

Abstract
The cellular mechanisms that modulate the redox state of p53 tumor suppressor remain unclear, although its DNA-binding function is known to be strongly inhibited by oxidative and nitrosative stresses. We show that human p53 is subjected to a new and reversible posttranslational modification, namely, S-glutathionylation in stressed states including DNA damage. First, a rapid and direct incorporation of biotinylated GSH or GSSG into the purified recombinant p53 protein was observed. The modified p53 had significantly decreased ability to bind its consensus DNA sequence. Reciprocal immunoprecipitations and a GST-overlay assay showed that p53 in tumor cells was marginally glutathionylated, however, the modification increased greatly after oxidant and DNA-damaging treatments. GSH-modification coexisted with the serine phophorylations in activated p53, and the thiol-conjugated protein was present in nuclei. When tumor cells treated with camptothecin or cisplatin were subsequently exposed to glutathione-enhancing agents, p53 underwent dethiolation accompanied by detectable increases in p21waf1 expression, relative to the DNA damaging drugs alone. Mass spectrometry of GSH-modified p53 protein identified the cysteines 124, 141 and 182, all present in the proximal DNA-binding domain, as the sites of glutathionylation. Biotinylated maleimide also reacted rapidly with Cys141, implying this to be the most reactive cysteine on p53 surface. The glutathionylatable cysteines were found to exist in a negatively-charged microenvironment in cellular p53. Molecular modeling studies located Cys124 and 141 to the dimer interface of p53 and showed glutathionylation of either residue would inhibit p53-DNA association, and also interfere with protein dimerization. These results show for the first time that shielding of reactive cysteines contributes to a negative regulation for human p53, and imply that such an inactivation of the transcription factor may represent an acute defensive response with significant consequences for oncogenesis.
The p53 gene product is a DNA sequence-specific transcription factor, which as a homotetramer, controls the expression of a wide-array of genes through direct binding with response elements (1). This best studied and probably most important function bestows human p53 with regulatory responses to a variety of cellular stresses, including DNA damage, nucleotide depletion, chemotherapeutic drugs, oxidative stress, and many aberrant growth signals (2, 3). A complex and diverse set of posttranslational modifications, such as the site-specific phosphorylations, ubiquitination and sumoylation govern the activation and stabilization of p53 protein in these functional transactions (4). However, the cellular mechanisms including covalent modifications, if any, that protect and modulate the p53 protein during the constant and recurring episodes of oxidative and nitrosoative stresses, which often initiate and promote carcinogenesis and many disease states (5) remain unknown. Several lines of evidence suggest that p53 is highly prone to oxidative inactivation. For example, the in vitro binding of p53 to its recognition sequences requires the presence of reductant such as 2-mercaptoethanol or dithiothreitol in the binding buffers, and is sensitive to oxidants such as H2O2, diamide (6). Target gene transactivation by p53 in human cells is affected by the pharmacological oxidizing and reducing agents (7). The expression of reporter genes driven by a p53-responsive promoter is also decreased by oxidative treatment (8). Hypoxia and nitric oxide-induced inactivation of p53-dependent transactivation are yet other examples (9, 10). The transactions of p53 are also sensitive to metal cations and cu2+/cu+ redox cycling (11). In contrast to the oxidation effects, the Ref-1 and thioredoxin redox modulators have been shown to reactivate oxidized p53 and stimulate p53 transactivation in cells (12, 13). Therefore, p53 resembles other redox-dependent transcription factors such as the NF-κB and AP-1 in these properties.
Majority of the redox-sensitive proteins contain one or more cysteines that exist as thiolate anions, also called reactive cysteines, which play crucial roles in redox signaling (14). The reactive cysteines are more nucleophilic, and therefore, are highly sensitive to attack by reactive oxygen and reactive nitrogen species (ROS and RNS) (15). ROS/RNS cause oxidation of protein thiols (PSH) in a step wise fashion involving the formation of thiyl radical (PS), sulfenic acid (PSOH), sulfinic acid (PSO2H), sulfonic acid (PSO3H) or S-nitrosothiol/S-nitrosated proteins (PSNO). All these forms except PSO3H can be stabilized within the protein environment and recycled, via disulfide bond intermediates, back to the thiol state (16). In this process called S-thiolation, low molecular weight thiols such as glutathione (GSH or GSSG) can form mixed disulfides with reactive cysteines or oxidized cysteine forms in proteins (17). This modification is readily reversible, because increases in GSH/GSSG ratio or enzymatic reactions involving protein disulfide isomerase, glutaredoxin, thioredoxin or sulfiredoxin can restore the protein sulfhydryls to their reduced state (18). Thus, glutathionylation of reactive cysteines in metabolic enzymes, kinases, phosphatases, and transcription factors has emerged as a central mechanism by which changes in the intracellular redox state may be transduced into functional cellular responses (18). Very much like phosphorylation, this modification can modulate enzyme activities, protein functions, and protein-protein interactions (17, 18).
Pertinent to this study, evidence for the involvement of cysteines in the redox modulation and DNA binding function of p53 has been demonstrated (19-22). Human p53 protein has 10 cysteine residues, all of which, interestingly, are located within the DNA binding domain (DBD)1, between amino acids, 100-300 (19). The positions of cysteines relative to the recognition loop in the tertiary structure of p53 are shown in Fig. S1 (Supporting Information) of this article. The cysteines 176, 238, 242, along with histidine 179 bind to a divalent zinc atom, which stabilizes the loop/helical structure of the core domain. Mutagenesis of these cysteines leads to complete loss of DNA binding, while replacement of cysteines 124, 135, 141, and 277 alter murine p53-affinity to DNA, suggesting that the latter group of cysteines may regulate the structural dynamics of DBD (19, 21). Nevertheless, which of these cysteines are most reactive and respond to oxidative insults in human cells is not known. Also, the consequences of these responses, if any, to p53 function and cell signaling remain unclear. In this report, we show that human p53 is a substrate for glutathionylation in vitro and in cells, and demonstrate the site-specific modification of three cysteines present in the proximal core domain disrupts p53 interaction with its target DNA. Evidence for the modulation of p53 function by glutathionylation in cells is also shown.
Materials and Methods
Cell lines and reagents
Human cancer cell lines, U87 malignant glioblastoma (U87MG) and HCT116 colon carcinoma, both of which harbor a wild-type p53 (23) were used. Most reagents, of the highest grade available, were obtained from Sigma Chemicals. Monoclonal antibodies to p53 (DO-1), and glutathione were purchased from Santa Cruz Biotechnology and Virogen (MA, USA), respectively.
Expression and purification of recombinant p53 Protein (rp53)
E. coli BL21(DE3) strain was transformed with a pRSET plasmid containing histidine-tagged wild-type human p53 coding sequence (24) kindly provided by Dr. Jennifer Nyborg, Colorado State University, USA. Protein expression was initiated by the addition of inducer (an IPTG alternative from MoleculA, VA, USA), which provided more soluble protein. rp53 in its native form was purified by metal chelation chromatography on Talon resin (BD Biosciences) as described previously (25). Dialyzed protein (90% homogeneity) was stored frozen in the presence of 25 μM β-mercaptoethanol. For glutathionylation studies described in this report, the protein preparations were dialyzed against 50 mM Tris-HCl (pH 8.0) at 4°C immediately before use.
Biotinylation of GSH, GSSG, and GST-Pi protein and their use for detection of glutathionylated p53
Biotinylation of GSH and GSSG was performed according to Sullivan et al. (26) using sulfo-NSH-biotin (Pierce Biochemicals). Human placental glutathione S-transferase protein (GST-Pi) was biotinylated for detecting glutathionylated p53 by the overlay assays as described previously (27). For this, purified GST-pi (0.5 mg) was immobilized on GSH-Sepharose for protecting the GSH-binding site, and the bound protein treated with sulfo-NSH-biotin. The beads were washed, biotinylated GST was eluted with 10 mM GSH followed by dialysis (27). After incubation of rp53 (0.5 μg with 5 mM biotinylated thiols (Biot-GSH, Biot-GSSG) in phosphate buffer (pH 7.5), 10 mM N-ethylmaleimide (NEM) was added to stop the reactions. The samples were run by non-reducing SDS-PAGE, and proteins transferred to PVDF membranes. The blots were soaked in 5% non-fat dry milk, and incubated with streptavidin-linked HRP (Pierce, 1:8,000 dilution), followed by washings and enhanced chemiluminiscence (ECL). The biotinylated GST protein retains ability to bind the glutathione moiety present in the mixed disulfide linkages formed by glutathionylation (27-29); a Far-western procedure described previously (27) was used for detecting GSH-linked p53. Briefly, the p53 protein was immunoprecipitated from control and drug-treated cells, the complexes were resolved by non-reducing SDS-PAGE followed by protein blotting. Next, the membranes were blocked with 5% BSA, and incubated with biotinylated GST-pi (30 μg/ml) for 10 h. After extensive washings, the blots were exposed to streptavidin- HRP (Strep-HRP) followed by ECL (27).
Combined immunoprecipitation-immunoblotting analysis
For immunoprecipitating the glutathione conjugated p53 protein, tumor cells were lysed at 4°C in Tris-buffered saline containing 1X protease cocktail (Sigma), 0.5 mM sodium vanadate and 0.5% NP-40 as described previously (25). Equal protein amounts in lysates from different treatments were precleaned with 5 μl protein-A agarose beads, and the proteins immunoprecipitated using monoclonal antibodies to p53 or GSH. The complexes were solubilized in non-reducing SDS-sample buffers, subjected to electrophoresis on 10% gels followed by western blot analysis using reciprocal antibodies.
Quantitation of p53-binding with target DNA by affinity-immunoblotting (DAI) and electrophoretic mobility shift assays (EMSA)
Binding of p53 to its consensus recognition sequences was examined by the biotin-oligo pull-down assay followed by western blotting (30). The ds- 5′TAC AGA ACA TGT CTA AGC ATG CTG GGG ACT 3′ and a mutant 5′TAC AGA ATC GCT CTA AGC ATG CTG GGG ACT 3′ oligomers were labeled with biotin at 5′ end on one strand (Integrated DNA Technologies). Equal amounts (1 μg) unmodified and glutathionated rp53 protein were allowed to bind with duplex DNA in a binding buffer [20 mM Tris-HCl, pH 7.2, 1 mM EDTA, 0.1% Triton X-100 and 4% glycerol, 1 μg poly(dI.dC), 100 mM NaCl] at 4°C for 30 min. The DNA-bound protein complexes were captured by the addition of 10 μl of streptavidin magnetic beads (Roche Diagnostics), washed three times with 1 M NaCl, eluted by boiling the beads in SDS-sample buffer, and electrophoresed. After protein transfer, western blot analysis for p53 was performed (30). For the EMSA, the p53 consensus and mutant oligonucleotides were end-labeled using T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP in 10× kinase buffer supplied with the enzyme. p53 recombinant priein, treated or untreated with GSH were preincubated in 5 μl of 5× binding buffer (20% glycerol, 5 mM MgCl2, 5 mM EDTA, 5 mM dithiothreitol, 500 mM NaCl, 50 mM Tris-HCl, 0.4 mg/ml calf thymus DNA), and 2 μg of poly(dI-dC) for 15 min followed by binding with labeled oligonucleotide for 30 min. Some samples were incubated with 200 ng of anti-p53 antibody before the addition of labeled probes. The proteins were separated by electrophoresis using a 4% native polyacrylamide gel and 40 mM Tris borate-EDTA (pH 7.5) as running buffer. Gels were dried and exposed to Kodak x-ray film overnight.
Analysis of glutathionylation-susceptibility of p53 protein in human cancer cells
Our recently published procedure on the ability of GSH-Sepharose to detect the redox changes in proteins (31) was applied. U87MG cells were treated with H2O2 (15 min exposure at 0.4 mM), and further postincubated in oxidant-free medium for 0.5 to 6 h. Extracts from these cells were prepared in PBS and equal protein amounts there from were then mixed with 0.1 ml GSH-Sepharose beads (31). The bound proteins were eluted with 10 mM DTT, resolved by SDS-PAGE and western blotted for p53.
