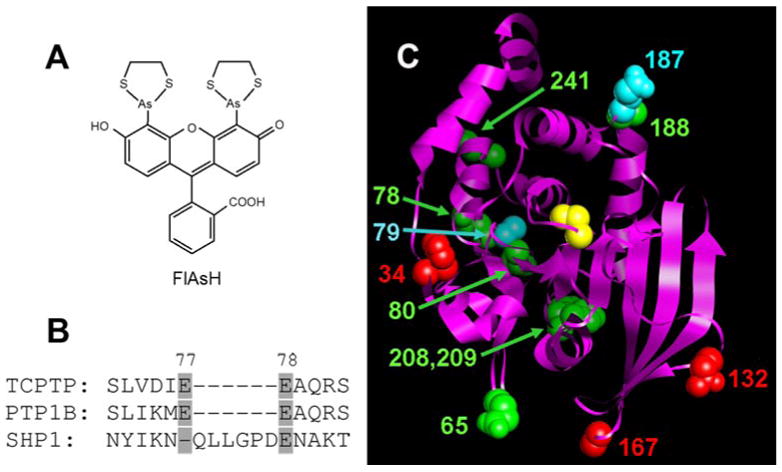
(A) Chemical structure of FlAsH. (B) Representative example of the strategy used to choose TetraCys-insertion sites in TCPTP: the SHP1 PTP domain contains a natural insertion between residues 77 and 78 that is not present in most other PTPs, suggesting the potential tolerance of this region for non-natural insertions. (C) Positions of TetraCys insertions modeled on to the TCPTP crystal structure (Residues 5-277; PDB code: 1L8K).10 The TCPTP catalytic domain is shown as a ribbon, with amino acids that correspond to the sites of insertions shown in space-filling representation and colored as follows: red for insertions that rendered TCPTP unstable or inactive; green for insertions associated with modest FlAsH sensitivity; and cyan for insertions that gave rise to strongly FlAsH-sensitive TCPTP activity. The position of the C-terminal insertion is not shown, as TCPTP’s C-terminal portion is not present in the only solved TCPTP structure.10 For perspective, TCPTP’s active-site catalytic cysteine (Cys216) is shown in yellow.
Many proteins have regions that can tolerate modest peptide insertions7; and insertional mutagenesis can be used to generate ligand-sensitive variants of a bacterial antibiotic-resistance enzyme, TEM-1.8 However, to our knowledge, no insertional mutagenesis on PTP catalytic domains has been reported. To identify potential sites in the PTP catalytic domain in which an insertion could be catalytically tolerated, we inspected a primary sequence alignment of all human classical PTPs.9 We avoided regions of conserved secondary-structural elements, focusing on loop regions that contain no highly conserved amino-acid residues. Furthermore, we targeted portions of the PTP domain in which nature allows for “insertions”—i.e., regions in which individual wild-type PTPs idiosyncratically contain peptides that are absent in most PTPs (Figure 1B). Based on our analysis, we identified eight regions of the TCPTP catalytic domain as candidate insertion sites, and we inserted TetraCys at twelve positions within these regions: at amino acid 34, 65, 78, 79, 80, 132, 167, 187, 188, 208, 209 and 241 (TCPTP-34, etc.; numbering is according to human TCPTP and corresponds to the position of the first cysteine in the TetraCys insert; Figure 1C). In addition, a mutant containing TetraCys at the C-terminal tail of TCPTP was prepared (TCPTP-CT).
A surprisingly large fraction of our insertion mutants expressed efficiently as soluble enzymes, showing that, given the constraints of our design, the PTP catalytic domain is amenable to peptide insertions. Ten out of twelve insertion mutants expressed at high levels (≥3 mg/L of culture) from E. coli and could be readily purified as His6-tagged proteins. (Only TCPTP-132 and TCPTP-167 failed to express.) To verify the incorporation of the TetraCys insertion, we incubated the TCPTP variants with FlAsH and measured the fluorescence intensities (510 nm excitation, 528 nm emission) of the resulting solutions. As expected, all of the purified mutants bound FlAsH, as demonstrated by increased fluorescence intensity, compared to a wild-type (WT) TCPTP control (Figure S1, Supplementary Information).
Insertion of the ligand-binding peptide had, for the most part, only small effects on TCPTP’s inherent catalytic activity when assayed in the absence of FlAsH. The majority of insertion mutants retained catalytic efficiencies [kcat/KM, measured with para-nitrophenyl phosphate (pNPP) as substrate] close to that of wild-type TCPTP (Table 1). Only TCPTP-34 expressed as a soluble enzyme but demonstrated severely compromised catalytic activity even in the absence of FlAsH (not shown). While the catalytic efficiencies of the active mutants vary from somewhat reduced to somewhat increased, all are within seven-fold of the wild-type value, well within the range of values that have been found to be useful in other ligand-sensitive-engineering approaches.11
Table 1.
