

Abstract
Glutamate mutase is one of a group of adenosylcobalamin-dependent enzymes that catalyze unusual isomerizations that proceed through organic radical intermediates generated by homolytic fission of coenzyme's unique cobalt-carbon bond. These enzymes are part of a larger family of enzymes that catalyze radical chemistry in which a key step is the abstraction of a hydrogen atom from an otherwise inert substrate. To gain insight into the mechanism of hydrogen transfer we previously used pre-steady state, rapid quench techniques to measure the α-secondary tritium kinetic and equilibrium isotope effects associated with the formation of 5’-deoxyadenosine when glutamate mutase was reacted with [5’-3H]-adenosylcobalamin and L-glutamate. We showed that both the kinetic and equilibrium isotope effects are large and inverse, 0.76 and 0.72 respectively. We have now repeated these measurements using glutamate deuterated in the position of hydrogen abstraction. The effect of introducing a primary deuterium kinetic isotope effect on the hydrogen transfer step is to reduce the magnitude of the secondary kinetic isotope effect to a value close to unity, 1.05 ± 0.08, whereas the equilibrium isotope effect is unchanged. The significant reduction in the secondary kinetic isotope effect is consistent with motions of the 5’-hydrogen atoms being coupled in the transition state to the motion of the hydrogen undergoing transfer, in a reaction that involves a large degree of quantum tunneling.
Keywords: enzyme, coenzyme-B12, protein-radical, isomerization, transition state, secondary isotope effects
Glutamate mutase is one of a group of adenosylcobalamin1 (AdoCbl, coenzyme B12) dependent enzymes that catalyze unusual carbon skeleton isomerisations. These rearrangements formally involve a 1,2 hydrogen atom migration and proceed through a mechanism involving carbon-based free radical intermediates (1-6). The initial steps of these reactions involve homolysis of the reactive cobalt-carbon bond of the coenzyme to form cob(II)alamin and 5’-deoxyadenosyl radical. The adenosyl radical then abstracts the migrating hydrogen from the substrate to form 5-deoxyadenosine and substrate radical (or protein radical in the case of AdoCbl-dependent ribonucleotide reductase). These steps have been studied in some detail for several B12 enzymes and in each case homolysis and hydrogen abstraction are found to be kinetically coupled (7-10), as evidenced by the appearance of a kinetic isotope effect on cobalt-carbon bond homolysis when the enzymes are reacted with deuterated substrates.
This observation implies that adenosyl radical can only be present in very low concentrations as a high energy intermediate that does not accumulate on the enzyme. Arguments have been advanced for a formally concerted mechanism for homolysis and hydrogen abstraction (11), although this is now considered to be less likely (12). In many cases these deuterium isotope effects are unusually large, in the range of 40−30, and the most likely explanation for this is that hydrogen transfer involves a large degree of quantum tunneling (13, 14).
We have recently described rapid quench experiments (15) to measure the α-secondary tritium isotope effects associated with the formation of 5’-deoxyadensosine under pre-steady state conditions, as illustrated in Figure 1. AdoCbl, tritium-labeled in the 5’-position, was used to measure both the secondary equilibrium and kinetic isotope effects on the formation of 5’-dA. The measurements were made at very short time intervals between 10 and 100 ms so that loss of tritium from the 5’-position was minimal. Both the kinetic and equilibrium isotope effects were found to be large and inverse, kH/kT = 0.76 ± 0.02, KH/KT = 0.72 ± 0.04. These results indicate that the 5’-C-H bonds become significantly stiffer in going from AdoCbl to 5’-dA, even though the 5’-carbon remains formally sp3 hybridized, and are consistent with the hydrogen transfer, as opposed cobalt-carbon bond cleavage, being the slower step. Classically, the large inverse kinetic isotope effect would be interpreted as indicating a late transition state. However, if quantum tunneling is occurring in the hydrogen transfer step, this conclusion cannot be drawn (16).
Figure 1.

Top: Isomerization reaction catalyzed by glutamate mutase, with the migrating hydrogen atom circled. Bottom: Secondary isotope effects associated with the homolysis of AdoCbl and the formation 5’-dA when hologlutamate mutase is reacted with deuterated L-glutamate.
To investigate whether quantum tunneling is likely to be important in the mechanism of glutamate mutase, we have repeated the secondary α-tritium isotope effect measurements for the formation of 5’-dA using deuterated glutamate to trigger homolysis of AdoCbl. This introduces a primary deuterium isotope effect at the 5’-carbon, which should not change the secondary isotope effect if this arises purely semi-classically (16). However, we observe that the secondary kinetic isotope effect is significantly reduced in magnitude, which implies that the motions of the primary and secondary hydrogen atoms are coupled together in the transition state and that the reaction involves a significant degree of quantum tunneling.
EXPERIMENTAL PROCEDURES
Materials
The purification from recombinant E. coli of the engineered single subunit glutamate mutase protein, GlmES, which has been used in this and previous mechanistic studies, has been described previously (17). L-[G,-3H]–glutamate, 250 Ci/mol, was purchased from Amersham Pharmacia Biotech and [8-14C]-adenosine 50 Ci/mol was purchased from American Radiochemicals. (2,3,3,4,4 D5)-L-glutamic acid was purchased from Cambridge Isotope Laboratories, Inc., AdoCbl was purchased from Sigma Chemical Company.