Mass spectrometry for identification of glutathionylation sites on p53 protein
In the first approach, rp53 samples (25 μg) treated and untreated with GSH (1 h, 37°C) were resolved by SDS-PAGE. The protein bands in gel slices were digested with trypsin (25ng/μl) in 25 mM NH4HCO3, peptides extracted, and reconstituted with 0.1% formic acid (32). On-line nano-liquid chromatography/electrospray ionization mass spectrometry (nano-LC ESI-MS/MS experiments) was performed on a QTOF API-US mass spectrometer (Micromass, UK) equipped with the CapLC system (Waters, Milford, MA). The stream select module was configured with an OPTI-PAK Symmetry 300 C18 trap column (Waters) connected in series with a nanoscale analytical column. Peptide samples (5 μl) were injected onto the trap column, cleaned-up and back-flushed to the analytical column at 0.5 ml/min using gradient elution. The gradient consisted of 5%–50% (v/v) solvent B containing 95% (v/v) acetonitrile and 0.1% (v/v) formic acid, in 45 minutes, followed by 50% B for 15 minutes and 50%–90% B in five min (solvent A: 5% (v/v) acetonitrile) and 0.1 (v/v) formic acid). The MS/MS spectra recorded for the doubly and triply charged molecular ions of peptides were searched against the non-redundant National Center for Biotechnology Information protein database using the MASCOT search engine (33).
In the second method, rp53 protein was exposed to 5 mM biotinylated maleimide (Sigma Chemicals) for 5 min for rapid alkylation of surface thiol groups as described previously (34). The samples were immediately filtered on P6-Biogel spin columns for removing the alkylator. The thiolated protein was trypsinized, the peptides were allowed to bind with soft-link monomeric avidin resin (Promega), and washed with 0.15 M NaCl (34). The bound peptides were eluted with 10% acetic acid (200 μl) and analyzed by MALDI-TOF using an Autoflex II instrument (Bruker, Bremen, Germany) and by MALDI TOF/TOF MS/MS (Matrix-assisted Laser Desorption/Ionization Time-of-Flight Tandem Mass Spectrometry) on a 4700 Proteomics Analyzer mass spectrometer (Applied Biosystems, Foster City, CA) in positive ion mode. The MALDI probe was spotted with 1 μl of a 1:1 (v/v) mixture of peptide sample and a saturated solution of α-cyano-4-hydroxycinnamic acid in 50% (v/v) acetonitrile. The conditions used for the operation of Autoflex II, acquisition of the data by MS/MS analysis were the same as described previously (35). The experimentally determined peptide fragment ion masses were matched, within a window of ± 0.1 Da, to theoretical fragment ion masses generated by in silico fragmentation of p53 peptides.
Labeling of cellular p53 by biotinylated maleimide
For demonstrating the rapid modification of reactive cysteines present on p53 protein surface, cell free extracts were prepared in 40 mM Tris-HCl, 1 mM PMSF, 3 mM benzamidine, 1 mM EDTA, and dialyzed against the same buffer for 30 min. Three hundred μg protein aliquots were exposed to 5 mM biot-maleimide, with and without 5 mM NEM treatment for 5-25 min. The samples were then subjected to immunoprecipitation of p53, and incorporation of biotin group was detected by western blot analysis using strep-HRP as described in an earlier section.
Nuclear localization of glutathionylated p53 protein
Cytoplasmic and nuclear fractions from control and diamide-treated cells were separated using an extraction kit (Pierce Biochemicals) according to manufacturer's instructions. Equivalent protein amounts from these fractions were immunoprecipitated using p53 antibodies. The immunocomplexes were run on SDS-polyacrylamide gels and Western blotted using anti-GSH antibodies.
Protein modeling
The crystal structure of three p53 core regions consisting of amino acids 92-292 with a segment of DNA containing the consensus sequence (36) was obtained from the protein data bank (PBD ID 1TSR). Its three-dimensional image was visualized and manipulated using the Sibyl 6.9 program (Tripos Inc., St. Louis, MO). In this crystal structure, three P53 monomers (A, B, C) are associated with a DNA duplex. Two of these (A and B) make direct contacts with the DNA. The third one (C) makes contact with the B monomer, but, not with DNA (36). Therefore, the C chain was not included in this study. Also, Cys182 seen in the B chain faces away from DNA duplex at the B-C dimer interface, therefore, glutathionation of Cys182 was not studied here. The coordinates for glutathione were taken from the reported crystal structure of proteins containing covalently bound glutathione (http://www.rcsb.org). Forty one glutathione structures were obtained from the protein data bank and analyzed for the conformations. We grouped the structures into two families of conformations; one, extended and the other, folded. To simulate glutathionation, we visually docked one conformation of each of the families of glutathione molecule and formed a covalent bond either with Cys124 or Cys141 of p53 crystal structure. The coordinates of the whole complex were subjected to energy minimization using the CFF91 force field of the simulation program DISCOVER (Accelrys Inc., San Diego, CA). A distance-dependent dielectric constant was used in calculating the electrostatic energy. The coordinates for the DNA duplex were kept fixed during the minimization, and minimization was terminated when the root-mean-square value was less than 0.001 kcal/Å
Statistical analysis
All experiments including the DNA binding assays for p53, IP/western analysis, and mass spectroscopy procedures were performed at least three times independent of each other. Results were assessed by Student's t test. Significance was defined as defined as P<0.05.
Results
Direct evidence for GSH and GSSG -incorporation into p53 protein
As a first step in characterizing p53 glutathionylation, we assayed the incorporation of GSH and GSSG into the accessible cysteines on p53 surface. A rapid and time dependent incorporation of Biot-GSSG (Fig. 1A) or Biot-GSH (Fig. 1B) was observed. Consistent with the thiol-exchange mechanism, GSSG was more efficient than GSH with the initial burst of labeling observed at 5 and 20 min respectively in this reaction. The disulfide linkages thus induced are labile in the presence of reducing agents. In agreement, glutathionylation of rp53 was fully reversed when the modified protein was exposed to DTT before SDS-PAGE (Fig. 1A, B). The blots showed equivalent p53 protein levels in all samples (lower panels of Fig. 1A, B), thus, validating the assay and glutathionylation kinetics.
Figure 1. Human p53 protein is a substrate for glutathionylation in vitro.
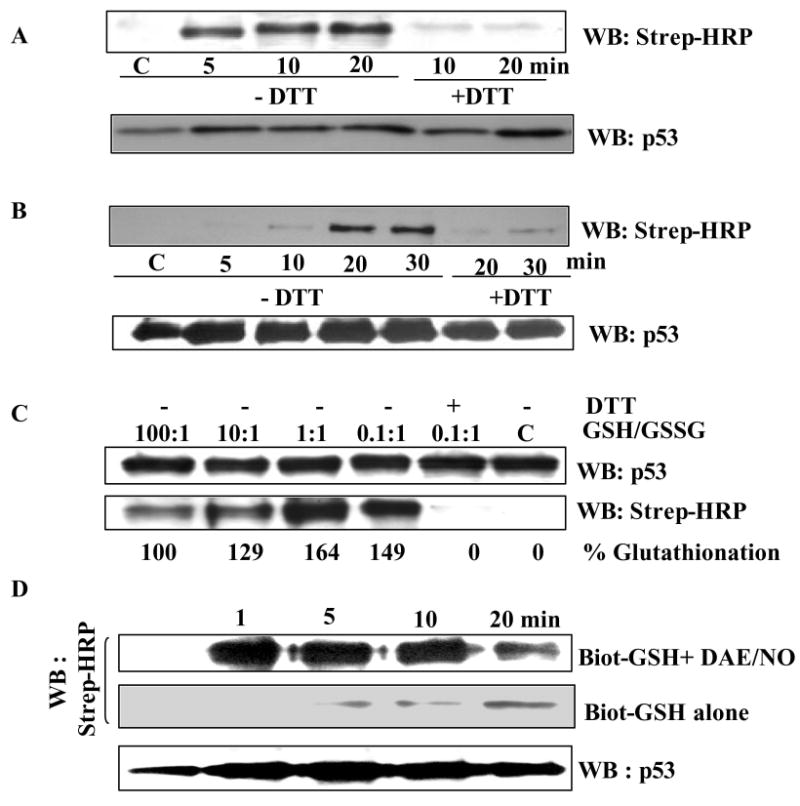
(A) Rapid incorporation of Biot-GSSG. rp53 (1 μg) was incubated with 5 mM Biot-GSSG in phosphate buffer at 37°C for times shown. Some samples were mixed with 10 mM DTT before loading on non-reducing SDS-Polyacrylamide gels. The blots were reacted with Strep-HRP to obtain the pattern shown. The kinetics of GSSG incorporation remained similar in three independent experiments (P<0.05). Loading of p53 protein on these blots was assessed by western analysis. (B) Kinetics of Biot-GSH incorporation into rp53. rp53 was exposed to 5 mM Biot-GSH, and samples were processed as described in legend to Fig. 1A. (C) p53 glutathionylation is regulated by redox conditions. rp53 protein was treated with mixtures of GSH and GSSG (0.01 to10 mM range) at different ratios in the presence of a constant amount of Biot-GSH (0.4 mM). The samples were incubated at 37°C for 10 min, terminated with the addition of NEM, and processed for blot analysis using Strep-HRP as described above. The lower panel shows the results obtained. DTT reversal of the glutathionation is shown in lane 5. The Blots were reprobed to assess p53 protein loading (upper panel). (D) DAE/NO (NO releasing agent) increases p53 glutathionylation. rp53 was incubated with Biot-GSH in the presence or absence of 0.25 mM DAE/NO, and the labeling was assayed through procedures described above.
Decreased GSH/GSSG ratios promote S-thiolation of p53
Protein glutathionylation is greatly enhanced by increased GSSG/GSH ratios that accompany cellular oxidative stress (17). Therefore, we examined Biot-GSH incorporation into rp53 the presence of varying GSH: GSSG ratios (100:1 to 0.1:1) that simulate mild to severe oxidative stress conditions in human cells. As shown in Fig. 1C, glutathionylation of p53 occurred at very low GSSG concentrations, and it increased significantly at higher GSSG/GSH ratios (65% increase at 1:1 ratio; lower panel of Fig. 1C), suggesting that p53 glutathionylation could occur in cells under physiological and moderately stressed conditions.
Influence of nitric oxide on p53 glutathionylation
While nitric oxide (NO) can modify the protein thiols directly by S-nitrosylation, it can also react with GSH to form GSNO, which, in turn can induce potent protein glutathionylation (17). Because the NO regulates p53 signaling (10, 37), we exposed rp53 to Biot-GSH in the presence or absence of the NO donor (DEA/NO; 0.25 mM). Fig. 1D shows that p53 was modified relatively faster and to a higher extent in the presence of NO donor, implying that nitrosative stress could induce glutathionylation of p53. This could occur through GSNO or the reaction of nitrosylated cysteines with GSH (17).
In vitro glutathionylation inhibits p53 binding to the recognition sequence
Because cysteines modulate p53 binding to DNA (6, 19), and glutathionylation modifies the cysteines, we used a DNA affinity immunoblotting assay (DAI) to assess the binding of GSH-modified p53 to the biotinylated consensus or mutated recognition sequences (30). The DAI assay combines sensitive biotin/streptavidin affinity-step and specific immunobloting to detect the DNA-bound p53 (30); therefore, this procedure was applied in most of our experiments. The western blot in Fig. 2A shows the results obtained in the initial assay. When rp53 was incubated with GSH for glutathionylation and DNA binding assays were performed without removing the thiol used for modification, no differences in the DNA binding of glutathionylated and non-glutathionylated p53 were apparent (compare lanes 1-3 in Fig. 2A); rapid dethiolation by the excess of thiol present in the reaction mixtures may explain this observation. In contrast, when p53 samples were gel-filtered to remove GSH before DNA binding, a consistent and reproducible reduction in the binding of glutathionylated p53 to its target DNA was observed (compare lanes 4 and 5 in Fig. 2A). As expected, exposure of the glutathionylated p53 to DTT restored the DNA binding to control levels (lane 6 in Fig. 2A). In the next series, we incubated rp53 was glutathionylated in the presence of GSH or GSSG. The modified protein was purified by gel filtration and DNA-binding reactions performed. The data in Fig. 2B shows that S-thiolation by both GSH and GSSG inhibited p53-specific DNA binding, GSSG was more efficient in this property, and that treatment of the thiolated protein with DTT reversed the inhibition of DNA binding in all cases. Fig. 2C depicts the DNA binding analysis of rp53 following glutathionylation at different GSH/GSSG ratios (100:1 to 0.1 to 1). The increased glutathionylation of p53 found under these oxidative stress-simulating conditions (see Fig. 1C) was clearly associated with diminished p53-DNA interactions. Overall, we observed a maximal reduction of 50% in p53 activity in glutathionylated samples.
