

Abstract
Bimolecular fluorescence complementation (BiFC) analysis enables direct visualization of protein interactions in living cells. The BiFC assay is based on the discoveries that two non-fluorescent fragments of a fluorescent protein can associate to form a fluorescent complex and that the association of the fragments can be facilitated by fusing them to two proteins that interact with each other. The non-covalent association of the fragments produces a bimolecular fluorescent complex. The specificity of bimolecular fluorescence complementation must be confirmed by parallel analysis of proteins in which the interaction interface has been mutated. It is not necessary for the interaction partners to juxtapose the fragments within a specific distance of each other since they can associate when they are tethered to a complex with flexible linkers. It is also not necessary for the interaction partners to form a complex with a long half-life or a high occupancy since the fragments can associate in a transient complex and un-associated fusion proteins do not interfere with detection of the complex. Many interactions can be visualized when the fusion proteins are expressed at concentrations comparable to their endogenous counterparts. The BiFC assay has been used for the visualization of interactions between many types of proteins in different subcellular locations and in different cell types and organisms. It is technically straightforward and can be performed using a regular fluorescence microscope and standard molecular biology and cell culture reagents.
Keywords: protein interaction, fluorescence complementation, complex formation, subcellular localization, cell culture, specificity
Introduction
Studies of protein interactions have provided fundamental insights into the regulation of cellular functions. Many methods have been developed to investigate protein interactions. Most methods that enable direct detection of protein interactions, such as co-purification and affinity precipitation assays, require removal of the proteins from their native environment. In contrast, methods that enable detection of protein interactions in cells, such as genetic suppressor analysis, generally rely on indirect consequences of the protein interactions.
The visualization of protein interactions in living cells provides the potential for direct detection of protein interactions with minimal perturbation of their normal environment. Many strategies for the visualization of protein interactions in cells have been developed. The most commonly employed are fluorescence resonance energy transfer (FRET) 1 and bimolecular fluorescence complementation (BiFC) 2. Others include fluorescence correlation spectroscopies 3, 4, image correlation spectroscopy 5 and complementation approaches using fragments of other proteins 6-8
Comparison of BiFC and FRET
FRET and BiFC analysis are fundamentally different approaches and have complementary advantages and disadvantages. FRET is based on the transfer of excitation energy between two fluorophores that are in close spatial proximity and have permissive relative orientations 1. This energy transfer results in a change in the fluorescence intensities and lifetimes of the two fluorophores. FRET analysis of a protein interaction requires quantitation of the change in the fluorescence intensity or lifetime of the donor and acceptor fluorophores in the presence versus the absence of energy transfer between the fluorophores.
BiFC is based on the association between fragments of a fluorescent protein when they are tethered in the same macromolecular complex (Figure 1) 2. Thus, the association produces a fluorescent complex from non-fluorescent constituents. BiFC analysis requires determination of the difference in fluorescence intensities produced by the association of the fluorescent protein fragments in the presence versus the absence of an interaction between the proteins fused to the fragments. BiFC analysis has been used for the visualization of interactions between many different proteins in many cell types and organisms (see Table 1).
Figure 1. Schematic diagram representing the principle of the BiFC assay.
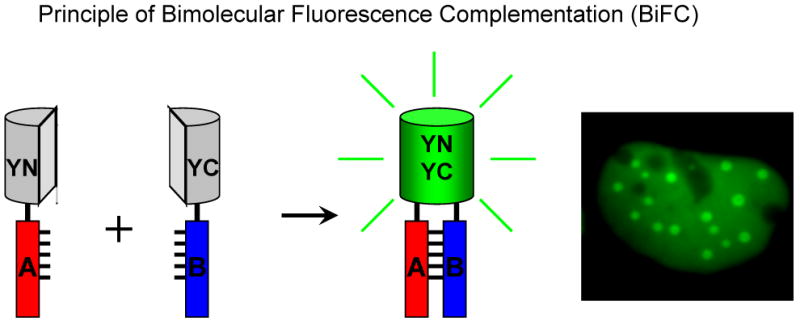
Two fragments (YN and YC) of the yellow fluorescent protein (YFP) are fused to two putative interaction partners (A and B). An interaction between the proteins facilitates association between the fragments to produce a bimolecular fluorescent complex. The image on the right shows BiFC analysis of the recruitment of a protein to subnuclear foci by dimerization with a mutated version. Image on the right acquired by Nirmala Rajaram25, Copyright © 2004, the American Society for Microbiology. All rights reserved.