Synthesis of [5’-3H]-AdoCbl and [8-14C]-AdoCbl
[5’-3H]-AdoCbl was synthesized enzymically by exchange of tritium from L-[G,-3H]–glutamate to unlabeled AdoCbl, and purified by reverse phase HPLC as described previously (18). [8-14C]-adenosylcobalamin was synthesized using established protocols described by Brown et. al, (19) from [8-14C]-adenosine that was purchased from Amersham; the radiolabelled material was first converted to 5’-chloroadenosine that was subsequently used to alkylate Cob(I)alamin generated by in situ reduction of hydroxocobalamin with zinc. The resulting [8-14C]-AdoCbl was purified by reverse phase HPLC.
Rapid quench flow
Experiments were performed at 10 °C using a HiTech RQF-63 apparatus. 80 μL of a solution containing 100 μM glutamate mutase, 120 μM [5’-3H, 8-14C]-AdoCbl (specific activity of each isotope ∼ 3,500 dpm/nmol) and 20 μM L-tryptophan, as an internal standard, in 50 mM potassium phosphate buffer, pH 7.0, was rapidly mixed with an equal volume of 20 mM L-glutamate in the same buffer. After various times reactions were quenched with 80 μL 5 % trifluoroacetic acid (TFA). 5’-dA was recovered by reverse phase HPLC (20), and the 14C:3H ratio of the 5’-dA determined by dual label scintillation counting. Isotope effects were calculated by comparing the ratio of 14C:3H in the recovered 5’-dA with that of the starting AdoCbl. The progress of the reaction with time was followed by monitoring the 14C content of the 5’-dA; the radioactivity of individual samples was normalized by reference to the tryptophan internal standard.
RESULTS
These experiments were performed under the same conditions as our previously published measurement of secondary isotope effects associated with the formation of 5’-dA using unlabeled glutamate as substrate (15). Holo-glutamate mutase was reconstituted with 14C,3H-labeled AdoCbl, and reacted with a saturating concentration (10 mM after mixing) of d5-L-glutamate at 10 °C. Under these conditions substrate binding is rapid and the rate of 5’-dA formation is independent of substrate concentration (7, 20). The reaction was allowed to proceed for various times between 7 and 80 ms, after which time the concentration of 5’-dA had essentially reached internal equilibrium on the enzyme. Previous work from our laboratory (21) has demonstrated that at times shorter than 80 ms exchange of hydrogen between 5’-dA and substrate, as evidenced by the formation of doubly-deuterated 5’-dA is minimal.
The time course for formation of H,H,D5’-dA and H,D,T5’-dA (the superscripts denote which hydrogen isotopes are present at the 5’-methyl group) were described by single exponential functions, as shown in Figure 2A. The data shown are from one experiment and represent the average of at least 3 independent measurements at each time point that were all made with the same batch of enzyme. Similar data were obtained from experiments on another batch of enzyme. The apparent first order rate constants for the formation of H,H,D5’-dA, and H,D,T5’-dA were determined as k(H,H,D-5’-dA) = 40 ± 8 s−1 and k(H,D,T-5’-dA) = 60 ± 10 s−1 respectively.
Figure 2.
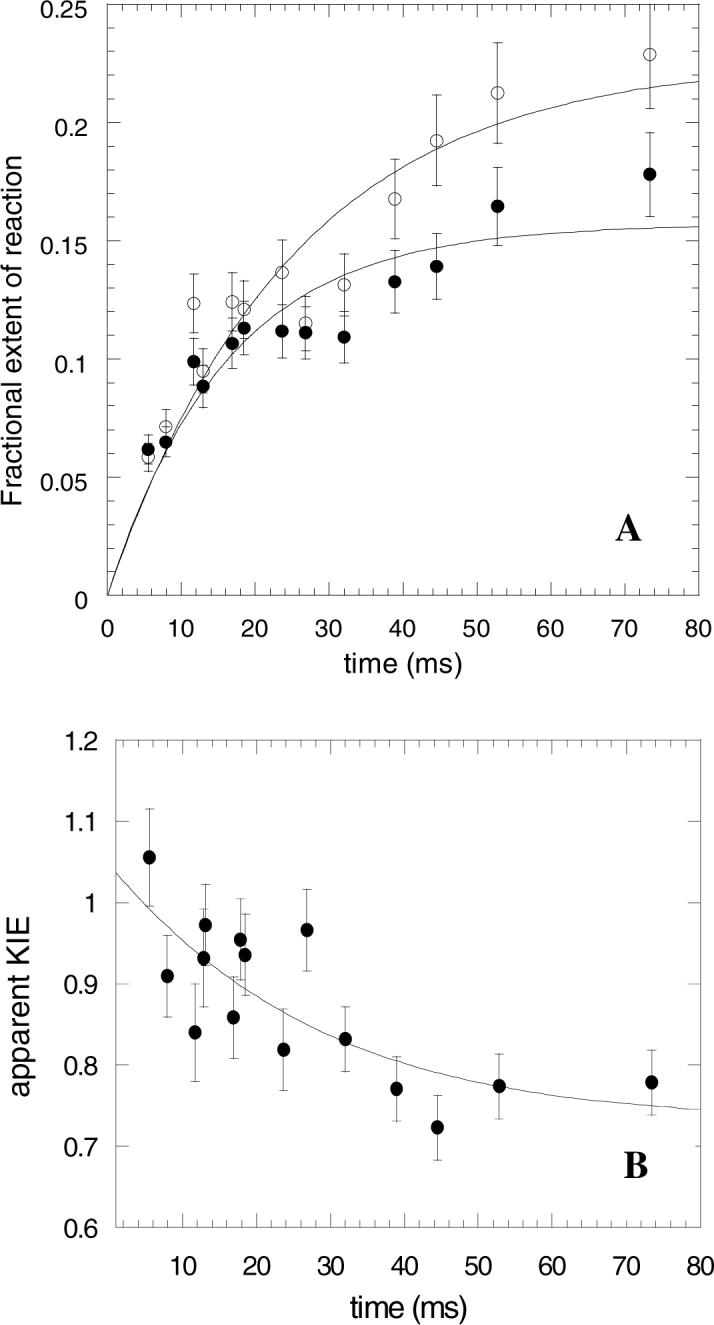
A Kinetics of 5’-dA formation (approach to internal equilibrium) for holo-glutamate mutase reacting with d5-L-glutamate (10 mM). (•) Formation of [5’-H]-dA, as determined by 14C counts; (o) formation of [5’-3H]-dA, as determined by tritium counts. Each time point represents an average of three independent measurements. B Variation of the apparent α-secondary tritium kinetic isotope effect on 5’-dA formation as a function of time, showing the fit of the data to Equation 2..