Kinetic Constants of TCPTP Insertion Mutants
| Protein | kcat(s−1) | KM for pNPP(mM) | kcat/KM(s−1mM−1) |
|---|---|---|---|
| WT TCPTP | 3.9±0.20 | 2.3±0.21 | 1.7 |
| TCPTP-65 | 2.8±0.13 | 2.9±0.16 | 1.0 |
| TCPTP-78 | 1.5±0.048 | 1.8±0.058 | 0.84 |
| TCPTP-79 | 5.0±0.043 | 1.2±0.046 | 4.1 |
| TCPTP-80 | 1.1±0.050 | 2.0±0.24 | 0.56 |
| TCPTP-187 | 0.21±0.0084 | 0.91±0.059 | 0.23 |
| TCPTP-188 | 0.25±0.012 | 1.1±0.12 | 0.23 |
| TCPTP-208 | 1.3±0.032 | 3.5±0.14 | 0.38 |
| TCPTP-209 | 0.41±0.017 | 1.8±0.13 | 0.23 |
| TCPTP-241 | 2.1±0.15 | 2.2±0.29 | 1.0 |
| TCPTP-CT | 1.2±0.034 | 2.2±0.084 | 0.54 |
When incubated with FlAsH, wild-type-TCPTP activity was unaffected, whereas the catalytic efficiencies of all nine active TetraCys insertion mutants and TCPTP-CT were reduced significantly (Figure 2; also, Table S1, Supplementary Information). In particular, the two TCPTP mutants that harbor the TetraCys insertions at positions 79 and 187 demonstrated specific FlAsH inhibition that was substantially more pronounced than in the other mutants (Figure 2A). The catalytic efficiency of TCPTP-79, which retains full activity in the absence of FlAsH, drops three-fold in the presence of the ligand (kcat/KM = 4.1 and 1.3 s−1mM−1, respectively; Figure 2C). With TCPTP-187, the FlAsH-induced inhibition is almost complete: the catalytic efficiency drops twelve-fold in the presence of FlAsH, from 0.23 to 0.019 s−1mM−1 (Figure 2D). This drop comprises predominately a drop in kcat (6-fold: 0.21 to 0.034 s−1), coupled with a modest rise in KM for pNPP (0.91 to 1.8 mM). The majority of mutants (TCPTP-65, -78, -80, -188, -208, -209, and -241) showed FlAsH-dependent inhibition that was at a level comparable to that of the C-terminal mutant, TCPTP-CT (Figure 2A). These results suggest that the simple, structure-independent binding of FlAsH to the PTP can modestly reduce enzymatic activity, whereas the more pronounced FlAsH-mediated inhibition is structure-specific. This is most starkly shown in the contrasting FlAsH responses of TCPTP-187 and TCPTP-188, which differ only by a one-amino-acid shift in the placement of TetraCys (Figure 2A).
Figure 2.
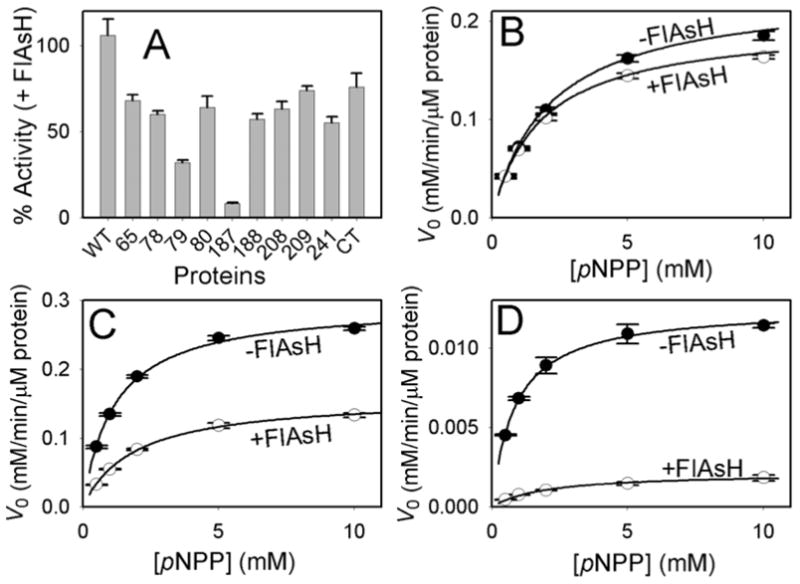
FlAsH-dependent inhibition of TCPTP insertion mutants. (A) The indicated TCPTP enzymes (2.5 μM) were incubated in the absence or presence of FlAsH (10 μM), diluted and assayed for activity with the artificial PTP substrate para-nitrophenyl phosphate (pNPP) at pH 7.0. “% Activity” represents the PTP catalytic efficiency (kcat/KM) in the presence of FlAsH divided by the control (no-FlAsH) catalytic efficiency of the same TCPTP enzyme. (B-D) Allosteric inhibition of TCPTP-79 and TCPTP-187 by FlAsH. The initial rates of wild-type TCPTP (B), TCPTP-79 (C), and TCPTP-187 (D) were measured at the indicated pNPP concentrations in the absence and presence of FlAsH as described in A.