Table 1.
Examples of protein interactions that have been visualized using the BiFC assay
| Category | Class | Proteins1 | Organism | References |
|---|---|---|---|---|
| Peptides | Coiled coil | Anti-parallel NZ-CZ | E. coli | 16 |
| Heat shock | Hsc70, Hsp90-TPR1, TPR2A, TPR2B | E. coli | 17 | |
|
| ||||
| Nuclear proteins | bZIP | Fos-Jun-ATF2; BATF-Jun; Maf-Sox | Mammalian cells | 2, 24, 25, 69 |
| Rel | p50,IkB-p65 | Mammalian cells | 2 | |
| bHLHZIP | Myc, Mxi1, Mad3, Mad4-Max, Mist-Mist | Mammalian cells | 10, 26 | |
| Bromodomain | AcH4-Brd2; SPA-1, P-TEFb-Brd4 | Mammalian cells | 27-29 | |
| Smad | PKB/Akt, Smad4-Smad3 | Mammalian cells | 30 | |
| IRF-Ets | IRF8-PU.1 | Mammalian cells | 32 | |
| Winged helix | AcFKH1-CPCR1 | A. chrysogenum | 33 | |
|
| ||||
| Ubiquitination | E3 ligase-substrate | Skp2-Myc | Mammalian cells | 23 |
| Grr1-Hof1 | S. cerevisiae | 34 | ||
| EID1-ASK1,2,4,9,13,14,15,SSK1 | S. alba, P. crispum | 35, 36, 70 | ||
| AtCUL1-ASK1,EID1, P0CA-ASK1 | ||||
| Peptide conjugates | Jun-Ub, Jun-SUMO1 | Mammalian cells | 12 | |
|
| ||||
| Plant pathogens | Type IV secretion | VirE2-VirD4 | A tumefaciens | 37, 71 |
| Host-pathogen | VirE2, VirF, H2A-AtVIP, VirE3 | N. tabacum | 38, 39, 41 | |
|
| ||||
| Signaling | MAP kinase network | MEKK3-IκBα; MEKK2-IκBβ: ERK1-p65; ERK2-p65 | Mammalian cells | 19 |
| PKB-PDK kinases | PKB/Akt, PDK1-hFt1 | Mammalian cells | 31 | |
| Heterotrimeric G proteins | Gβ1-Gγ7 | D. discoideum, Mammalian cells | 42, 43 | |
| Phospholipases | PLCβ2-PLCδ1 | Mammalian cells | 18 | |
| Apoptosis | Bif1-Bax, TRAF6-Src | Mammalian cells | 44, 65 | |
| Photosensitivity | FpsA-FpsA | A. nidulans | 45 | |
|
| ||||
| Enzyme complexes | ACCS | ACS1, ACS4 - ACS6, ACS7, ACS8 | E. coli | 46 |
| P450 | P450C2, P450E1-P450 reductase; P4502C2-BAP31 | Mammalian cells | 47, 48 | |
|
| ||||
| Membrane proteins | Integrin signaling | Integrin αIIbβ3, Syk-Src | Mammalian cells | 49 |
| Arf GTPases | Arf1, Arf3, Arf4, Arf5-GBF1 | Mammalian cells | 50 | |
| Lectin-glycoprotein | MCFD2, Cathepsin-ERGIC53 | Mammalian cells | 51 | |
| Cytokine receptors | gp130 – LIFR, gp130 | Mammalian cells | 52 | |
| APP processing | APP-Notch2, APP | Mammalian cells | 67 | |
|
| ||||
| Nucleic acid binding | RNA binding | IMP, FMRP, hStau1, IRP1, PTB1-RNA; Nef-Nef; NXF1-Y14 | Mammalian cells | 53,54, 55 |
| DNA binding | Zif268, PBSII-DNA | In vitro | 72 | |
|
| ||||
| Plant proteins | Transcription factors | FIE-MEA; bZIP63-bZIP63; LSD1-LSD1; bHLH1-OFP1; SAD, BPBF-GAMYB; LIP19-OBF19; GRP23-RBP36B | N. benthamiana, N. tabacum, A. thaliana, Allium sp. | 56-60, 64 |
| Protein modification | PFTα-PFTβ; T143c-T143c | N. benthamiana, A. thaliana | 56, 57 | |
| Flowering | FD-FT | N. benthamiana | 61 | |
| Plastid division | MinD1-MinE1; FtsZ1,ARC6-FtsZ2 | N. tabacum | 62 | |
| Enzyme complex | AtSufE-AtSufS, AtNifS | N. tabacum | 66 | |
Protein pairs that have been tested are separated by a dash. In cases where several protein pairs have been tested, the alternative partners are separated by a comma. Different combinations of proteins that have been tested are separated by semi-colons.