Figure 2. GSH or GSSG-modified p53 exhibits significantly decreased binding to consensus DNA sequence.
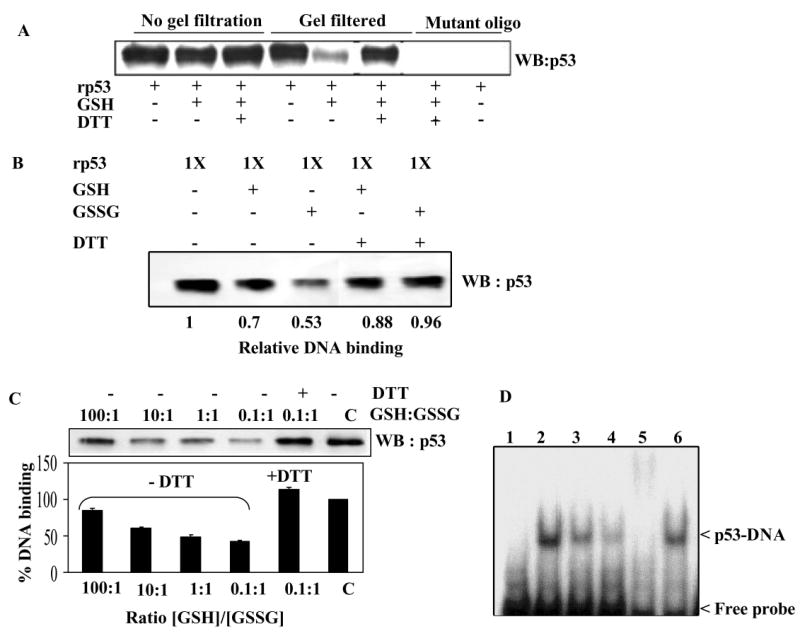
(A) Reduction in the DNA binding activity of rp53 after GSH conjugation. The limited amounts of mercaptoethanol present in rp53 preparations was removed by brief dialysis and used for DNA binding studies described here (See Methods). rp53 was incubated with and without GSH for 30 min. One set of samples (lanes 1-3) were subjected to DNA-affinity immunoblotting assay (DAI) assay without removing the GSH (labeled ‘no gel filtration’). The other set were subjected to gel filtration on Bio-gel P6 spin columns prior to the binding. DAI was performed with biotinylated normal or mutant target sequences for p53. DNA-bound p53 levels are shown. (B) DNA binding assays of rp53 following glutathionylation with GSH or GSSG. rp53 protein at 0.5 μg was incubated with the thiols in sodium phosphate buffer (30 mM, pH 7.5), all samples were then gel-filtered, and DAI assays performed. In some samples, the gel-filtered protein was exposed to DTT before the addition of ds- target DNA. (C) rp53 was incubated with differing GSH/GSSG ratios (100:1 to 0.1:1) for 30 min. All samples were gel-filtered and DAI assays performed. Densitometry of the p53 bands was performed to calculate the % DNA binding shown in the bar graph. Similar results were obtained in three separate experiments (P<0.05). (D) Effect of glutathionylation on the EMSA of p53 binding to its recognition sequence. rp53 was incubated with GSH or GSSG followed by gel-filtration as above, and EMSA performed as described in Methods. Reactions in lanes 1-6 contained 32P-labeled consensus recognition sequence. Lanes: 1, untreated rp53 incubated with mutant oligo; 2, untreated rp53 incubated with consensus oligo; 3, rp53 incubated with GSH; 4, rp53 incubated with GSSG; 5, untreated rp53 incubated with p53 antibody (DO1) before adding the probe to show supershift; 6, rp53 incubated with GSSG, but treated with 5 mM DTT for 10 min prior to probe addition.
Electrophoretic mobility shift assays (EMSA) yielded similar data on p53 binding to DNA. As shown in Fig. 2D, the rp53 protein bound specifically to the consensus recognition sequence (lane 2) and showed super shift in the presence of p53 antibodies (lane 5). GSH-treated (lane 3) and GSSG-treated (lane 4) p53 bound the DNA to a lesser extent, which was reversed by DTT (lane 6). Together, these results strongly suggest that changes in cellular redox inhibit p53 function through S-thiolation.
p53 protein is glutathionylated in human tumor cells
To demonstrate the p53 modification in cells, we used IP/western blot analyses, a GST-overlay assay (27) and GSH-affinity chromatography for assessing glutathionylation-susceptibility of the protein (31). First, for reciprocal IP/western, HCT116 colon cancer cells were treated with CPT to increase p53 levels, bulk glutathionated proteins or p53 were immunoprecipitated using specific antibodies to GSH and p53 respectively, the IPs were electrophoresed on non-reducing SDS-gels, and the resulting Western blots were probed with reciprocal antibodies. Fig. 3A shows that p53 was detected among the proteins precipitated by anti-GSH antibodies and p53 present in its IP was recognized by GSH antibodies. Exposing the IPs to DTT before SDS-PAGE slightly reduced the protein mass. These data clearly suggest that small amounts of p53 exist in a thiolated state in human cancer cells. Evidence from GST-overlay assays and GSH-affinity chromatography for this premise is presented in the sections to follow.
Figure 3. Detection of glutathionylated p53 protein in human tumor cells and its upregulation after DNA damage.
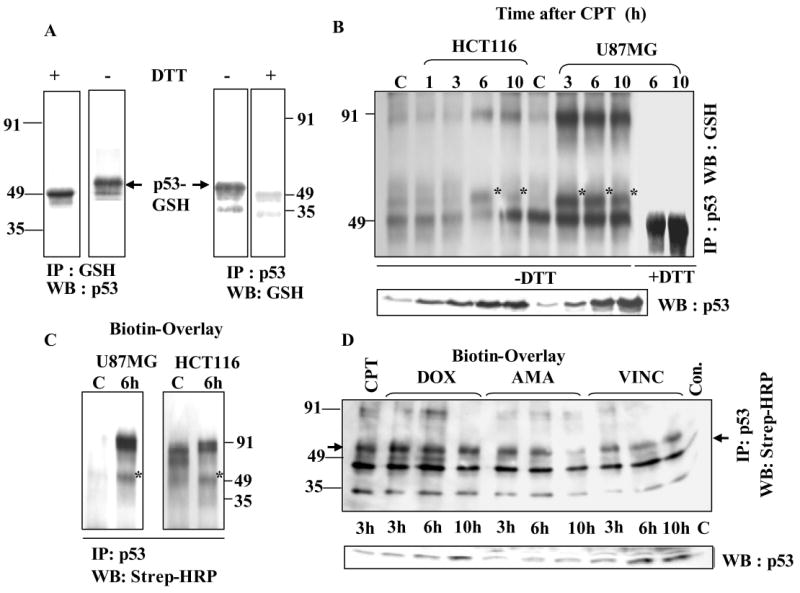
(A) Evidence from Reciprocal IP /western analysis. HCT116 cells were treated with 25 μM CPT. Extracts (500 μg protein) were immunoprecipitated using antibodies to p53 or GSH. The IPs were electrophoresed under non-reducing conditions, blotted, and reciprocal antibodies were used for p53 detection. Serial dilutions of the cell extracts followed p53 IP provided similar results (not shown). Wherever indicated, the immunoprecipitates were treated with 10 mM DTT for 10 min before SDS-PAGE. DTT-treated sample shows a slightly faster migrating monomer in the left panel. GSH antibodies did not recognize p53 after DTT exposure in the right panel. (B) CPT-induced DNA damage is accompanied by a significant increase in glutathionylated p53 levels. HCT 116 and U87MG cells were treated with 25 μM and 2.5 μM CPT respectively. At each time point, p53 present in 500 μg protein samples was Immunoprecipitated and processed for western analysis using GSH antibodies GSH-linked p53 bands are indicated with an *. C = control. The immunoprecipitates in the last two lanes were treated with DTT before electrophoresis. The lower panel of this figure represents a direct western blot obtained after SDS-PAGE of cell extracts. (C) Detection of GSH-linked p53 by Biot-GST overlay. p53 IPs from control and 6 h CPT-treated U87MG and HCT116 cells were electrophoresed on non-reducing SDS-gels and blotted. The membranes were incubated with Biot-GST protein followed by Strep-HRP staining as described in Methods. (D) Ability of anticancer drugs to generate glutathionylated p53 in U87MG cells. Cells were exposed to doxorubicin (DOX, 10 μM), amsacrine (AMA, 25 μM), vincristine (VINC, 15 μM) for times indicated. p53 present in the cell extracts (500 μg protein) was immunoprecipitated, and the resulting blots were subjected to Biot-GST overlay assay, whose results are shown in the upper panel. The arrows on the left and right point to the position of GSH-linked p53 band, which was not present in the untreated control (Con). The identity of lower band is not known. The lower panel of this figure shows the kinetics of p53 accumulation assessed by western blot analysis of extracts from cells treated with the chemotherapy agents (DOX, AMA, and VINC).
Ability of anticancer drugs to induce glutathionation of p53
For testing the possibility of p53 conjugation with GSH in cellular response to DNA damage, the U87MG and HCT116 cells were treated with camptothecin (CPT) for times specified and p53 was immunoprecipitated. The Western blots generated there from were probed with antibodies to GSH. As expected, CPT increased p53 expression in a time dependant manner (direct western shown in lower panel of Fig. 3B). Upper panel of Fig. 3B shows that glutathionated p53 (bands marked with an *) appeared after DNA damage with different kinetics in the two cell lines. In HCT116 cells, marginal levels of GSH-modified of p53 appeared 6 h after damage followed by its decline at 10 h; however, in U87MG cells its levels were significantly higher at 3 and 6 h followed by decrease at 10 h. The 100 kDa protein band recognized by GSH antibodies appears to be p53 dimer, cross-linked by a disulfide bond. DTT treatment before PAGE led to the loss of the monomer and dimer protein bands, validating the specificity of the anti-GSH antibodies (last two lanes in Fig. 3B). Far-western analysis (overlay) using Biot-GST (27) was also used to verify the presence of GSH-conjugated p53 after DNA damage. For this, p53 immunocomplexes were electrophoresed, the resulting blots were incubated with Biot-GST protein, which retains the ability to bind protein-linked GSH residues (27); the protein-bound biotin groups were then detected by strep-HRP staining. Fig. 3C shows that both in U87MG and HCT116 cells, this method detected increased levels of glutathionylated p53 after DNA damage.
Many studies have shown that various anticancer drugs induce ROS including oxygen free radicals, and cause oxidative stress (38). Therefore, we chose agents with these properties, namely, doxorubicin (DOX), and amsacrine (m-AMSA), which possess an anthracycline ring capable of quinone-semiquinone inter conversions and vincristine (VINC), a non-DNA binder, and analyzed kinetics of p53 thiolation in U87MG cells using the Biot-GST overlay. This method specifically detects the glutathionylated p53, but not the unmodified p53, relying on the recognition of GSH moieties present in the mixed disulfides by biotin-linked GST (27-29), and the subsequent detection of biotin groups in the sandwich by Steptavidin-conjugated HRP. The data in Fig. 3D reveal that while untreated control (far right lane) had no modified p53, the three anticancer drugs generated the glutathionylated p53 with different kinetics. Thus, when the total p53 expression levels (assessed by direct western analysis) were highest at 10 h post-doxorubicin and post-m-AMSA treatments (Fig. 3D, lower panel), the thiolated p53 was at its lowest, and in case of vincristine, the modified and unmodified p53 rose in parallel. Efforts were made to assess the extent of p53 glutathionylation in cells after DNA damaging and oxidative treatments described in this report; for this, we compared the band intensities of total p53 and glutathionylated p53 from a range of equivalent protein amounts in four separate experiments, and quantitating the bands shifted by glutathionylation (see Fig. 3A). These calculations showed that a maximum of 6-8% of p53 generated was thiolated during cell stress. The coexistence of glutathionated p53 with its unmodified form implies that a part of the p53 induced after DNA damage may remain inactive, and that reversible S-thiolation may replenish active p53 as its biological transactions are pursued.