Previously, when we measured the apparent rate constant for the formation of 5’-dA with protium in the primary position, we found k(5’-dA) = 53 ± 5 s−1 (for non-tritiated 5’-dA) (15). Therefore, we might have expected to see a larger primary isotope effect on the apparent rate constant with deuterium in the primary position, as in other published experiments we have concluded that kH/kD for the reaction is about 2 (21). In this case, comparing k(5’-dA) with k(H,H,D-5’-dA) (no influence of tritium in the secondary position) gives a smaller primary isotope effect of about 1.5. However, this study was not designed to measure primary isotope effects and so this number must be treated with caution. The lower precision with which can measure k(H,H,D-5’-dA) leads to a large uncertainty in the primary KIE. The two sets of experiments were conducted at different times with different batches of enzyme, and small variations in experimental conditions may lead to significant systematic error. Furthermore, for our determination of the primary deuterium isotope effect (21) we used HPLC (rather than radioactivity) to quantify the amount of 5’-dA produced in the reaction, and this difference in experimental design could also introduce some systematic error.
Lastly, we note that we have recently measured the intrinsic primary deuterium isotope effect for transfer of hydrogen from methylaspartate to 5’-dA by an internal competition experiment; the isotope effect is about 4. (M. Yoon, K. Hakansson, E.N.G.M. unpublished data) Thus we are quite confident that the primary deuterium isotope effects are indeed much smaller than those we originally measured by stopped flow spectroscopy (7), for reasons we have discussed previously (21).
It is also important to note that it is the relative magnitudes of k(H,H,D-5’-dA) and k(H,D,T-5’-dA), rather than the absolute values that are important in determining the secondary isotope effects, as we discuss below. Also, as we have discussed previously (7, 15), because the reaction is reversible, the apparent rate constant for the formation of 5’-dA is a function of both the forward and reverse rate constants associated with the equilibrium between the E:AdoCbl:S and E:5’-dA:Cbl(II):S• complexes (species I and III in Figure 1), such that k(5’-dA) = kf + kr.
The apparent secondary isotope effect, KIEapp was calculated by comparing the ratio of 3H to 14C counts in 5’-dA with that of the starting AdoCbl. The isotope effect This is plotted as a function of time in Figure 2B. Much of the error present in the measurement of the apparent rate constants for H,H,D5’-dA, and H,D,T5’-dA formation cancel out in the calculation of KIEapp. Thus although there is some scatter in the data, it is clear that at shorter times the apparent isotope effect is diminished in magnitude, and tends towards unity, whereas at longer times it tends towards a value of ∼ 0.7. This behavior is in contrast to that observed when the experiment was conducted with unlabelled glutamate and the apparent isotope effect was essentially invariant with time (15).
Calculation of secondary isotope effects
As we have discussed previously, because these measurements are made under single turnover conditions they not rely on competition between labeled and unlabeled molecules for the isotope effect to be manifested (15). Instead, tritiated and unlabeled AdoCbl molecules are reacting in parallel reactions under identical conditions, and therefore the ratio of H,H,D[5’-dA]t to H,D,T[5’-dA]t as a function of time is described by Equation 1. (in which the subscripts attached to the rate constants denote the hydrogen isotopes present at the secondary position).
| (1) |
Equation 1 can be rewritten to describe how the apparent kinetic isotope effect, KIEapp, varies as a function of time, by rearranging it and replacing the terms (kf + kr) by the corresponding apparent rate constants for the formation of H,H,D5’-dA and H,D,T5’-dA, k(H,H,D-5’-dA) and k(H,D,T-5’-dA) respectively; this gives Equation 2.
| (2) |
The values for these apparent rate constants can be determined from the data in figure 2A, as discussed above. Extrapolation of the data to t = 0 gives the secondary kinetic isotope effect, kf (H)/kf (T), on 5’-dA formation, whereas extrapolation to t = ∞ gives the secondary equilibrium isotope effect, KH/KT, on 5’-dA formation.