FRET enables, in principle, instantaneous monitoring of protein interactions whereas BiFC produces a signal after a delay required for the chemical reactions that generate the fluorophore. BiFC theoretically allows detection of interactions at lower protein concentrations and is predicted to be affected less by changes in cellular conditions that can alter the fluorescence intensities and lifetimes of fluorescent proteins. FRET requires close spatial proximity between the fluorophores whereas BiFC requires that the fluorescent protein fragments have the dynamic flexibility to associate in the protein complex. FRET requires that a large fraction of the fluorescent proteins associate with each other whereas BiFC can detect an interaction involving only a small subset of each fusion protein. Each approach is therefore applicable for different purposes. The fundamental principles and recent applications of the BiFC assay have been reviewed 9.
Applications of the BiFC assay
The BiFC assay is applicable for visualization of the steady-state distributions of complexes formed between virtually any combination of proteins in a wide variety of cell types and organisms. Although the approach is applicable for studies of protein interactions in a wide variety of organisms (see Table 1), the chemistry of fluorophore formation requires molecular oxygen, making this approach unsuitable for use in organisms that are obligate anaerobes. The proteins must be able to accept fusions to fluorescent protein fragments without disruption of their functions. The complex must tolerate stabilization of the interaction by the association between the fluorescent protein fragments without changes in function or deleterious effects for the cell. The BiFC assay is particularly valuable for determining the subcellular locations of protein interactions, which can provide insight into the biological functions of newly discovered protein complexes. Fragments of several proteins that can associate when they are brought together by an interaction between proteins fused to the fragments have been identified. These include fragments of ubiquitin, β-galactosidase and dihydrofolate reductase 6-8. The advantage of BiFC analysis is that association between fragments of fluorescent proteins produces a complex with intrinsic fluorescence, eliminating the need for exogenous stains and enabling direct detection of the protein complex. We have developed several extensions of the BiFC assay that enable visualization of multiple protein interactions in the same cell 10, 11 as well as covalent protein modifications 12. Recent studies have also suggested that BiFC analysis can be used to determine the topology of membrane proteins 13 and for high throughput screening for the effects of small molecules on protein complexes 14. Future efforts will undoubtedly identify many new applications for fluorescence complementation.
Limitations of the BiFC assay
The BiFC assay has several characteristics that limit its applicability and should temper interpretations based on results from this assay. One limitation of the BiFC approach is the time required for fluorophore maturation, which reflects the chemical reactions required for formation of the cyclic fluorophore. This prevents real-time detection of rapid changes in interactions using the BiFC assay. It is possible that some of the chemical reactions required for fluorophore formation occur in the isolated fragments, accelerating fluorescence complementation under some conditions 15.
Bimolecular fluorescent complex formation is also likely to affect the dynamics of complex dissociation and partner exchange. Under many in vitro conditions, formation of the bimolecular fluorescent complex is essentially irreversible 2, 16, 17. However, in some experiments, rapid changes in the fluorescence signal have been observed 18, 19. It is possible that bimolecular fluorescent complex formation is reversible under these conditions, but it is difficult to exclude the possibility that other processes, such as protein degradation, affect the signal in living cells. The signal from fluorescence complementation has also been shown to decrease under conditions predicted to reduce complex formation under some in vitro conditions 15, 18. However, dissociation of the fusion proteins has not been directly demonstrated.