Tumor cell exposure to oxidants induces p53 glutathionylation
Because the extent of protein glutathionylation increases significantly after oxidative stress (17, 18), we subjected the U87MG cells to acute oxidative stress by exposing to moderate concentrations of H2O2, tert-butyl hydroperoxide (TBH), diamide for 15 min, and assessed the glutathionylation of p53 protein. First, p53 IPs prepared from the oxidant-treated cells were electrophoresed, and the resulting western blots were probed with antibodies to GSH. The acute oxidant stress resulted in a concentration dependent increase of glutathionated p53 (Fig. 4A). The oxidants used here have been shown previously to increase protein glutathionylation (39, 40), which could occur through oxidation of reactive cysteines in proteins or oxidation of glutathione, and subsequent formation of mixed disulfides (17). In the second method, the blots containing the resolved p53 IPs were exposed to Biot-GST protein followed by western analysis using strep-HRP; this procedure confirmed the increased levels of modified p53 after H2O2 treatment (Fig. 4B).
Figure 4. Oxidative stress increases glutathionylated p53 levels in human tumor cells as determined by IP/western, Biot-GST-overlay, and glutathionation-susceptibility assays.
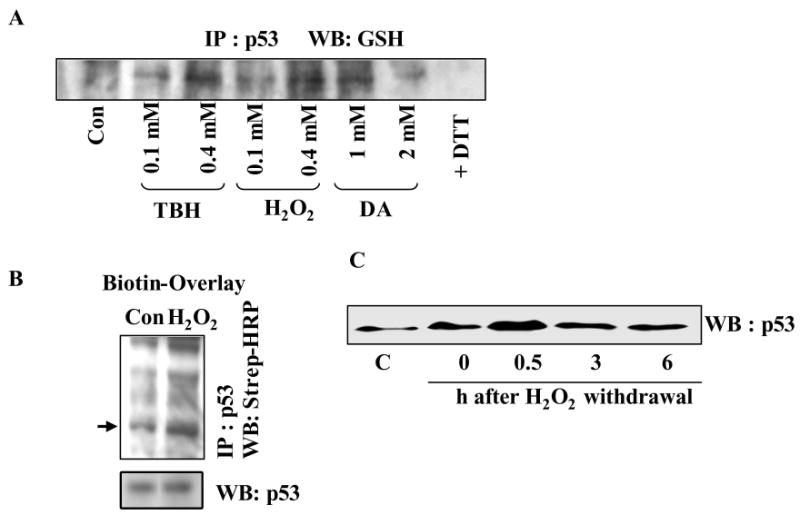
(A) Oxidants increase p53 glutathionylation in U87MG cells. Cells were treated with diamide (DA), H2O2 and TBH at concentrations indicated for 15 min. p53 was immunoprecipitated, and the IPs were processed for western analysis using GSH antibodies. In the last lane, p53 IP from 2 mM diamide-treated cells was exposed to 5 mM DTT before electrophoresis. (B) p53 immunoprecipitates from control and H2O2 (0.4 mM, 15 min) treated cells were electrophoresed, and the resulting blot was subjected to Biot-GST overlay assays. Arrow points to the glutathionated p53 band. Direct western blot of p53 is shown in the lower panel. (C) Changes in glutathionylation susceptibility of p53 after H2O2 and DNA-damaging treatments. U87MG cells were exposed to H2O2 (0.4 mM for 15 min) and further postincubated in oxidant-free medium for times shown. Equal protein amounts were then mixed with 0.1 ml GSH-Sepharose beads (ref. 31). All of the bound proteins were eluted with 10 mM DTT, resolved by SDS-PAGE and western blotted for p53.
Changes in glutathionylation-susceptibility of p53 protein following tumor cell exposure to oxidizing agents
Recently, we demonstrated that proteins with reactive cysteines can bind to immobilized glutathione matrices (GSH or GSSG) through disulfide bond formation in a reaction that mimics intracellular glutathionylation, and that such a binding can reflect alterations in cysteine redox status for specific proteins (31). Therefore, we induced acute oxidative stress by exposing U87MG cells to 0.5 mM H2O2 for 15 min followed by their post-incubation in oxidant-free medium up to 6 h. At each time point, equal protein amounts (1 mg in cell extracts) were chromatographed on GSH- Sepharose, and assessed the p53 protein levels present in the bound fractions (eluted by DTT) by Western analysis (31). p53 became markedly susceptible for glutathionylation 30 min after H2O2, which declined at later times (3 and 6 h), possibly, due to intracellular dethiolation (Fig. 4C). Both the early oxidation forms of p53 (sulfenic and sulfinic acids), as well as the glutathionylated p53 possess the ability to interact with GSH, and this may explain the increased binding of the p53 protein to the affinity-matrix in H2O2-treated cells. Collectively, the data presented in Figures 3 and 4 reveal for the first time that p53 exists as a glutathionylated protein at low levels in cells, however, the modification increases markedly after DNA damaging or oxidizing treatments.
Coexistence of glutathionylation with activating phosphorylations in p53 induced after DNA damage
It is well established that p53 induced by different stress stimuli is exquisitely modulated by various posttranslational modifications including phosphorylation, acetylation, sumoylation and others, most of which regulate the tumor suppressor in a positive manner to accomplish its biological functions (4, 41). To verify the presence of glutathionylation in activated p53, HCT116 cells were treated with CPT, and p53 isolated there from was assessed simultaneously for the presence of GSH, serine phosphorylations, and the target genes transactivated by p53. Immunoprecipitates of p53 protein prepared from these cells were western blotted in parallel using anti-p53 or anti-GSH antibodies. Fig. 5A shows that p53 levels were increased as expected, and the GSH conjugation of the p53 was initially increased followed by a gradual decrease. p53 protein present in the same samples showed increased phosphorylation at serine 20 and serine 392 (Fig. 5B), both of which have been previously shown to accompany the DNA damage response (42, 43). Further, the levels of p21waf1, MDM2, and Bax proteins, whose gene transcription is well known to be activated by p53 (1, 44) were significantly elevated (Fig. 5B). The studies performed in an identical setting suggest that p53 increased in response to DNA damage was not only phosphorylated, but was also marginally glutathionylated, which constitutes a negative regulatory mechanism for the tumor suppressor.
Figure 5. Glutathionylation coexists with the positive-regulatory phosphorylations in activated p53, the modified protein is nuclear and functionally less active.
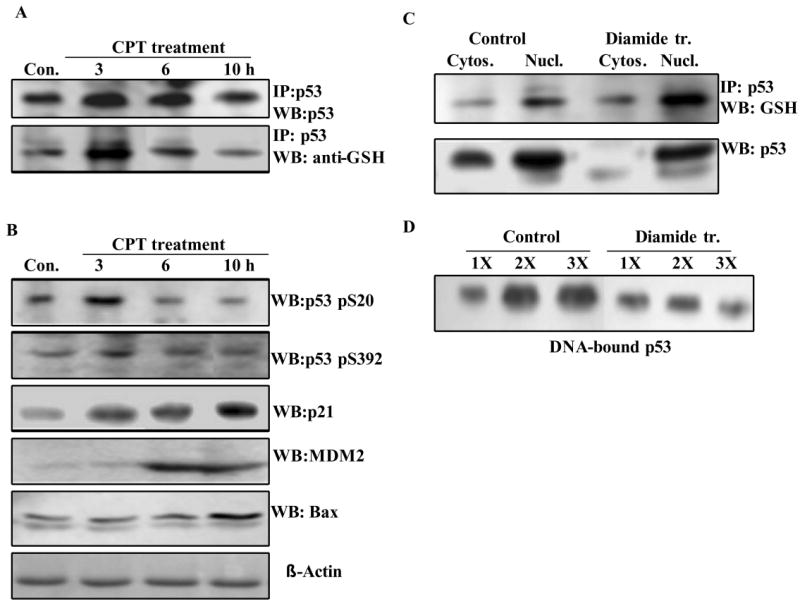
(A) HCT116 cells were treated with 25 μM CPT up to 10 h. At times specified, p53 present in the cell extracts (300 μg in all samples) was immunoprecipitated. The IPs were electrophoresed on non-reducing SDS-gels, and blotted. Parallel blots were probed using anti-p53 or anti-GSH antibodies. The extracts used for IP were also resolved by SDS-PAGE and blotted. These blots were incubated with p53 phospho-specific antibodies (phosphoserine 20 and phosphoserine 392), or antibodies to hMDM2, p21waf1 and human Bax proteins. The antigens were detected by routine western blotting. (B) Localization of glutathionylated p53 in nuclei. HCT 116 cells were treated with 1 mM diamide for 2 h. The cytosolic and nuclear fractions from control and diamide treated cells were separated using an extraction kit. Equal protein amounts (300 μg) from these samples were subjected to immunoprecipitation of p53 protein. The IPs were electrophoresed and Western blotted using anti-GSH antibodies to obtain the pattern shown in the upper panel. The levels of p53 in the cytosolic and nuclear fractions were assessed by direct Western blotting in the lower panel. (C) Binding of p53 to its consensus recognition sequence in nuclear extracts from diamide-treated cells. HCT116 cells were treated with 1 mM diamide for 3 h. Nuclear extracts were prepared using the Pierce extraction kit. Binding of p53 to its recognition sequence in the presence of increasing nuclear protein, 100 μg (1X), 200 μg (2X) and 300 μg (3X) was assessed by the DNA-affinity immunoblotting (DAI) procedure as described in Experimental Procedures.
Glutathionylated p53 is present in the nuclei and exhibits inefficient binding to DNA
Because p53 functions exclusively in nuclei, it was of interest to examine the subcellular location of the GSH-modified tumor suppressor. HCT 116 cells were subjected to acute oxidative stress (1 mM diamide for 2 h), the cytosolic and nuclear fractions were separated, p53 present in the extracts was immunoprecipitated, and western blotting using antibodies to GSH was performed. The data in Fig. 5C (upper panel) clearly show that nuclear p53 was slightly increased by diamide and it was glutathionylated. The lower panel of this figure displays the p53 levels assessed by direct Western blot analysis; the predominant nuclear location of p53 is again evident.
Next, the binding of p53 to its consensus recognition sequence in the nuclear extracts from control and diamide-treated HCT116 cells was determined by the DAI procedure. While p53 from untreated controls showed a protein concentration-dependent-increase in DNA binding, its counterpart in diamide-treated cells showed reduced DNA binding, which did not increase in the presence of additional nuclear protein (Fig. 5D). We suggest that oxidation of p53 protein and its glutathionylation mediate this functional inactivation.
Ability of glutathione-enhancing agents to dethiolate cellular p53 after DNA damage
Proof of glutathionylation as a physiologically relevant mechanism requires the demonstration of its reversibility, and functional changes in its substrates in cellular milieu. We sought to answer this question by inducing glutathionation in p53 through DNA damage, and then incubating the cells with cysteine prodrugs for increasing the intracellular GSH content, and examining the possible deglutathionylation of p53. The levels of p21waf1 protein, whose gene is transactivated by p53, were also assessed in this setting. N-Acetylcysteine (NAC) and glutathione ethyl ester (GEE) cell permeable non-toxic compounds that rapidly elevate GSH levels in cells (45, 46). In the first experiment, the HCT116 cells were exposed to CPT for 2 h, and subsequently incubated in the presence or absence of NAC for 5 h. Figure 6A shows that p53 was significantly glutathionylated in response to CPT alone, and lost about 50% of the modification when cells were incubated with NAC. Loss of glutathionylation was accompanied by a marginal, but, reproducible 30% increase in p21waf1 protein levels.
Figure 6. Augmentation of intracellular glutathione levels using N-acetylcysteine (NAC) or glutathione ethyl ester (GEE) results in the dethiolation of p53 in cells treated with DNA damaging or oxidizing agents.