To determine kf (H)/kf (T), the data in Figure 2B were fitted to Equation 2, using the values for k(H,H,D-5’-dA) and k(H,D,T-5’-dA) determined from the data in Figure 2A, and allowing kf (H)/kf (T) to float. Similarly good fits could be obtained if the initial values for k(H,H,D-5’-dA) and k(H,D,T-5’-dA) were also allowed to vary during the fitting process. Although the values for k(H,H,D-5’-dA) and k(H,D,T-5’-dA) are not be determined with any precision by fitting to Equation 2, the value of kf (H)/kf (T) is not very sensitive to these parameters. The value for the secondary kinetic isotope effect at the 5’-carbon when deuterium is in the primary position, kf (H)/kf (T), is close to unity, 1.05 ± 0.08. The equilibrium secondary isotope effect, KH/KT, is calculated by allowing t → ∞ in Equation 2 and is therefore given by kf (H)/kf (T) × (k(H,H,D-5’-dA) / k(H,D,T-5’-dA)). This gives a value for KH/KT of 0.70 ± 0.14 when deuterium is in the primary position.
DISCUSSION
The primary and secondary isotope effects measured for the formation of 5’-dA in glutamate mutase are summarized in Table 1. The secondary tritium isotope effects measured with protium in the primary position are among the larger secondary effects measured for an enzyme reaction, whereas with deuterium in the primary position the kinetic isotope effect is close to unity. These measurements were made in the pre-steady state, so that the isotope effects should not be affected by the kinetics of substrate binding or product dissociation. Changing the isotope in the primary position to deuterium should not alter substrate binding or product dissociation and is thus very unlikely to be the reason that the secondary kinetic isotope effect is diminished.
TABLE 1.
Isotope effects measured for the formation of 5′-dA from AdoCbl and L-glutamate catalyzed by glutamate mutase
We present two plausible explanations for the unusual kinetic behavior that we have encountered. First we explore whether a change in the rate-determining step upon substitution of deuterium for protium in the primary position, in conjuction with a conformational change in the protein might adequately account for our data. Secondly, we consider whether hydrogen tunneling and coupled motion in the transition state could result in the change in secondary isotope effects, an explanation that we find more appealing.
First we note that in these experiments the tritium at the 5’-carbon reports upon two bond making/breaking steps: the first involved in breaking the Co-C bond of AdoCbl (step I → II in Figure 1), the second involved in hydrogen transfer from glutamate to 5’-dA (step II → III in Figure 1). The overall equilibrium isotope effect, which is simply the product of the equilibrium isotope effect for each step, is large, inverse and, as expected, independent of whether protium or deuterium is in the primary position. This implies that the 5’-hydrogens of 5’-dA make stiffer bonds to carbon than do the 5’-hydrogens of AdoCbl, and, as discussed previously (15), suggests that the equilibrium isotope effect on the conversion of I to II is closer to unity than might be expected based simply on the changes in bonding involved.
The secondary kinetic isotope effect is determined by the differences in 5’-C-H bonding between AdoCbl and the transition state for the overall reaction. Again, this will reflect contributions from both steps I → II and II → III, however a large body experimental data from this and other AdoCbl-dependent enzymes strongly suggest that the second step in the reaction (II → III) is the slower step (7, 9, 10, 21).
Change of rate-determining step?
By introducing deuterium at the primary position we expect to slow the hydrogen transfer step (II → III) through the action of the primary isotope effect, but this should not affect the transition state of the Co-C bond cleavage reaction (I → II). In doing so, we should make the second step more rate-determining and thus isolate the secondary kinetic isotope effect associated with the conversion of II → III from that associated with the conversion of I → II. This would be predicted to have either no effect on the magnitude of the secondary kinetic effect, if it is already full expressed, or actually make it more inverse if it is partially suppressed by other steps.
However, it is possible that a more complex kinetic scheme involving a change in the rate-determining step when deuterium is in the primary position might lead to the apparent diminution in the kinetic isotope effect. This could occur if a three-step process was involved consisting of a) cobalt-carbon bond homolysis (I → II); b) hydrogen transfer between substrate and coenzyme (II → III); and c) a conformational change in the protein that stabilizes the resulting substrate radicals (III → III*), which are then trapped by the acid quench. If the protein conformation change was rate-determining with protiated substrate, and allowed a rapid pre-equilibrium to be established in the hydrogen transfer step, this could explain why the values of the apparent secondary kinetic and equilibrium isotope effects are very similar with protiated substrate. In effect the experiment would only measure the secondary equilibrium isotope effect. However, if step II → III became (partially) rate determining with deuterated substrate, then the secondary kinetic isotope effect would be now be manifested (distinct from the equilibrium isotope effect), and if this was relatively small it could account for why the kinetic isotope effect is very different with deuterated substrate. The conformational change would also mask the intrinsic primary kinetic isotope effect resulting in a relatively small deuterium isotope effect being measured. This kinetic mechanism is illustrated by the free energy profile shown in Figure 3.
Figure 3.