Finally, fluorescent protein fragments have a finite ability to associate with each other independent of an interaction between proteins fused to the fragments. This major source of background signal in the BiFC assay varies depending on the identities of the fusion proteins and their levels of expression. Generally, this problem can be alleviated by expression of the fusion proteins at concentrations approximating their endogenous counterparts. In cases where this may not be possible, alternative fusions can be tested for the specificity of fluorescence complementation.
Strengths of the BiFC assay
There are many characteristics of the BiFC assay that make it useful for the study of protein interactions. First, it enables direct visualization of protein interactions and does not rely on their secondary effects. Second, the interactions can be visualized in living cells, eliminating potential artifacts caused by cell lysis or fixation. Third, the proteins are expressed in a relevant biological context, ideally at levels comparable to those of their endogenous counterparts. This increases the likelihood that the results reflect the properties of native proteins, including potential effects of post-translational modifications. Fourth, the BiFC assay does not require stoichiometric complex formation but can be used to detect interactions between subpopulations of each protein. Finally, BiFC does not require specialized equipment, apart from an inverted fluorescence microscope equipped with objectives that allow imaging of fluorescence in cells. The direct detection of bimolecular complex formation requires no post-acquisition image processing for interpretation of the data.
This protocol focuses on the visualization of protein interactions in cultured mammalian cells using the BiFC assay 2, but the general principles described are applicable to many other experimental systems. Although this protocol describes the use of transiently transfected plasmid vectors, it is likely that other expression strategies (retroviruses, lentiviruses etc.) can be used with only slight modifications to the protocol.
Materials
Reagents
Plasmid vectors for expression of fusion proteins (also see below)
DNA encoding N-terminal fragment of fluorescent protein (see Table 2)
DNA encoding C-terminal fragment of fluorescent protein (see Table 2)
DNA encoding proteins of interest (interaction partners, wildtype)
DNA encoding mutated proteins of interest that do not interact with each other (negative controls)
Cells that can be transfected with plasmid DNA (preferably adherent, monolayer cell line)
Materials for cell culture
Materials for cell transfection
Tissue culture vessels appropriate for experiment: e.g. cluster plates, slide chambers, or glass coverslips
Additional reagents and equipment for expressing proteins in mammalian cells, mammalian cell culture, and immunoblotting
Table 2.
Combinations of fluorescent protein fragments recommended for BiFC analysis.
| Fusions* | Purpose | Excitation filter | Emission filter | Reference |
|---|---|---|---|---|
| A-YN155
B-YC155 |
A-B interaction | 500/20 nm | 535/30 nm | 2 |
| A-YN173
B-YC173 |
A-B interaction | 500/20 nm | 535/30 nm | 11 |
| A-VN155
B-VC155 |
A-B interaction | 500/20 nm | 535/30 nm | 20 |
| A-VN173
B-VC173 |
A-B interaction | 500/20 nm | 535/30 nm | 20 |
| A-CN155
B-CC155 |
A-B interaction | 436/10 nm | 470/30 nm | 11 |
A and B correspond to proteins whose interaction is to be tested. YN155 corresponds to residues 1-154 of EYFP72. YC155 corresponds to residues 155-238 of EYFP. YN173 corresponds to residues 1-172 of EYFP. YC173 corresponds to residues 173-238 of EYFP. VN155 corresponds to residues 1-154 of Venus 22. VC155 corresponds to residues 155-238 of Venus. VN173 corresponds to residues 1-172 of Venus. VC173 corresponds to residues 173-238 of Venus. CN155 corresponds to residues 1-154 of ECFP 21. CC155 corresponds to residues 155-238 of ECFP.