(A) HCT 116 cells were exposed to 25 μM camptothecin for 2 h to induce p53 glutathionation. These cells and untreated controls were washed, and then incubated with 7.5 mM NAC (found to be optimal in separate experiments) for 5 h. p53 present in equivalent protein amounts (400 μg) was then immunoprecipitated, and western blotted using antibodies to GSH. Cell extracts (50 μg) were immunoblotted in parallel for detection of p21waf1 protein. (B) Following HCT116 cell treatment with tert-butyl hydroperoxide (TBH, 0.2 mM for 15 min) or camptothecin (CPT, 25 μM, 2 h), cells were washed and incubated with 4 mM GEE in the medium for 5 h. p53 immunoprecipitates prepared from cell extracts (400 μg protein) were processed for western blotting for the detection of p53 itself and glutathionated p53. p21waf1 protein levels were assessed by direct western blotting. (C) Following cisplatin treatment (cispt, 20 μM for 2 h), HCT116 cells were incubated in the presence or absence of 7.5 mM NAC for 5 h. Con. = Untreated control cells. P53 immunoprecipitates were western blotted using GSH antibodies for detection of glutathionated p53. Direct immunoblotting was performed for determining p53 and p21waf1 protein levels. (D) Effect of GSH depletion on p53 glutathionation. HCT116 cells were treated with buthionine sulfomine (BSO, 100 μM for 20 h). p53 immunoprecipitates from control and BSO-treated cells were western blotted using antibodies to GSH.
Similar experiments were performed to examine the dethiolation of p53 in the presence of GEE after tumor cell treatment with an oxidant or CPT (Fig. 6B). The wt p53 present in HCT116 cells was slightly glutathionylated after oxidative stress induced by TBH and DNA damage by CPT, which were reduced by 50 to 75% in the presence of GEE. Again, the p21waf1 protein levels were slightly increased (10-40%) after deglutathionylation of p53 (Fig. 6B). Cisplatin (cispt), a clinically used anticancer agent which damages DNA by platination and formation of intrastrand crosslinks induced a 3-fold increase in p53 levels, and a discernible increase in the glutathionation of p53 over untreated controls (Fig. 6C). Incubation of cisplatin-treated cells with NAC reduced the glutathionylation of p53 to the levels found in untreated controls. Approx. 10% increased levels of p21waf1 protein accompanied the dethiolation of p53 in this setting. Collectively, the data presented in this section show that thiolation and dethiolation is a dynamic process for human p53 with potential to impact its functions.
Depletion of cellular GSH downregulates p53 glutathionylation
Glutathione plays a key role in cellular defense response and its reduction has been shown to both activate and suppress apoptotic stimuli (47). To gain insight into the redox signaling of p53 in the context of present study, we examined the effect of GSH deficiency on glutathionylation of p53. For this, HCT116 cells were exposed for 20 h to 50 μM buthionine sulfoximine (BSO), which lowers cellular GSH by irreversibly inhibiting the rate-limiting enzyme γ-glutamylcysteine synthetase in its biosynthesis (48). Tumor cells retained approximately 10% of their GSH content compared to controls after this treatment (not shown). The steady-state glutathionylation of cellular p53 as determined by the immunoprecipitation of p53 followed by western analysis using anti-GSH antibodies showed a marked reduction in BSO-treated cells (Fig. 6D). A reduction of the GSH (and GSSG), which are substrates for glutathionylation may to explain these results, however, depletion of the tripeptide is likely to generate oxidative stress signals, and more studies are required to determine the consequences of decreased p53 glutathionylation in GSH-deficient cells.
Identification of cysteines 124, 141, and 182 as the sites of glutathionylation in rp53 protein by mass spectrometry
For obtaining biochemical evidence on the identity of reactive cysteines that conjugate with GSH, we performed electrospray-ionization-tandem mass spectrometry (ESI-MS/MS). The p53 protein treated with GSH was run on non-reducing SDS gels and the comassie stained protein band was digested in-gel with trypsin. Search of the MS/MS spectra against the NCBI protein database using MASCOT produced 11 matches to tryptic fragments of human p53, accounting for 43% of the protein sequence. Further examination of the peptide signals produced the identification of three tryptic p53 cysteine GSH-conjugates. Table 1 displays the sequence of these peptides designated T6, T8 and T14, and their elemental composition. The cysteine residues 124, 141 and 182 were conjugated with GSH in peptides T6, T8 and T14 respectively. The isotopic distributions of these glutathionated peptides are shown in Fig. S2 in Supporting Information. The masses, calculated and measured for these GSH-linked peptides differed only by 20 ppm (Table 1).
Table 1.
Sequence and elemental composition of the p53 glutathionylated tryptic peptides identified by ESI-MS/MS
| p53
Peptide |
Peptide sequence | Elemental formula of the glutathione adduct | Calculated monoisotopic peak | Measured monoisotopic peak | Error ppm |
|---|---|---|---|---|---|
| T6 | 121SVTCTYSPALNK132 | C65 H105 N17 O25 S2 | 794.8533 | 794.8671 | 17.3 |
| T8 | 140TCPVQLWVDSTPPPGTR156 | C92 H143 N25 O31 S2 | 1070.999 | 1080.0212 | 20.5 |
| T14 | 182CSDSDGLAPPQHLIR196 | C77 H124 N24 O29 S2 | 957.4282 | 957.4460 | 18.5 |
The sequences of the GSH-modified tryptic peptides of p53 obtained from the in-gel digests are shown. The amino acid residue numbers corresponding to human p53 protein are shown in superscript. The cysteines modified are shown in bold.
To further confirm the identity of reactive cysteines that undergo glutathionylation in p53 protein, rp53 was treated with the SH-alkylator, biotinylated maleimide for 5 min for selective labeling of the exposed cysteines (34). The biotin-labeled peptides were purified from the tryptic digest, and the MALDI spectrum recorded showed a strong signal at m/z = 2305.0867 (see Fig. S3 in Supporting Information), whose sequence was identical to the T8 peptide, recorded earlier by ESI-MS/MS for the glutathionated rp53 (Table 1). To accurately assess the biotin maleimide attachment to Cys141 found in this peptide, tandem mass spectrometry was performed after low-energy and high energy CID fragmentation. The MS/MS fragmentation spectrum displayed in Fig.7 reveals the breakage pattern of this Cys-biotinylated peptide under low-energy CID conditions. The presence of the biotin tag is clearly indicated by the two abundant diagnostic ions at m/z = 227.1 and 259.1. A prominent b2 ion present at m/z = 656.25 indicates that the biotin tag is located on the first two N-terminal amino acids; this notion is also confirmed by the presence of y15 ion at m/z 1649.80. Fragmentation of the singly charged protonated molecular ion of the T8 peptide under high-energy CID conditions showed a modified y16 fragment ion containing a portion of the biotin-maleimide tag with a mass of 199.1 Da (see Fig. S4 in Supporting Information). The cleavage of the biotin tag occurred simultaneously with the cleavage of the peptide backbone in this case. The results from the low and high-energy CID MS/MS spectra unambiguously demonstrate the presence of biotin probe on Cys141 from T8 peptide of p53. Taken together, evidence obtained from ESI and MALDI mass spectrometry clearly prove that cysteines 124. 141 and 182 in human p53 are facile targets for GSH modification, and Cys141 is probably the most reactive among them, and consequently, is likely be highly prone for glutathionylation in cells.
Figure 7. QTOF MS/MS spectrum of T8 biotinylated peptide.
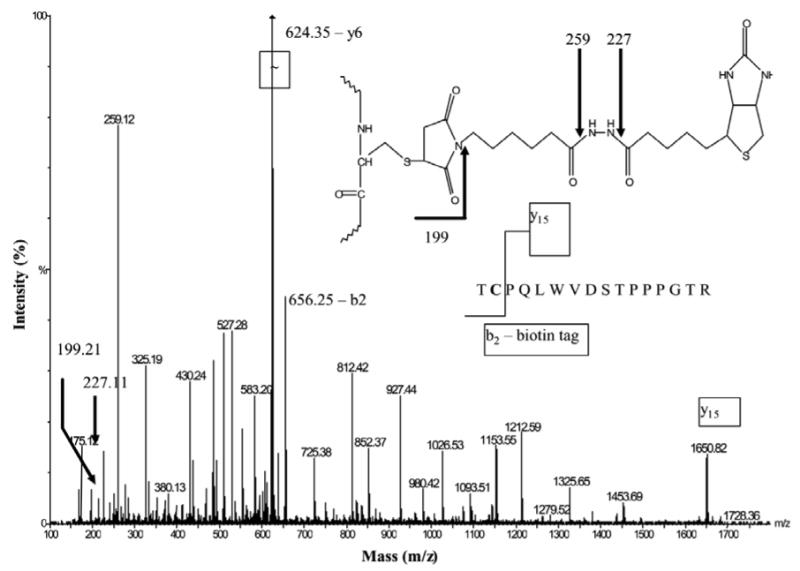
rp53 was treated with biotin-maleimide, trypsinized, and the biotin containing peptides were purified on monomeric avidin resin (32). The doubly protonated molecular ion at m/z = 1153.03 (T8 peptide of p53, 140TCPVQLWVDSTPPPGTR156) was fragmented in the collision cell of a QTOF instrument under low-energy conditions (42 eV). Three fragmentation ions (m/z = 199.2, 227.1 and 259.1) indicate the presence of the biotin tag pictured in the inset. The presence of the y15 ion m/z = (1649.80) and a b2 (m/z = 656.25) ion containing the whole biotin-maleimide tag, locates the biotin tag on Cys141.
Evidence that glutathionylation sites in cellular p53 are likely to be same as found in vitro
Majority of the reactive cysteines targeted for glutathionation are located on protein surface, possess lower pka, and are readily accessible to solvents, oxidants, and sulfhydryl-reactive agents (34). Two approaches were undertaken to test whether the cysteines identified as sites of glutathionylation in the rp53 protein (Table 1 and Fig. 7) represent the residues prone for this modification in cellular p53 as well. Cysteines 124, 141 and 182 identified by mass spectrometry have been confirmed as exposed residues by us (see Fig. 9B) and others (21, 22). The first approach involved the analysis of protein microenvironment near these cysteines in the native crystal structure of human p53 (36). The reactivity of Cys residues within proteins is variable and depends on the local charge in the immediate vicinity, which determines their pKa (14). The pKa depends on the charge interactions of the thiol with the negatively charged side chains of nearby amino acids. We assessed the microenvironment of all seven cysteines present in the non-zinc binding cluster in the DBD of the p53 crystal structure (Table 2). The results clearly show that cysteines 124, 141, and 182 which we identified as targets for glutathionation are located in close proximity of one or two charged residues, which is likely to impart a thiol anionic character on these cysteines. In contrast, the other cysteines which were not in a charged environment (Cys135 and 229), or whose charges were negated through salt bridge formation (Cys275) or hydrogen bonding with DNA (Cys277) were not detected as targets for GSH modification. Further, the presence of two charged residues, Lys139 and His233 near Cys141 validates its identity as probably the most reactive cysteine, borne out in our mass spectrometry studies.
Figure 9. Protein cross-linking and structural modeling of S- glutathionated p53.
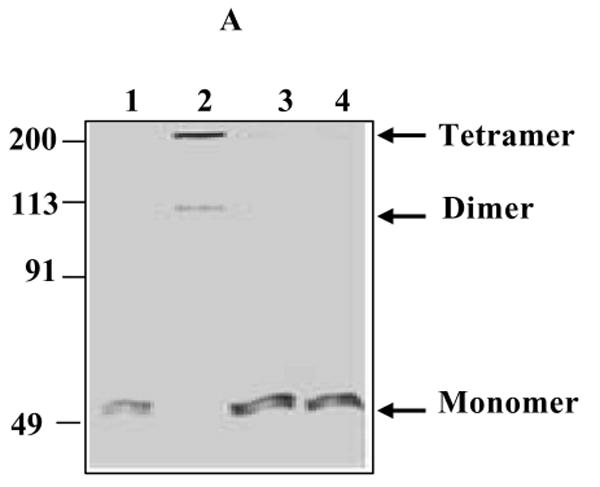

(A) Glutaraldehyde cross-linking and gel analysis of the oligomeric structure of glutathionated p53. rp53 (5 μg) was treated with 5 mM GSH or GSSG for 1 h. The thiols present in the samples were removed by gel-filtration on Bio-gel P6 spin columns. Glutaraldehyde was then added to the eluted proteins to 0.1% concentration, and incubated for 10 min. Next, SDS-sample buffer containing 2-mercaptoethanol was added, and the samples were resolved by SDS-PAGE, and the gel stained with Comassie Blue. Lanes: 1, untreated rp53; 2, cross-linked rp53; 3, rp53 incubated with GSH, and then cross-linked; 4, rp53 incubated with GSSG, and then cross-linked. (B) A ribbon representation of the crystal structure of p53-DNA complex. The p53 chains A and B found in the crystal structure (1TSR) are colored magenta and yellow respectively. These monomers form a dimer and stably associate with the consensus sequence in the crystal structure. Cys124 and Cys141 are present at the dimerization interface and are shown as color coded ball- and –sticks. Note the close proximity of these residues in the native structure. (C) Schematic ribbon representation of the energy-minimized model of p53 glutathionated at Cys124. Procedures used for protein modeling are described in Experimental Procedures. Structural alterations due to the modification resulted in the retraction of the DNA presenting loop that harbors Lys120 in the B monomer (yellow, displaced loop marked with **). Changes also occurred at the dimer interface loop, and elsewhere in the molecule (some are marked with an *). The deformation at the protein interface was determined to inhibit the dimerization. Modeling of a glutathione to Cys141 induced similar perturbations, and the spatial closeness of cysteines 124 and 141 indicated that both may not be thiolated in a single molecule.