Inspection of the known TCPTP crystal structure10 suggests that FlAsH-induced inhibition of TCPTP-79 and TCPTP-187 is allosteric, due either to a FlAsH-induced conformational change or, possibly, inhibition of a conformational change that is necessary for activity. Remarkably, the insertional position in TCPTP-79 (Ala79) falls in a loop region that is more than 20 Å removed from the TCPTP’s catalytic cysteine (Cys216) in the wild-type enzyme; it is thus difficult to hypothesize a specific mode of action for TCPTP-79 inhibition. Although the 187 insertion position (Glu187) is also distal from Cys216 (17 Å at the closest atom-to-atom distance), the allosteric mechanism of TCPTP-187 is easier to posit: Glu187 lies at the end of the conserved PTP-activation loop (“WPD loop”). Binding of FlAsH to TCPTP-187 may impede proper closure of the activation loop, in a manner that is consistent with inhibitors that target a natural allosteric site of PTP1B.4
One drawback of our decidedly “non-rational” mutagenic approach is a relatively low hit rate: the majority of mutants showed only weak ligand sensitivity. However, our TCPTP findings will guide a more directed extension of allosteric-site engineering to other PTPs. No criteria in our insertion-site selection were specific to the sequence of TCPTP: the vast majority of classical PTPs lack the idiosyncratic natural inserts that were used as guides for our mutagenesis (35 of 37 human PTPs for TCPTP-79, and 34 of 37 for TCPTP-187). Insertion of TetraCys, or other ligand-biding peptides, may therefore represent a general strategy for engineering allosteric-control elements in PTPs, facilitating the chemical-genetic analysis of a critical family of cell-signaling enzymes in cells and model organisms.
Supplementary Material
Supplementary Figure S1 and Table S1, as well as complete experimental and synthetic protocols.
Acknowledgments
This research was supported by the National Institutes of Health (1 R15 GM071388-01A1), Research Corporation (CC6372), and Amherst College. The authors thank Anna Marie Lone for early expression work on the TCPTP mutants, Prof. Patricia B. O’Hara and her laboratory for help with fluorescence experiments, and Prof. David E. Hansen for helpful comments on the manuscript.
References
- 1.(a) Hoffman HE, Blair ER, Johndrow JE, Bishop AC. J Am Chem Soc. 2005;127:2824–2825. doi: 10.1021/ja043378w. [DOI] [PubMed] [Google Scholar]; (b) Blair ER, Hoffman HE, Bishop AC. Bioorg Med Chem. 2006;14:464–471. doi: 10.1016/j.bmc.2005.08.025. [DOI] [PubMed] [Google Scholar]; (c) Bishop AC, Blair ER. Bioorg Med Chem Lett. 2006;16:4002–4006. doi: 10.1016/j.bmcl.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 2.Bialy L, Waldmann H. Angew Chem Int Ed. 2005;44:3814–3839. doi: 10.1002/anie.200461517. [DOI] [PubMed] [Google Scholar]
- 3.Hardy JA, Wells JA. Curr Opin Struct Biol. 2004;14:706–715. doi: 10.1016/j.sbi.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 4.(a) Wiesmann C, Barr KJ, Kung J, Zhu J, Erlanson DA, Shen W, Fahr BJ, Zhong M, Taylor L, Randal M, McDowell RS, Hansen SK. Nat Struct Mol Biol. 2004;11:730–737. doi: 10.1038/nsmb803. [DOI] [PubMed] [Google Scholar]; (b) Hansen SK, Cancilla MT, Shiau TP, Kung J, Chen T, Erlanson DA. Biochemistry. 2005;44:7704–7712. doi: 10.1021/bi047417s. [DOI] [PubMed] [Google Scholar]
- 5.Griffin BA, Adams SR, Tsien RY. Science. 1998;281:269–272. doi: 10.1126/science.281.5374.269. [DOI] [PubMed] [Google Scholar]
- 6.Bourdeau A, Heinonen KM, Brunet DV, Tailor P, Lapp WS, Tremblay ML. Topics Curr Gen. 2004;5:185–200. [Google Scholar]
- 7.Hayes F, Hallet B. Trends Microbiol. 2000;8:571–577. doi: 10.1016/s0966-842x(00)01857-6. [DOI] [PubMed] [Google Scholar]
- 8.Erster O, Penso J, Liscovitch M. Mol Biol Cell (suppl) 2004;15:469A. [Google Scholar]
- 9.Andersen JN, Mortensen OH, Peters GH, Drake PG, Iversen LF, Olsen OH, Jansen PG, Andersen HS, Tonks NK, Moller NP. Mol Cell Biol. 2001;21:7117–7136. doi: 10.1128/MCB.21.21.7117-7136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iversen LF, Moller KB, Pedersen AK, Peters GH, Petersen AS, Andersen HS, Branner S, Mortensen SB, Moller NP. J Biol Chem. 2002;277:19982–19990. doi: 10.1074/jbc.M200567200. [DOI] [PubMed] [Google Scholar]
- 11.Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, Wood JL, Morgan DO, Shokat KM. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1 and Table S1, as well as complete experimental and synthetic protocols.