Equipment
Inverted fluorescence microscope equipped with:
Sensitive CCD camera
20× to 100× objectives
Filters for visualization of fluorescent proteins: YFP, Venus (excitation 500 ± 10 nm; emission 535 ± 15 nm); and CFP, Cerulean (excitation 436 ± 5 nm; emission 470 ± 15 nm)
Software for instrument control and data analysis
Facilities for cell culture
Procedure
1. Select the fluorescent protein fragments to be used
Several combinations of fluorescent protein fragments support bimolecular fluorescence complementation 11; those recommended for BiFC analysis are listed in Table 2. For most purposes, fragments of YFP truncated at residue 155 (designated YN155 and YC155) are recommended, as they exhibit a relatively high complementation efficiency when fused to many interaction partners, yet produce low fluorescence when fused to proteins that do not interact with each other 2. Fragments of YFP truncated at residue 173 (designated YN173 and YC173) can also be used 11, and may exhibit a different efficiency of complementation due to differences in the steric constraints imposed by tethering of the fragments to the protein complex. Fragments of Venus (a mutated GFP with high fluorescence intensity) 22 truncated at either residue 155 or 173 (designated VN155 and VC155 or VN173 and VC173 respectively) produce a significantly brighter fluorescent signal when fused to specific interaction partners 20. However, these fragments also produce a brighter signal when fused to proteins that do not selectively interact with each other 20. These fragments have the great advantage that the bimolecular fluorescent complex is readily detectable at 37°C, which avoids the incubation at 30°C that is generally necessary to detect complementation using YFP fragments. Other combinations of fluorescent protein fragments can also be used, especially when using BiFC analysis for the visualization of multiple protein complexes in the same cell 11.
2. Determine the sites where the fluorescent protein fragments can be fused to the putative interaction partners
Determine the positions of the fusions empirically to fulfill three criteria.
First, ensure that the fusions allow the fragments of the fluorescent proteins to associate with each other if the putative partners interact. Information about the structure and location of the interaction interface may be useful to determine optimal positions for the fusions. However, this information is not essential since fusions that can be used for BiFC analysis can be identified by screening multiple combinations of fusion proteins for fluorescence complementation. One strategy for the identification of fusion proteins that allow bimolecular fluorescence complementation is to fuse each of the fluorescent protein fragments to the N- and C-terminal end of each interaction partner, and to test for complementation in all eight combinations that contain both fragments of the fluorescent protein (Figure 2).
Second, confirm that fusions do not affect the localization or the stabilities of the proteins by comparing the localization and expression levels of the fusion proteins with those of wildtype proteins lacking the fusions; indirect immunofluorescence and immunoblot analyses can be used.
Third, test the fusion proteins for all known functions of the endogenous proteins to ensure that the fusions do not affect the functions of the proteins under investigation.
Figure 2. Combinations of fusion proteins to be tested for bimolecular fluorescence complementation.
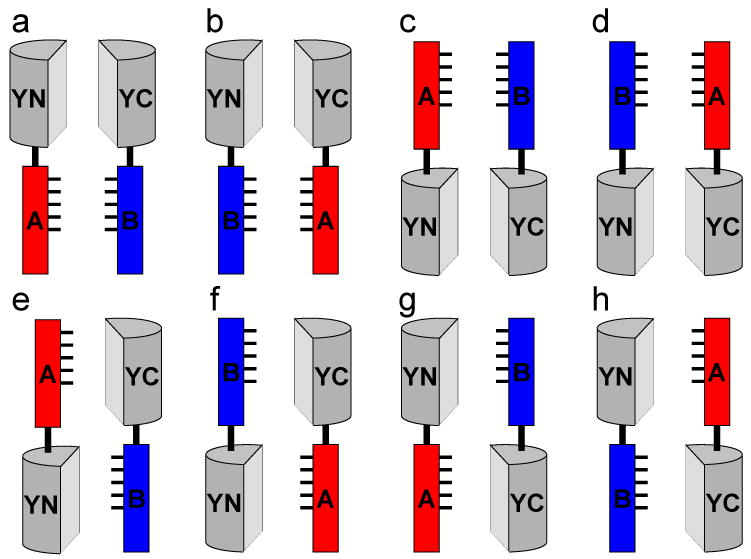
Fusion proteins that produce optimal signal must generally be empirically determined. Multiple combinations of fusion proteins should be tested for bimolecular fluorescence complementation. Amino- and carboxyl-terminal fusions can be used to test eight distinct combinations (a through h). Although it may appear that combinations e through h might not be favorable for bimolecular complex formation, this will depend on the precise structures and flexibilities of the fusion proteins, which are difficult to predict. For true interaction partners, it is virtually always possible to find fusion proteins that produce a detectable signal.