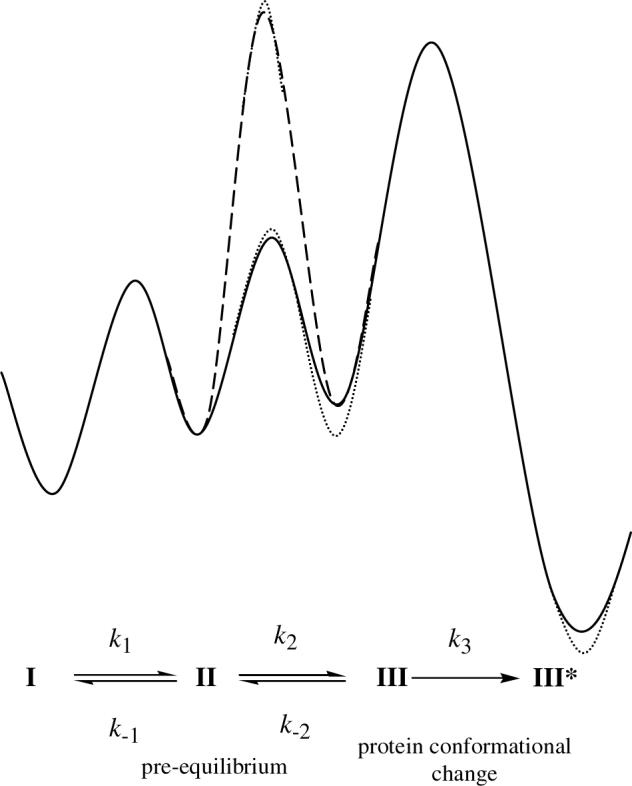
Kinetic scheme illustrating a three-step process involving a hypothetical rate-determining protein conformational changes that stabilizes post-homolysis radical intermediates. The enzyme-bound species I, II and III are those indicated in Figure 1; III* represents the conformational change in the protein. The hypothesized change in rate-determining step when deuterated substrate is used is indicated by a dashed line. The secondary kinetic and equilibrium isotope effects are indicated by dotted lines. For discussion see the text.
Whereas such a mechanism would explain the current isotope data, it requires an extra step be inserted at this point in the mechanism, i.e. a protein conformational change, which adds complexity and is aesthetically less pleasing. The crystal structure of glutamate mutase with substrate and coenzyme bound indicates that the entire catalytic cycle can be accomplished with minimal movement of protein side chains (22), suggesting that at a rate-determining protein conformational change is unlikely. Also, for this mechanism to explain the data it requires that the isotope effect be large enough to significantly change the rate-determining step, and that the hypothetical protein conformational change occurs at rate that is just right to allow the isotope effect to change the rate-determining step. This imposes a significant constraint on this mechanism, so although it cannot be rigorously excluded it we think it less likely.
Hydrogen tunneling and coupled motion?
Kinetic secondary isotope effects arise mainly from differences in bending modes between the ground and transition states. The secondary kinetic effect is often interpreted with respect to the equilibrium secondary isotope effect, which reflects differences in the bending modes between the reactant and product ground states. A secondary kinetic isotope effect of 1 representing a very early transition state, resembling the structure of the reactants, whereas a secondary isotope effect equal to the equilibrium isotope effect indicates a very late transition state resembling the structure of the products. Furthermore, substitution of a heavy isotope at the primary position should not change the secondary kinetic isotope effect. However, this interpretation is only valid in the absence of hydrogen tunneling, i.e. the in semi-classical regime; these assumptions break down if tunneling and/or coupled motion are involved in the transition state.
In general, isotope effects obey the “Rule of the Geometric Mean” which in effect states that multiple isotopic substitutions tend to be independent, i.e. there are no isotope effects on isotope effects. As Schowen has pointed out, there are cases where this rule breaks down for isotope effects, and the failure of the rule is mechanistically significant (16). In this case the rule is broken because the secondary isotope effect is reduced by the substitution of a heavy isotope at the primary position. As discussed below, this observation is consistent with the motion of the hydrogen atoms in the secondary position (which move from planar sp2 geometry to tetrahedral sp3 geometry around the 5’-carbon) being coupled to that of the hydrogen atom undergoing transfer in a transition state that involves a significant degree of quantum tunneling (23, 24). This is illustrated for glutamate mutase in Figure 4.
Figure 4.
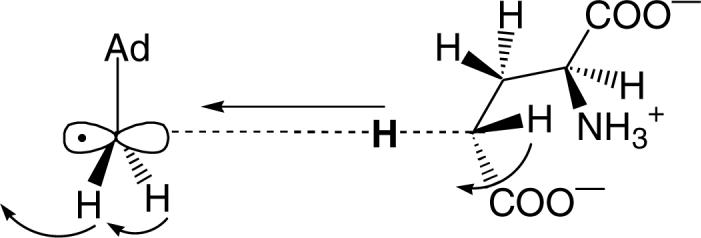
Illustration of the coupled motions involved in the transition state for hydrogen transfer between 5’-dA radical and glutamate.
The phenomenon of hydrogen tunneling in enzymes has been studied in a number of enzymes (25-30), in particular it was first identified in studies of hydride transfer between NAD+ and various alcohols catalyzed by yeast alcohol dehydrogenase (YADH) (30-33). For this enzyme, Klinman and colleagues demonstrated tunneling and coupled motion using labeled benzylalcohols as substrates, for which both the primary and secondary isotope effects deviated from the Swain-Schaad exponential relationship that relates the kH/kT to the kD/kT isotope effect2 (33). Moreover, theoretical studies have shown that to reproduce the large secondary isotope effects (which in YADH are larger than predicted by semi-classical models) both coupling of the secondary hydrogens to the reaction coordinate and quantum tunneling are necessary (34).