Table 2.
Microenvironment of non-zinc binding cluster of cysteines in p53-DNA crystal structure
| Cysteine site | Neighboring amino acid(s) with positive charge | Inter-residue distance (Å) | Reactive Cys?* | Other comments |
|---|---|---|---|---|
| Cys124 | Lys139 | 7.3 | Yes | - |
| Cys135 | - | - | No | No positively charged residues within 8 Å |
| Cys141 | Lys139
His233 |
6.3
5.5 |
Yes | - |
| Cys182 | His178
His179 |
4.1
5.7 |
Yes | - |
| Cys229 | - | - | No | No positively charged residues within 10 Å |
| Cys275 | Arg273 | 3.6 | No | Arg273 forms a salt bridge with Asp281, therefore, it is likely to exert less influence on Cys275. |
| Cys277 | Arg280 | 4.1 | No | Cys277 and Arg280 respectively form one and two hydrogen bonds with guanine in DNA. This will reduce the charge effects on Cys277. |
The crystal structure of p53-DNA complex (1TSR) was imported from the Protein Data Bank and the native protein structure was visualized using the Argus Lab 4.0.1 molecular modeling and graphics program (http://www.arguslab.com), which also allowed measurement of inter-residue distances.
= Residues found to be reactive in this study. The microenvironment of the zinc-binding cysteines 176, 238 and 242 was not examined as they may be less accessible for redox modifications.
Second, to gain further insight into the reactive cysteines in cellular p53, we studied the kinetics of Biotin-maleimide incorporation into the cellular p53. The glutathionylatable reactive cysteines are generally located on protein surfaces, and are solvent accessible; these thiols are readily alkylated by agents such as the biotinylated maleimide (34). Therefore, the extent of biotin-maleimide incorporation is a good indicator of cysteine reactivity, and hence, a measure of glutathionylation potential. Therefore, we assessed the alkylator incorporation into the p53 protein. For this, dialyzed HCT116 cell extracts were exposed to biotin-maleimide for 5-25 min in the presence or absence of 5 mM NEM, the wt p53 present was analyzed by immunoprecipitation-western blot analysis using strep-HRP. Figure 8 shows a rapid incorporation of biotin maleimide into cellular p53, the kinetics of which are very similar to Biot-GSSG labeling of rp53 observed in Fig. 1A. These results, together with the association of charged environment surrounding Cys124, 141, and 182 (Table 2) strongly suggest that the same cysteines are likely to be glutathionation targets in cellular p53 protein.
Figure 8. Rapid incorporation of Biot-maleimide into cellular p53.
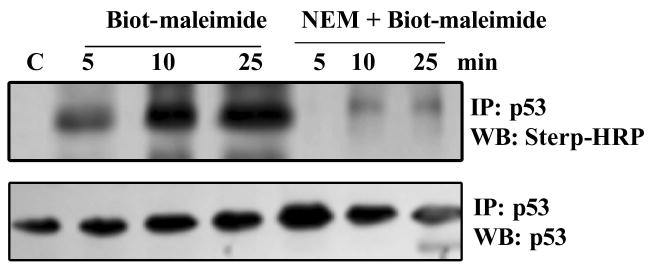
Biotin-labeled maleimide incorporation into the endogenous p53 protein in HCT116 cells was determined as described in Methods. Briefly, HCT116 cell extracts were exposed to 5mM Biot-maleimide with or without NEM pretreatment. 5 mM NEM was also added to terminate the reactions. p53 immunoprecipitates were prepared from these extracts and western blotted using the Strep-HRP reagent.
Protein electrophoresis reveals that glutathionylation inhibits p53 oligomerization
Because the Cys124 and 141 are located on p53 surface, in the beta sheet behind the loop-sheet-helix (LSH) structure of p53 that contacts the DNA (ref. 21, 34 and Fig. S1 in Supporting Information), we surmised that glutathionylation of these residues may inhibit p53 binding to DNA through structural perturbations and/or altering the protein oligomeric state. Therefore, we used glutaraldehyde to crosslink the rp53 protein that had been treated or untreated with GSH. The samples were then resolved by SDS-PAGE. The gel stained with Comassie blue shown in Fig. 9A reveals that while untreated p53 was cross-linked in its oligomeric state by glutaraldehyde (lane 2), glutathionated p53 failed to form these oligomers, after cross-linking (lanes 3 and 4). Exposure of the GSSG-treated rp53 to DTT prior to glutaraldehyde-treatment restored the protein oligomerization (not shown). Since p53 in its native state exists and binds its target sequences as a tetramer (dimer of a dimer), these data suggest that GSH-conjugation to the reactive cysteines introduces structural perturbations that hinder the dimeric association of p53 protein. The data are also consistent with the reported inability of cysteine-oxidized p53 to form oligomers (22, 49).
Simulation of glutathionylation in the crystal structure of p53-DNA complex demonstrates the inhibition of DNA-binding and disruption of protein dimerization
To gain a molecular and structural understanding by which glutathionylation inhibits p53 binding to DNA, we bonded the GSH molecule to cysteines 124, and 141 in the crystal structure of p53-DNA deduced by Cho et al. (36; PDB ID code: 1TSR). This structure has been used for modeling cancer mutations found in p53 and their DNA interactions (50), and validated recently for study of p53 subunit interactions (51). It shows three copies of p53 monomers (A, B, and C) crystallized with the DNA-consensus element. Of these, the A and B chains form a dimer as shown in Fig. 9B and bind DNA, interacting with the major grooves in one turn of the duplex. Although there is some interaction between B and C, the C monomer does not contact DNA. The DBD of monomer B (yellow ribbon in Fig. 9B) makes several hydrogen bonds with the consensus site (GGGCA), with lysine 120 contacting the second guanine. Cys182, which we found to be a site for glutathionylation is located in the H1 α-helix of the protein, faces away from the DBD in the crystal structure (Fig. S1 and ref. 34), and is seen at the B-C dimer interface. Our modeling indicated that Cys182 modification will not affect the DNA binding with p53. Interestingly, the Cys124 and Cys141, the two other sites for glutathionylation lie in very close proximity in the native structure of p53 at the dimer interface (marked in Fig. 9B). Due to the spatial closeness of Cys124 and Cys141, it appears that only one of these residues can be modified by glutathionylation, but not both. Figure 9C shows the same interface in the crystal structure after linking a GSH molecule with Cys124. Energy minimization showed that Cys124 modification not only inhibits the formation of the dimer but also distorts the loop containing Lys120 which normally interacts with DNA (see the displacement of yellow monomer from DNA in Fig. 9C). Therefore, p53 interaction with DNA as well as the dimer formation between the monomers will not be possible after glutathionylation of Cys124. Identical structural impairment was observed after bridging a GSH molecule to Cys141 in p53 crystal structure, implying that thiolation of either of these reactive cysteines could alter p53 protein structure by blocking its oligomerization during oxidative stress. The protein modeling data together with different biochemical approaches described provide strong proof that structural alterations downregulate the p53 protein during the harmful condition that occurs when there is an excess of free radicals, a decrease in antioxidant levels, or both.
Discussion
Despite the significant contributions of p53 to antioxidant mechanisms that control genetic stability (52), its transcription factor activity is highly prone to oxidative inactivation during physiological and pathological stresses that generate ROS and RON, and their by-products, H2O2, and hydroxyl radicals, which all arise as an inevitable consequence of aerobic metabolism. Additionally, the UV, γ-rays, hypoxia, chemotherapy drugs and inflammatory cytokines contribute significantly to cellular oxidative stress, and p53 modulation (3, 53). This study provides first and convincing evidence for the modification of specific cysteines by GSH conjugation in human p53 protein, and a molecular explanation for the resulting inhibition of the tumor suppressor after oxidative stress. Several lines of evidence show that p53 is subjected to glutathionylation, in vitro and in cells exposed to DNA damaging agents, oxidants of endogenous (H2O2) and exogenous (diamide, TBH) origin. The results also show that glutathionylation imparts structural effects on p53 protein.
Significant levels of glutathionylation was observed for p53 at physiological thiol concentrations (Fig. 1C), and this was stimulated by increased GSSG/GSH ratios in vitro, and oxidative/ nitrosative stressed cells. While the augmentation of glutathionated p53 in oxidant treated cells was expected, its increase after DNA damage was intriguing and warrants discussion. Most of the posttranslational modifications p53 accrues after genomic injury regulate it positively by increasing the transcriptional factor competency, so that the check point functions are pursued appropriately. However, our results show that S-thiolation is a negative regulatory mechanism for p53, and it coexists with the positive-regulatory phosphorylations. Further, the glutathionylated p53 was present in the nucleus, suggesting an important role in downregulating its functional activities.
In our hands, the DNA damaging anticancer drugs (CPT and cispt) at concentrations routinely used by researchers for p53 studies resulted in approximately 6 to 10% of total p53 being glutathionylated. The modification reached much higher levels during oxidative stress. We suggest that p53 glutathionation in vivo could be significant, and may vary depending on the cell type, stress level, spatial considerations, nuclear microenvironment, and GSH abundance. The modified p53 (both recombinant and cellular) was less efficient in binding to DNA. Therefore, it appears that a small proportion of p53 generated in stressed cells becomes thiolated, remains inactive as a transcription factor, and coexists with the unthiolated active form. The molecular basis of p53 glutathionylation to divergent stimuli is currently unclear. However, given the paradigm that stress signals are transmitted to p53 by posttranslational modifications (4, 41), we propose that p53 thiolation is an acute cellular response to various stresses including genomic injury, redox imbalance, and other adverse stimuli. Similar to p53, the conserved cysteines in other transcription factors such as the c-jun /AP-1, NF-κB, and Pax-8 also undergo glutathionylation, leading to their functional inactivation (54-56). p53, being a key player in the expression of many proapototic genes (PUMA, Bax etc), it is tempting to speculate that inactivation of the tumor suppressor through glutathionylation may represent an acute adaptation strategy to suppress the apoptotic signals generated in stressed cells. This defensive response may facilitate a reprogramming of gene expression for cytoprotection, and/or prevent cell death (57, 58). Further studies are required to test this hypothesis. Nevertheless, the glutathionated p53 may serve as a small pool of inactive suppressor protein, which could be drawn upon as the situation demands by conversion to a competent transcription factor through non-enzymatic or enzymatic dethiolation. Consistent with this notion, we found that p53 thiolation (following DNA damage or oxidant stress) can be reversed by increasing the intracellular GSH levels. The dethiolation of p53 achieved in the presence of NAC or GEE was accompanied by a modest increase in p21waf1 protein as well. The known interactions of p53 with the thiol-exchange proteins (Ref-1, thioredoxin reductase) (12, 13) are also consistent with this premise. The negative regulation of p53 glutathionylation bears some analogy with the MDM2-p53 feed back loop (2), in which MDM2 transactivated by p53 functions to downregulate the tumor suppressor through ubiqutination-dependent proteolysis.