3. Select linkers to connect the fragments to the proteins of interest
The linkers must provide flexibility for independent motion of the fluorescent protein fragments and the interaction partners, allowing the fragments to associate when the proteins interact. We have used the RSIAT and RPACKIPNDLKQKVMNH linker sequences in many fusion constructs used for BiFC analysis 2, 11. These linkers have been used for the visualization of interactions between many structurally unrelated proteins. The sequence AAANSSIDLISVPVDSR encoded by the multiple cloning sites of the pCMV-FLAG vector (Sigma) has also been successfully used as a linker in many BiFC experiments. A peptide sequence designed to be flexible such as (GGGS)n can also be used, although it can potentially affect the degradation of the fusion protein. Although these linker sequences have worked well for the proteins examined previously, it is possible that linkers of a different length or sequence are optimal for BiFC analysis of interactions between other proteins.
4. Select a cell culture system
Choose a cell culture system that represents the biological context to be investigated, and allows efficient introduction of DNA into a large fraction of the cells. Cells that grow as an adherent monolayer are generally easier to image. The BiFC assay has been used for the analysis of protein interactions in many mammalian cell lines including COS-1, HEK293, HeLa, Hep3B, αTN4, and NIH3T3 cells as well as in intact organisms 2, 10, 12, 18, 19, 23-67.
5. Select a strategy for expression of the fusion proteins
Choose either transient expression (A) or stable expression (B) strategies, based on the purpose of the experiment.
A). Transient overexpression of the fusion proteins
This approach may be adequate to determine if a pair of proteins can interact in cells and to determine the subcellular location of the complex. To minimize protein mislocalization and formation of non-native complexes due to overexpression, express the fusion proteins at levels comparable to the endogenous proteins. This can be achieved by using plasmids with weak promoters, by transfecting small amounts of plasmid DNA and by observing the cells as soon as signal is detectable.
B) Expression in stable cell lines
More reproducible levels of expression can be obtained by using inducible expression vectors integrated into the genomes of stable cell lines. This allows for the control of protein expression at relatively uniform levels in the entire cell population and replication of experiments at constant expression levels, independent of transfection efficiency and other factors that are difficult to control in transient assays.
6. Design controls to determine if complementation reflects a specific protein interaction
As fluorescent protein fragments are able to form fluorescent complexes with a low efficiency in the absence of a specific interaction, it is essential to include negative controls in each experiment. Spontaneous complementation is generally reduced when the fragments are fused to proteins that do not interact with each other; appropriate negative controls are fusion proteins in which the interaction interface has been mutated and fused to the fluorescent protein fragments in a manner identical to the wildtype fusion proteins (Figure 3) 2, 10. Compare the level of expression and localization of the mutated and wild-type fusion proteins by immunoblot and indirect immunofluorescence analyses (using any standard or commercially available method). Quantify and compare the efficiencies of fluorescence complementation between the wild-type and mutated proteins. If there is no prior knowledge of the location or the structural nature of the interaction interface, it is possible to screen for mutations that alter the efficiency of bimolecular fluorescence complementation, and thereby determine if the complementation reflects a specific interaction. The BiFC assay can therefore be used to determine whether two proteins interact in cells without prior knowledge of the location or the structural nature of the interaction interface.
Figure 3. Determination of the specificity of bimolecular fluorescence complementation by mutational analysis.