Of particular relevance to the present case, is the observation that introducing deuterium at the primary position reduced the secondary tritium kinetic isotope effect measured in YADH from a value of 1.35 to 1.10 (33). A similar diminution of the secondary isotope effect in response to deuteration at the primary position has been observed with formate dehydrogenase (35). In glutamate mutase we observe a qualitatively similar phenomenon, with the secondary tritium isotope effect being deflated from 0.76 to about 1.05. The precision of our measurements is somewhat less than the those made on dehydrogenases, because our measurements have to be made under single turn-over conditions, whereas as most secondary isotope effects are measured under steady state conditions.
This behavior is predicted for cases where coupled motion and hydrogen tunneling are involved, as tunneling behavior will propagate from the primary hydrogen to secondary hydrogens and inflate the value of the secondary kinetic isotope effects beyond the semi-classical limit. When a heavy isotope is present in the primary position, tunneling is reduced, or eliminated, and the secondary kinetic isotope effects are reduced to within the semi-classical limit (23, 34). Note that this behavior has the effect of reducing the primary kinetic isotope effect, which is “stolen” by the coupled motion of the secondary hydrogens; for example, in YADH kH/kD primary is 3 − 5, depending upon substrate, values that are well with the semi-classical range.
The secondary kinetic isotope effect measured with deuterium in the primary position is more likely to represent the “true” value of the isotope effect; i.e. the semi-classical value unaffected by quantum tunneling. As such, this should provide a more reliable indication of the position of the transition state along the reaction coordinate. In the absence of tunneling, secondary kinetic isotope effects may be interpreted with respect to the secondary equilibrium isotope effect; the value of the kinetic isotope effect should lie somewhere between unity, reflecting an early transition state, and the equilibrium isotope effect, reflecting a late transition state (16).
The fact that the secondary isotope effect measured for glutamate mutase reacting with deuterated glutamate is close to unity suggests an early transition state for the reaction, or more specifically, that rehybridization of the 5’-carbon, from planar to tetrahedral, occurs after the transition state for hydrogen transfer. This is in accord with the position of the transition state predicted from Hammond's postulate; i.e. that the transition state will resemble the structure of the higher energy reactant. In this case the 5’-dA radical is of significantly higher energy than the glutamyl radical formed by hydrogen abstraction, and we would expect the transition state structure to be closer to that of 5’-dA radical. We note that the opposite conclusion would be drawn from the large inverse secondary kinetic isotope effect measured with protium in the primary position, if tunneling was not suspected!
The existence of hydrogen tunneling in the glutamate mutase reaction has been suspected because of the very large primary isotope effects (kH/kD > 20) on the formation of cob(II)alamin observed with several AdoCbl-dependent enzymes. In particular, Banerjee and coworkers have conducted an investigation of the temperature dependence of the primary deuterium isotope effects in methylmalonyl-CoA mutase and showed that the isotope effects on the Arrhenius factors and activation energies are consistent with hydrogen tunneling in this enzyme (13). However, coupled motion has not been observed before for an AdoCbl-dependent enzyme, and indeed the secondary isotope effect measurements necessary to diagnose coupled motion have not been conducted for other AdoCbl-dependent enzymes.
Although we originally reported a very large primary deuterium isotope effect for the formation of cob(II)alamin and 5’-dA upon reaction of hologlutamate mutase with deuterated substrates (kH/kD = 28 with glutamate and 35 with methylaspartate) (7), we have recently had cause to revise our interpretation of the stopped-flow data upon which these numbers were based. As we described recently, (21) rapid quench measurements of the deuterium isotope effect, combined with mass spectral analysis of 5’-dA formed in the reaction, indicate that the intrinsic primary isotope effect is likely much smaller, see Table 1.
As Klinman has pointed out (23, 25), hydrogen tunneling does not necessarily have to occur with coupled motion of the secondary hydrogen atoms. For example, bovine serum amine oxidase (BSAO) exhibits a very large primary kinetic isotope effect for proton abstraction from the Schiff base of the amine, and the isotope effect on the Arrhenius factor is significantly less than one, consistent with tunneling. However, the secondary isotope effects for this enzyme are in the normal range, which appears to rule out coupled motion. This raises the intriguing possibility that in AdoCbl-dependent enzymes two different mechanisms of hydrogen transfer may be operating. In methylmalonyl-CoA mutase, in which the intrinsic deuterium isotope effects approach 50 at low temperatures (13), quantum tunneling of the hydrogen atom undergoing transfer may occur without coupled motion. Whereas in glutamate mutase, where the primary isotope effects appear to be much lower, quantum tunneling of the primary hydrogen may be coupled to the movement of the secondary hydrogen atoms.
ACKNOWLEDGEMENT
We thank Christel Fox for help with protein purification, and the reviewers of this manuscript for helpful comments and suggestions.
Footnotes
This research was supported by a grant from the National Institutes of Health, GM 59227 to E.N.G.M.
The abbreviations used are: AdoCbl, adenosylcobalamin; 5’-dA, 5’-deoxyadenosine; H,H,D5’-dA, 5’-deoxyadenosine specifically containing protium and deuterium in the 5’-position; H,D,T5’-dA, 5’-deoxyadenosine specifically containing tritium and deuterium in the 5’-position
Later work on horse liver alcohol dehydrogenase mutants demonstrated that steric interactions between protein and substrate also change the exponential relationship between secondary kH/kT and kD/kT isotope effects. This suggests that the secondary isotope effect may also be sensitive to the more rigorous constraints that deuterium tunneling in the primary position imposes on the geometry of the reactants (30).