Human p53 possesses two clusters of cysteines in the DBD, which, interestingly, is devoid of any other posttranslational modifications. A striking feature of these cysteines is that they are all located in one area of p53 between the β-sandwich and the specific DNA-binding loops and helix (19). One cluster of three cysteines coordinates a zinc atom. The other cluster, harboring the cysteines 124, 135, 141, and 277 is located adjacent to the zinc–binding site in or near the loop-sheet-helix (LSH) region that makes contacts with DNA (19, 34, and Fig. S1). The cysteines in the non-zinc binding cluster are nicely positioned to modulate the access of the LSH to the p53 recognition site. Of these, the cysteines at positions 124, 141, and 277 have been hypothesized previously to mediate the redox regulation of p53, particularly, because, of their surface accessibility (21, 53). In agreement, our mass spectrometry studies provided direct evidence for the site-specific glutathionylation of cysteines 124, 141 and 182 in the rp53 protein.
The protein cross-linking of glutathionated p53 (Fig. 9A) and molecular modeling studies (Fig.9B and C) provided new information on the core domain interactions and structural perturbations induced by glutathionylation for p53. A discussion of how p53 DBD interacts with DNA and contributes to the oligomerization of the protein is relevant in the context of our findings. p53 binds to DNA as a symmetrical tetramer consisting of two head-to-head (→←→←) dimers, with each dimer occupying a half-site of its recognition sequence (59). Although a single dimer is sufficient for DNA-binding, interaction of the second dimer with the DNA dramatically enhances the binding affinity (>50-fold) and stabilizes the tetramer (49, 59). This cooperative dimer-dimer interaction occurs even in the absence of the tetramerization domain (36, 60, 51), demonstrating the existence of an efficient core-core interactions and this has been the subject of many studies (60-62). Evidence indicates that p53-DBD contributes significantly to the quaternary structure despite the fact that the tetramerization domain is structurally independent (60). For example, replacement of the p53 tetramerization domain with the dimerization domain of GCN4 transcription factor bestows a near wild-type p53 tumor suppression activity in vivo (63). Our studies with p53-DNA crystal structure revealed for the first time that Cys124 and 141, present in a β–sheet just beneath the recognition loop are at the dimer interface (Fig. 9B, C, and S1). Further, the short H1 helix of p53 (Pro177-Cys182, the latter being a site of glutathionylaton), which resides near the zinc atom has been shown to play an essential role in the intermolecular DBD dimerization; mutation of cys182 (C182A) reduces the diffusion coefficient of complexation with consensus DNA (60, 61). Interestingly, a previous study, based on theoretical considerations, hypothesized that Cys182 of p53 could be a target for glutathionylation (22). These authors based on molecular modeling, concluded that GSH conjugation with Cys182 would inhibit the dimerization of p53 (22). However, because Cys182 is juxtaposed to the recognition loop in the crystal structure, our studies did not support this possibility. Nevertheless, our data revealed that glutathionylation of either Cys124 or Cys141 could occur in a single p53 molecule, but not together due to their closeness and steric hindrance. The energy-minimized model of S-glutathionated p53 (either at Cys124 or 141) suggested that the modification would result in retraction of the recognition loop from DNA and a dimer interface will not form. These data agree with the previous reports showing the failure of oxidized p53 to form tetramers (22, 49), and provide a molecular explanation for inhibition of p53 function by S-thiolation. Our results hint that cysteines 124 and 141 play important roles in core domain architecture of p53, and invite further structural studies to assess their contributions.
In summary, our observations shed light on new and understudied aspects of p53 redox regulation. The critical redox-sensitive cysteines in p53 were shown to reside in the proximal DNA binding domain. In a reducing environment, these residues appear to organize the DBD and facilitate subunit interactions. However, when confronted by oxidative insult and other stresses, the reactive cysteines or their early oxidation products appear primed to undergo conjugation with GSH. While this modification may protect the critical and conserved cysteine residues of p53 (which are rarely mutated in human cancers) from irreversible oxidations, it also inhibits various p53 functions, albeit temporarily; the non-enzymatic and enzymatic thiol-exchange mechanisms have the ability to reduce the mixed disulfide linkages and restore p53 function. Our results predict that a non-glutathionatable p53 protein may escape the redox-regulation, and may become more active than wild-type p53 in response to oxidative stress; consistent with this notion, Cys141Arg has been shown to be a dominant mutation for p53 (64), and Cys124Ser mutation increase the DNA-binding compared to wild-type p53 (20). Overall, the findings provide novel insight into the biochemical mechanisms that affect the p53 protein during cellular stress and possess clear relevance to different aspects of p53 in tumor biology, anticancer therapy as well as targeted chemoprevention. For example, whether a persistent S-thiolated state of p53 initiates oncogenic events, whether p53 mutant proteins are susceptible for glutathionylation, and whether S-thiolated p53 is a target for enhanced degradation, are a few questions that emerge from this study.
Supplementary Material
Locations of cysteine residues in the three-dimensional ribbon structure of p53 protein (Figure S1), mass spectra of thiolated peptides from p53 (Figure S2), MALDI-TOF spectrum of TCPVQLWVDSTPPPGTR peptide (T8) labeled with biotin-maleimide (Figure S3), MS/MS spectrum of the T8 peptide (biotin-maleimide conjugated molecular ion at m/z = 2305.1 on the 4700 proteomics analyzer (Figure S4). This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank Dr. Jennifer Nyborg (Colorado State University) for the his-tagged p53 expression vector.
Footnotes
This work was supported by Grants from NIH (RO1 CA97343) and Women's Health Research Institute of Amarillo to KSS. Part of this study was presented at the 97th annual meeting of the American Association for Cancer Research, Washington, DC, April 1-5, 2006.
Abbreviations: Biot-GSH, biotinylated glutathione; Biot-GSSG, biotinylated glutathione-disulfide; Biot-GST, biotinylated glutathione S-transferase; CID, collision-induced dissociation; Cispt, cisplatin; CPT, camptothecin; DA, diamide; DAE/NO, 2,2-diethyl-1-nitroso-oxyhydrazine sodium salt; DAI, DNA- affinity immunoblotting; DBD, DNA-binding domain; DTT, dithiothreitol; ECL, enhanced chemiluminiscence; EMSA, electrophoretic mobility shift assay; ESI, electrospray ionization; GEE, glutathione monoethyl ester; IP, immunoprecipitation; MALDI, Matrix-assisted laser desorption/ionization; NAC, N-acetyl-L-cysteine; NEM, N-ethylmaleimide; rp53, human recombinant p53 protein; Strep-HRP, streptavidin-linked horse radish peroxidase; TBH, tert-butyl hydroperoxide; TOF, time of flight; WB; Western blot.
References
- 1.Mills AA. p53: link to the past bridge to the future. Genes Develop. 2005;19:2091–2099. doi: 10.1101/gad.1362905. [DOI] [PubMed] [Google Scholar]
- 2.Inoue T, Wu L, Stuart J, Maki CG. Control of p53 nuclear accumulation in stressed cells. FEBS Lett. 2005;579:4978–4984. doi: 10.1016/j.febslet.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 3.Strosznajder RP, Jesko H, Banasik M, Tanaka S. Effects of p53 inhibitor on survival and death of cells subjected to oxidative stress. J Physiol Pharmacol. 2005;4:215–221. [PubMed] [Google Scholar]
- 4.Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4:793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- 5.Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cellular Physiol. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- 6.Hainaut P, Milner J. Redox modulation of p53 conformation and sequence-specific DNA binding in vitro. Cancer Res. 1993;53:4469–4473. [PubMed] [Google Scholar]
- 7.Verhaegh GW, Richard MJ, Hainaut P. Regulation of p53 by metal ions and by antioxidants: dithiocarbamate down-regulates p53 DNA-binding activity by increasing the intracellular level of copper. Mol Cell Biol. 1997;17:5699–5706. doi: 10.1128/mcb.17.10.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merwin JR, Mustacich DJ, Pearson GD, Merril GF. Reporter gene transactivation by human p53 is inhibited in thioredoxin reductase null yeast by a mechanism associated with thioredoxin oxidation and independent of changes in the redox state of glutathione. Carcinogenesis. 2002;23:1609–1615. doi: 10.1093/carcin/23.10.1609. [DOI] [PubMed] [Google Scholar]
- 9.Hammond EM, Giaccia AJ. The role of p53 in hypoxia-induced apoptosis. Biochem Biophys Res Commun. 2005;331:718–725. doi: 10.1016/j.bbrc.2005.03.154. [DOI] [PubMed] [Google Scholar]
- 10.Cobbs CS, Whisenhunt TR, Wesemann DR, Harkins LE, Van Meir EG, Samanta M. Inactivation of wild-type p53 protein function by reactive oxygen and nitrogen species in malignant glioma cells. Cancer Res. 2003;63:8670–8673. [PubMed] [Google Scholar]
- 11.Wu HH, Thomas JA, Momand J. p53 protein oxidation in cultured cells in response to pyrrolidine dithiocarbamate: a novel method for relating the amount of p53 oxidation in vivo to the regulation of p53-responsive genes. Biochem J. 2000;351:87–93. doi: 10.1042/0264-6021:3510087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanson S, Kim E, Deppert W. Redox factor 1 (Ref-1) enhances specific DNA binding of p53 by promoting p53 tetramerization. Oncogene. 2005;24:1641–1647. doi: 10.1038/sj.onc.1208351. [DOI] [PubMed] [Google Scholar]
- 13.Seemann S, Hainaut P. Roles of thioredoxin reductase 1 and APE/Ref-1 in the control of basal p53 stability and activity. Oncogene. 2005;24:3853–3863. doi: 10.1038/sj.onc.1208549. [DOI] [PubMed] [Google Scholar]
- 14.Claiborne A, Mallett TC, Yeh JI, Luba J, Parsonage D. Structural, redox, and mechanistic parameters for cysteine-sulfenic acid function in catalysis and regulation. Adv Protein Chem. 2001;58:215–276. doi: 10.1016/s0065-3233(01)58006-7. [DOI] [PubMed] [Google Scholar]
- 15.Claiborne A, Yeh JI, Mallett TC, Luba J, Crane E, Charrier VJ, Parsonage D. Protein-sulfenic acids: diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry. 1999;38:15407–15416. doi: 10.1021/bi992025k. [DOI] [PubMed] [Google Scholar]
- 16.Eaton P, Jones ME, McGregor E, Dunn MJ, Leeds N, Byers HL, Leung KY, Ward MA, Pratt JR, Shattock MJ. Reversible cysteine-targeted oxidation of proteins during renal oxidative stress. J Am Soc Nephrol. 2003;14:S290–S296. doi: 10.1097/01.asn.0000078024.50060.c6. [DOI] [PubMed] [Google Scholar]
- 17.Klatt P, Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur J Biochem. 2000;267:4928–4944. doi: 10.1046/j.1432-1327.2000.01601.x. [DOI] [PubMed] [Google Scholar]
- 18.Ghezzi P, Bonetto V, Fratelli M. Thiol-disulfide balance: from the concept of oxidative stress to that of redox regulation. Antioxid Redox Signal. 2005;7:964–972. doi: 10.1089/ars.2005.7.964. [DOI] [PubMed] [Google Scholar]