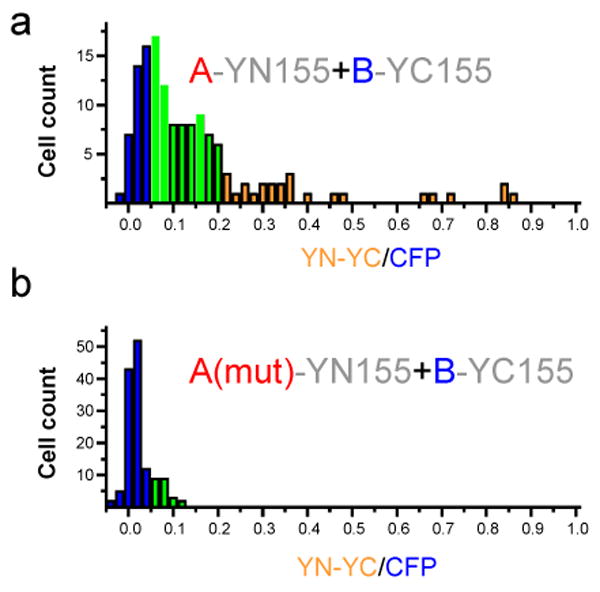
The requirement for a specific protein interaction for fluorescence complementation should be tested by examining the effects of mutations that prevent the association of the interaction partners (data adapted from 2, reproduced with permission from Elsevier. All rights reserved.). The wild type (a) and mutated (b) interaction partners should be expressed at equal concentrations together with an internal reference (i.e. CFP). The fluorescence intensities produced by bimolecular fluorescence complementation (YN-YC) and the internal reference (CFP) are measured in individual cells. The distribution of ratios between the fluorescence intensities in individual cells are plotted in each histogram. The colored bars indicate cells with different ratios of YN-YC to CFP fluorescence. When the proteins interact (a), the fluorescence intensity of YN-YC will increase, resulting in a higher fluorescence ratio. The interaction may occur in all or in a subset of cells. When no interaction is observed (b), most cells will have a very low fluorescence ratio.
Use some of the numerous fusion proteins whose interactions have been visualized using the BiFC assay as positive controls (see Table 1). However, the failure to detect fluorescence complementation between the proteins under investigation does not demonstrate the absence of an interaction (see ANTICIPATED RESULTS).
7. Prepare plasmids encoding fusion proteins
Construct plasmid expression vectors, using the appropriate vectors, by fusing the sequences encoding the selected fluorescent protein fragments (see Table 2) to the sequences encoding the proteins of interest. Any standard cloning techniques can be used. Whenever possible, test fusions to both the N- and C-terminal ends of the proteins to be investigated (Figure 2). Construct negative control plasmids that encode mutated non-interacting variants of the proteins (see step 6) using the same strategy. Positive controls should be included to ensure that a known interaction can be detected (see step 6).
8. Prepare cells for transfection
Seed cells the day before transfection at an appropriate density. This density should allow for cell proliferation over the course of the experiment while taking into consideration the effects of cell growth and density on the interaction under investigation. Cluster plates are convenient for processing multiple transfections in parallel. If short-working-distance objectives will be used to visualize the interaction, grow the cells in slide chambers or on glass coverslips.
9. Transfect cells
Transfect cells (using the optimal procedure for the cells) with appropriate amounts (e.g. 0.25 μg) of the BiFC plasmids encoding the fusion proteins when an appropriate confluency (e.g. ∼50%) is reached. In parallel, transfect cells with the negative and positive control plasmids. For quantitation of the efficiency of fluorescence complementation, all plasmids should be co-transfected with the same amount of an internal control plasmid (e.g. a plasmid that expresses CFP, see Fig. 3)
10. Allow time for fusion protein expression and fluorophore maturation
Grow cells under conditions appropriate for the cell-type until fluorescence is detected (12 to 36 hr). If necessary, incubate the cells at 30°C with 5% CO2 to promote maturation of the fluorophore and to increase the signal. Results obtained under low temperature conditions should be interpreted with care, as incubation at a lower temperature could alter protein interactions.
11. Remove dead cells
Wash the cells once with an amount of PBS sufficient to remove dead cells and cell debris, then add fresh medium.