REFERENCES
- 1.Frey PA, Hegerman AD, Reed GH. Free radical mechanisms in enzymology. Chem. Rev. 2006;106:3302–3316. doi: 10.1021/cr050292s. [DOI] [PubMed] [Google Scholar]
- 2.Banerjee R. Radical carbon skeleton rearrangements: Catalysis by coenzyme B12-dependent mutases. Chem. Rev. 2003;103:2083–2094. doi: 10.1021/cr0204395. [DOI] [PubMed] [Google Scholar]
- 3.Marsh ENG, Drennan CL. Adenosylcobalamin-dependent isomerases: new insights into structure and mechanism. Curr. Opin. Chem.l Biol. 2001;5:499–505. doi: 10.1016/s1367-5931(00)00238-6. [DOI] [PubMed] [Google Scholar]
- 4.Frey PA. Radical mechanisms of enzymatic catalysis. Ann. Rev. Biochem. 2001;70:121–148. doi: 10.1146/annurev.biochem.70.1.121. [DOI] [PubMed] [Google Scholar]
- 5.Banerjee R. Radical Peregrinations Catalyzed by Coenzyme B12-Dependent Enzymes. Biochemistry. 2001;40:6191–96198. doi: 10.1021/bi0104423. [DOI] [PubMed] [Google Scholar]
- 6.Marsh ENG. Coenzyme B12-dependent glutamate mutase. Bioorg. Chem. 2000;28:176–189. doi: 10.1006/bioo.2000.1168. [DOI] [PubMed] [Google Scholar]
- 7.Marsh ENG, Ballou DP. Coupling of cobalt-carbon bond homolysis and hydrogen atom abstraction in adenosylcobalamin-dependent glutamate mutase. Biochemistry. 1998;37:11864–72. doi: 10.1021/bi980512e. [DOI] [PubMed] [Google Scholar]
- 8.Licht SS, Lawrence CC, Stubbe J. Thermodynamic and kinetic studies on cobalt-carbon bond homolysis by ribonucleotide triphosphate reductase: the importance of entropy in catalysis. Biochemistry. 1999;34:1234–1242. doi: 10.1021/bi981886a. [DOI] [PubMed] [Google Scholar]
- 9.Padmakumar R, Banerjee R. Evidence that cobalt-carbon bond homolysis is coupled to hydrogen atom abstraction from substrate in methylmalonyl-CoA mutase. Biochemistry. 1997;36:3713–8. doi: 10.1021/bi962503g. [DOI] [PubMed] [Google Scholar]
- 10.Bandarian V, Reed GH. Isotope effects in the transient phases of the reaction catalyzed by ethanolamine ammonia-lyase: Determination of the number of exchangeable hydrogens in the enzyme-cofactor complex. Biochemistry. 2000;39:12069–12075. doi: 10.1021/bi001014k. [DOI] [PubMed] [Google Scholar]
- 11.Licht SS, Booker S, Stubbe JA. Studies on the catalysis of carbon-cobalt bond homolysis by ribonucleoside triphosphate reductase: Evidence for concerted carbon-cobalt bond homolysis and thiyl radical formation. Biochemistry. 1999;38:1221–1233. doi: 10.1021/bi981885i. [DOI] [PubMed] [Google Scholar]
- 12.Chen DW, Abend A, Stubbe J, Frey PA. Epimerization at carbon-5 ' of (5 ' R)-[5 ',H- 2]adenosylcobalamin by ribonucleoside triphosphate reductase: Cysteine 408-independent cleavage of the Co-C5 ' bond. Biochemistry. 2003;42:4578–4584. doi: 10.1021/bi030018x. [DOI] [PubMed] [Google Scholar]
- 13.Chowdhury S, Banerjee R. Evidence for quantum mechanical tunneling in the coupled cobalt-carbon bond homolysis-substrate radical generation reaction catalyzed by methylmalonyl-CoA mutase. J. Am. Chem. Soc. 2000;122:5417–5418. [Google Scholar]
- 14.Bahnson BJ, Klinman JP. Hydrogen Tunneling in Enzyme Catalysis. Methods Enzymol. 1995;249:373–397. doi: 10.1016/0076-6879(95)49042-6. [DOI] [PubMed] [Google Scholar]
- 15.Cheng M-C, Marsh ENG. Pre-steady state measurement of intrinsic secondary tritium isotope effects associated with the homolysis of adenosylcobalamin and the formation of 5'-deoxyadensosine in glutamate mutase. Biochemistry. 2004;43:2155–2158. doi: 10.1021/bi036122w. [DOI] [PubMed] [Google Scholar]
- 16.Suhnel J, Schowen RL. In: Enzyme mechanism from isotope effects. Cook PF, editor. CRC Press; Boca Raton, FL: 1991. pp. 3–35. [Google Scholar]
- 17.Chen HP, Marsh ENG. Adenosylcobalamin-dependent glutamate mutase: examination of substrate and coenzyme binding in an engineered fusion protein possessing simplified subunit structure and kinetic properties. Biochemistry. 1997;36:14939–45. doi: 10.1021/bi971374g. [DOI] [PubMed] [Google Scholar]