- 19.Rainwater R, Parks D, Anderson ME, Tegtmeyer P, Mann K. Role of cysteine residues in regulation of p53 function. Mol Cell Biol. 1995;15:3892–3903. doi: 10.1128/mcb.15.7.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buzek J, Latonen L, Kurki S, Peltonen K, Laiho M. Redox state of tumor suppressor p53 regulates its sequence-specific DNA binding in DNA-damaged cells by cysteine 277. Nucleic Acids Res. 2002;30:2340–2348. doi: 10.1093/nar/30.11.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hainaut P, Mann K. Zinc binding and redox control of p53 structure and function. Antioxid Redox Signal. 2001;3:611–623. doi: 10.1089/15230860152542961. [DOI] [PubMed] [Google Scholar]
- 22.Sun XZ, Vinci C, Makmura L, Han S, Tran D, Nguyen J, Hamann M, Grazziani S, Sheppard S, Gutova M, Zhou F, Thomas J, Momand J. Formation of disulfide bond in p53 correlates with inhibition of DNA binding and tetramerization. Antioxid Redox Signal. 2003;5:655–665. doi: 10.1089/152308603770310338. [DOI] [PubMed] [Google Scholar]
- 23.Srivenugopal KS, Shou J, Mullapudi SR, Lang FF, Rao JS, Ali-Osman F. Enforced expression of wild-type p53 curtails the transcription of the O(6)-methylguanine-DNA methyltransferase gene in human tumor cells and enhances their sensitivity to alkylating agents. Clin Cancer Res. 2001;7:1398–1409. [PubMed] [Google Scholar]
- 24.Van Orden K, Giebler HA, Lemasson I, Gonzales M, Nyborg JK. Binding of p53 to the KIX domain of CREB binding protein. A potential link to human T-cell leukemia virus, type I-associated leukemogenesis. J Biol Chem. 1999;274:26321–26328. doi: 10.1074/jbc.274.37.26321. [DOI] [PubMed] [Google Scholar]
- 25.Mullapudi SR, Ali-Osman F, Shou J, Srivenugopal KS. DNA repair protein O6-alkylguanine-DNA alkyltransferase is phosphorylated by two distinct and novel protein kinases in human brain tumour cells. Biochem J. 2000;351:393–402. [PMC free article] [PubMed] [Google Scholar]
- 26.Sullivan DM, Wehr NB, Fergusson MM, Levine RL, Finkel T. Identification of oxidant-sensitive proteins: TNF-alpha induces protein glutathiolation. Biochemistry. 2000;39:11121–11128. doi: 10.1021/bi0007674. [DOI] [PubMed] [Google Scholar]
- 27.Cheng G, Ikeda Y, Iuchi Y, Fuji J. Detection of S-glutathionylated proteins by glutathione S-transferase overlay. Arch Biochem Biophys. 2005;435:42–49. doi: 10.1016/j.abb.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 28.Armstrong RN. Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem Res Toxicol. 1997;10:2–18. doi: 10.1021/tx960072x. [DOI] [PubMed] [Google Scholar]
- 29.Listowsky I. Proposed intracellular regulatory functions of glutathione transferases by recognition and binding to S-glutathiolated proteins. J Pept Res. 2005;65:42–46. doi: 10.1111/j.1399-3011.2004.00209.x. [DOI] [PubMed] [Google Scholar]
- 30.Liu Y, Asch H, Kulesz-Martin MF. Functional quantification of DNA-binding proteins p53 and estrogen receptor in cells and tumor tissues by DNA affinity immunoblotting. Cancer Res. 2001;61:5402–5406. [PubMed] [Google Scholar]
- 31.Niture SK, Velu CS, Bailey NI, Srivenugopal KS. S-thiolation mimicry: quantitative and kinetic analysis of redox status of protein cysteines by glutathione-affinity chromatography. Arch Biochem Biophys. 2005;444:174–184. doi: 10.1016/j.abb.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 32.Schvchenko A, Wilm M, Vorm M, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 33.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 34.Hamnell-Pamment Y, Lind C, Palmberg C, Bergman T, Cotgreave IA. Determination of site-specificity of S-glutathionylated cellular proteins. Biochem Biophys Res Commun. 2005;332:362–369. doi: 10.1016/j.bbrc.2005.04.130. [DOI] [PubMed] [Google Scholar]
- 35.Wen B, Doneanu CE, Gartner CA, Roberts AG, Atkins WM, Nelson SD. Fluorescent photoaffinity labeling of cytochrome P450 3A4 by lapachenole: identification of modification sites by mass spectrometry. Biochemistry. 2005;44:1833–1845. doi: 10.1021/bi048228c. [DOI] [PubMed] [Google Scholar]
- 36.Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 37.Schonhoff CM, Daou MC, Jones SN, Schiffer CA, Ross AH. Nitric oxide-mediated inhibition of Hdm2-p53 binding. Biochemistry. 2002;41:13570–13574. doi: 10.1021/bi026262q. [DOI] [PubMed] [Google Scholar]
- 38.Sarangarajan R, Apte SP, Ugwu SO. Hypoxia-targeted bioreductive tyrosine kinase inhibitors with glutathione-depleting function. Anticancer Drugs. 2006;17:21–24. doi: 10.1097/01.cad.0000185179.34486.a3. [DOI] [PubMed] [Google Scholar]
- 39.Wang J, Boja ES, Tan W, Tekle E, Fales HM, English S, Mieyal JJ, Chock PB. Reversible glutathionylation regulates actin polymerization in A431 cells. J Biol Chem. 2001;276:47763–47766. doi: 10.1074/jbc.C100415200. [DOI] [PubMed] [Google Scholar]
- 40.Borges CR, Geddes T, Watson JT, Kuhn DM. Dopamine biosynthesis is regulated by S-glutathionylation. Potential mechanism of tyrosine hydroxylase inhibition during oxidative stress. J Biol Chem. 2002;277:48295–48302. doi: 10.1074/jbc.M209042200. [DOI] [PubMed] [Google Scholar]
- 41.Xu Y. Regulation of p53 responses by post-translational modifications. Cell Death Differ. 2003;10:400–403. doi: 10.1038/sj.cdd.4401182. [DOI] [PubMed] [Google Scholar]
- 42.Shieh SY, Taya Y, Prives C. DNA damage-inducible phosphorylation of p53 at N-terminal sites including a novel site, Ser20, requires tetramerization. EMBO J. 1999;18:1815–1823. doi: 10.1093/emboj/18.7.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keller DM, Lu H. p53 serine 392 phosphorylation increases after UV through induction of the assembly of the CK2.hSPT16.SSRP1 complex. J Biol Chem. 2002;277:50206–50213. doi: 10.1074/jbc.M209820200. [DOI] [PubMed] [Google Scholar]
- 44.Thornborrow EC, Patel S, Mastropietro AE, Schwartzfarb EM, Manfredi JJ. A conserved intronic response element mediates direct p53-dependent transcriptional activation of both the human and murine bax genes. Oncogene. 2002;21:990–999. doi: 10.1038/sj.onc.1205069. [DOI] [PubMed] [Google Scholar]
- 45.De Flora S, Cesarone CF, Balansky RM, Albini A, D'Agostini F, Bennicelli C, Bagnasco M, Camoirano A, Scatolini L, Rovida A, et al. Chemopreventive properties and mechanisms of N-Acetylcysteine. The experimental background. J Cellular Biochem Supplement. 1995;22:33–41. doi: 10.1002/jcb.240590806. [DOI] [PubMed] [Google Scholar]
- 46.Drake J, Sultana R, Aksenova M, Calabrese V, Butterfield DA. Elevation of mitochondrial glutathione by gamma-glutamylcysteine ethyl ester protects mitochondria against peroxynitrite-induced oxidative stress. J Neurocsi Res. 2003;74:917–927. doi: 10.1002/jnr.10810. [DOI] [PubMed] [Google Scholar]
- 47.Sen CK. Cellular thiols and redox-regulated signal transduction. Curr Top cell Regul. 2000;36:1–30. doi: 10.1016/s0070-2137(01)80001-7. [DOI] [PubMed] [Google Scholar]
- 48.Bailey HH, Mulcahy RT, Tutsch KD, Arzoomanian RZ, Alberti D, Tombes MB, Wilding G, Pomplun M, Spriggs DR. Phase I clinical trial of intravenous L-buthionine sulfoximine and melphalan: an attempt at modulation of glutathione. J Clinical Oncol. 1994;12:194–205. doi: 10.1200/JCO.1994.12.1.194. [DOI] [PubMed] [Google Scholar]
- 49.Delphin C, Cahen P, Lawrence JJ, Baudier J. Characterization of baculovirus recombinant wild-type p53. Dimerization of p53 is required for high-affinity DNA binding and cysteine oxidation inhibits p53 DNA binding. Eur J Biochem. 1994;223:683–692. doi: 10.1111/j.1432-1033.1994.tb19041.x. [DOI] [PubMed] [Google Scholar]
- 50.Martin ACR, Facchiano AM, Cuff AL, Hernandez-Boussard T, Olivier M, Hainaut P, Thornton JM. Integrating mutation data and structural analysis of the TP53 tumor-suppressor protein. Human Mutat. 2002;19:149–164. doi: 10.1002/humu.10032. [DOI] [PubMed] [Google Scholar]
- 51.Ma B, Pan Y, Gunasekaran K, Venkataraghavan RB, Levine AJ, Nussinov R. Comparison of the protein-protein interfaces in the p53-DNA crystal structures: towards elucidation of the biological interface. Proc Natl Acad Sci US A. 2005;102:3988–3993. doi: 10.1073/pnas.0500215102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nature Med. 2005;11:1306–1313. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu HH, Sherman M, Yuan YC, Momand J. Direct redox modification of p53 protein: potential sources of redox control and potential outcomes. Gene Ther Mol Biol. 1999;4:119–132. [Google Scholar]
- 54.Klatt P, Molina EP, Lacoba MCD, Padilla CA, Martinez-Galisteo E, Barcena JA, Lamas S. Redox regulation of c-Jun DNA binding by reversible S-glutathiolation. FASEB J. 1999;13:1481–1490. doi: 10.1096/fasebj.13.12.1481. [DOI] [PubMed] [Google Scholar]
- 55.Pineda-Molina E, Klatt P, Vazquez J, Marina A, Lacoba MGD, Perez-Sala D, Lamas S. Glutathionylation of the p50 subunit of NF-kappaB: a mechanism for redox-induced inhibition of DNA binding. Biochemistry. 2001;40:14134–14142. doi: 10.1021/bi011459o. [DOI] [PubMed] [Google Scholar]
- 56.Cao X, Kambe F, Lu X, Kobayashi N, Ohmori S, Seo H. Glutathionylation of two cysteine residues in paired domain regulates DNA binding activity of Pax-8. J Biol Chem. 2005;280:25901–25906. doi: 10.1074/jbc.M411443200. [DOI] [PubMed] [Google Scholar]
- 57.Dandrea T, Bajak E, Warngard L, Cotgreave IA. Protein S-glutathionylation correlates to selective stress gene expression and cytoprotection. Archives Biochem Biophys. 2002;406:241–252. doi: 10.1016/s0003-9861(02)00462-9. [DOI] [PubMed] [Google Scholar]
- 58.Winyard PG, Moody CJ, Jacob C. Oxidative activation of antioxidant defence. Trends Biochem Sci. 2005;22:207–210. doi: 10.1016/j.tibs.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 59.McLure KG, Lee PWK. How p53 binds DNA as a tetramer. EMBO J. 1998;17:3342–3350. doi: 10.1093/emboj/17.12.3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ho WC, Fitzgerald MX, Marmorstein R. Structure of the p53 core domain dimer bound to DNA. J Biol Chem. 2006;281:20494–20502. doi: 10.1074/jbc.M603634200. [DOI] [PubMed] [Google Scholar]
- 61.Dehner A, Kessler H. Diffusion NMR spectroscopy: folding and aggregation of domains in p53. Chembiochem. 2006;6:1550–1565. doi: 10.1002/cbic.200500093. [DOI] [PubMed] [Google Scholar]
- 62.Veprinstev DB, Freund MV, Andreeva A, Rutledge SE, Tidow H, Canadillas JMP, Blair CM, Fersht AR. Core domain interactions in full-length p53 in solution. Proc Natl Acad Sci USA. 2006;103:2115–2119. doi: 10.1073/pnas.0511130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pitenpol JA, Tokino T, Thiagalingam S, El-diery WS, Kinzler KW, Vogelstein B. Sequence-specific transcriptional activation is essential for growth suppression by p53. Proc Natl Acad Sci USA. 1994;91:1998–2002. doi: 10.1073/pnas.91.6.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bischoff JR, Casso D, Beach D. Human p53 inhibits growth in Schizosaccharomyces pombe. Mol Cell Biol. 1992;12:1405–1411. doi: 10.1128/mcb.12.4.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Locations of cysteine residues in the three-dimensional ribbon structure of p53 protein (Figure S1), mass spectra of thiolated peptides from p53 (Figure S2), MALDI-TOF spectrum of TCPVQLWVDSTPPPGTR peptide (T8) labeled with biotin-maleimide (Figure S3), MS/MS spectrum of the T8 peptide (biotin-maleimide conjugated molecular ion at m/z = 2305.1 on the 4700 proteomics analyzer (Figure S4). This material is available free of charge via the Internet at http://pubs.acs.org.