- 18.Marsh ENG. Tritium isotope effects in adenosylcobalamin-dependent glutamate mutase: implications for the mechanism. Biochemistry. 1995;34:7542–7. doi: 10.1021/bi00022a030. [DOI] [PubMed] [Google Scholar]
- 19.Brown KL, Cheng S, Zou X, Li J, Chen GD, Valente EJ, Zubkowski JD, Marques HM. Structural and enzymatic studies of a new analogue of coenzyme B-12 with an alpha-adenosyl upper axial ligand. Biochemistry. 1998;37:9704–9715. doi: 10.1021/bi980707m. [DOI] [PubMed] [Google Scholar]
- 20.Chih HW, Marsh EN. Pre-steady-state kinetic investigation of intermediates in the reaction catalyzed by adenosylcobalamin-dependent glutamate mutase. Biochemistry. 1999;38:13684–91. doi: 10.1021/bi991064t. [In Process Citation] [DOI] [PubMed] [Google Scholar]
- 21.Cheng M-C, Marsh ENG. Isotope effects for deuterium transfer between substrate and coenzyme in adenosylcobalamin-dependent glutatmate mutase. Biochemistry. 2005;44:2686–2691. doi: 10.1021/bi047662b. [DOI] [PubMed] [Google Scholar]
- 22.Gruber K, Reitzer R, Kratky C. Radical shuttling in a protein: Ribose pseudorotation controls alkyl-radical transfer in the coenzyme B12 dependent enzyme glutamate mutase. Angew. Chem. Intl, Ed.n. 2001;40:3377–+. doi: 10.1002/1521-3773(20010917)40:18<3377::aid-anie3377>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 23.Klinman JP. Hydrogen tunneling and coupled motion in enzyme reactions. CRC Press; Boca Raton FL: 1991. [Google Scholar]
- 24.Karsten WE, Hwang C-C, Cook PF. α-secondary tritium kinetic isotope effects indicate hydrogen tunneling and coupled motion occur in the oxidation of L-malate by NAD-malic enzyme. Biochemistry. 1999;38:4398–4402. doi: 10.1021/bi982439y. [DOI] [PubMed] [Google Scholar]
- 25.Kohen A, Klinman JP. Enzyme catalysis: Beyond classical paradigms. Acc. Chem. Res. 1998;31:397–404. [Google Scholar]
- 26.Kohen A, Cannio R, Bartolucci S, Klinman JP. Enzyme dynamics and hydrogen tunnelling in a thermophilic alcohol dehydrogenase. Nature. 1999;399:496–9. doi: 10.1038/20981. [DOI] [PubMed] [Google Scholar]
- 27.Liang ZX, Klinman JP. Structural bases of hydrogen tunneling in enzymes: progress and puzzles. Curr. Opin. Struct. Biol. 2004;14:648–655. doi: 10.1016/j.sbi.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 28.Masgrau L, Basran J, Hothi P, Sutcliffe MJ, Scrutton NS. Hydrogen tunneling in quinoproteins. Arch. Biochem. Biophys. 2004;428:41–51. doi: 10.1016/j.abb.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 29.Sutcliffe MJ, Scrutton NS. A new conceptual framework for enzyme catalysis - Hydrogen tunneling coupled to enzyme dynamics in flavoprotein and quinoprotein enzymes. Eur. J. Biochem. 2002;269:3096–3102. doi: 10.1046/j.1432-1033.2002.03020.x. [DOI] [PubMed] [Google Scholar]
- 30.Nagel ZD, Klinman JP. Tunneling and dynamics in enzymatic hydride transfer. Chem. Rev. 2006;106:3095–3118. doi: 10.1021/cr050301x. [DOI] [PubMed] [Google Scholar]
- 31.Cook PF, Oppenheimer NJ, Cleland WW. Secondary deuterium and nitrogen-15 isotope effects in enzyme catalyzed reactions. Chemical mechanism of liver alcohol dehydrogenase. Biochemistry. 1981;20:1817–1825. doi: 10.1021/bi00510a016. [DOI] [PubMed] [Google Scholar]
- 32.Welsh KM, Creighton DJ, Klinman JP. Transition-State Structure in the Yeast Alcohol-Dehydrogenase Reaction - the Magnitude of Solvent and Alpha-Secondary Hydrogen Isotope Effects. Biochemistry. 1980;19:2005–2016. doi: 10.1021/bi00551a001. [DOI] [PubMed] [Google Scholar]
- 33.Cha Y, Murray CJ, Klinman JP. Hydrogen Tunneling in Enzyme-Reactions. Science. 1989;243:1325–1330. doi: 10.1126/science.2646716. [DOI] [PubMed] [Google Scholar]
- 34.Huskey WP, Schowen RL. Reaction Coordinate tunneling in hydride transfer reactions. J. Am. Chem. Soc. 1983;105:5704–5704. [Google Scholar]
- 35.Hermes JD, Morrical SW, O'Leary MH, Cleland WW. Variation of Transition-State Structure As a Function of the Nucleotide in Reactions Catalyzed By Dehydrogenases .2. Formate Dehydrogenase. Biochemistry. 1984;23:5479–5488. doi: 10.1021/bi00318a016. [DOI] [PubMed] [Google Scholar]