12. Observe cells
Image the cells using an inverted fluorescence microscope. When using cells grown on plastic, a long-working-distance objective is convenient, but produces lower signal (due to the lower numerical aperture) than a short-working distance objective. A 20× objective is useful for observing large numbers of cells and can provide general subcellular localization information, whereas 60× or 100× objectives can be necessary for detailed localization within subcellular compartments. For detection of complementation between YFP or Venus fragments an excitation filter with 500 ± 10 nm transmission and an emission filter with 535 ± 15 nm transmission are appropriate. Confirm that fluorescent cells are alive by comparing their morphology to that of non-transfected cells. Cells grown on coverslips can be fixed and individual proteins can be visualized by indirect immunofluorescence analysis. Use protocols for fixation and immunofluorescence that have been established for the cell line and antibody to be used.
13. Establish the levels of protein expression
Compare the levels of fusion protein expression with those of the endogenous proteins by immunoblot analysis. The cultures used for imaging can be subsequently processed for immunoblotting or separate cultures can be prepared in parallel. Use protocols for immunoblotting that have been established to work for the cell line and antibody to be used. Use the ratio between the intensities of the bands corresponding to the transfected and endogenous proteins together with the transfection efficiency to estimate the relative levels of transfected and endogenous proteins. Ideally, the amount of transfected protein should not exceed that of the endogenous proteine in the cells.
14. Analyze the data
Compare the intensities and numbers of fluorescent cells observed when the cells are transfected with the wildtype interaction partners with those observed when the cells are transfected with the negative control constructs. For quantitative analysis of the efficiencies of fluorescence complementation, divide the fluorescence intensities produced by fluorescence complementation by the fluorescence intensities produced by intact fluorescent protein in individual cells (see Fig. 3). Higher fluorescence intensity and an increased number of fluorescent cells for the wild type proteins is consistent with a specific interaction.
Timing
The construction of plasmid vectors for the expression of fusion proteins can be accomplished in a few days once the design for the vectors has been completed. Preparation of cells that express the fusion proteins can vary a great deal from days in transient transfection experiments to months or years for the production of stable cell lines or transgenic organisms. In transient expression experiments, fluorescence from specific interactions can generally be detected between 12 and 30 hours after transfection. In the case of complementation between YFP or CFP fragments, this generally requires a short incubation at 30°C to facilitate fluorophore maturation. In the case of complementation between Venus fragments, this step is generally not necessary. Longer incubation should be avoided since this may result in higher expression of fusion proteins and complementation due to nonspecific interactions. Images can be recorded and analyzed in less than an hour for each combination of fusion proteins.
Anticipated Results
Interpretation of results from BiFC analysis
If fluorescence is detected when wildtype proteins are expressed, and this signal is eliminated or significantly reduced by single amino acid substitutions that prevent the interaction, it is likely that the bimolecular fluorescence complementation represents a specific interaction between the proteins fused to the fragments of fluorescent proteins.
If mutations that are known to eliminate the interaction do not reduce or eliminate the fluorescence, then the bimolecular complementation is due to a nonspecific interaction between the fusion proteins. If this is the case, the BiFC assay may not be an appropriate assay for the study of the two proteins. If the levels of expression of the fusion proteins were higher than those of their endogenous counterparts, modify the protocol or use another expression system to achieve levels of protein expression more consistent with those of the endogenous proteins. Alternatively, a different combination of fusion proteins or linkers should be tested.
If no fluorescence complementation is detected in the BiFC assay it does not prove the absence of an interaction, even if coexpression of the same fusion proteins with other interaction partners results in bimolecular fluorescence complementation. Different combinations of BiFC constructs or alternative approaches should be employed. Fluorescence in a small subpopulation of cells is difficult to interpret, since it may represent complementation due to nonspecific interactions, or it may reflect a regulated interaction that occurs only in response to signaling in a subset of cells. Further studies are necessary to distinguish these possibilities.
The fluorescence intensity produced by bimolecular fluorescence complementation in living cells is generally less than 10% of that produced by intact fluorescent proteins. It is likely that only a subset of the fragments associate with each other since the fluorescence intensity of BiFC complexes produced in vitro is comparable to that of intact fluorescent proteins 2. Several variants of the BiFC assay have been developed that enable visualization of multiple protein interactions in the same cell as well as covalent protein modifications 9, 11, 67
Table 3.
Troubleshooting table.
| STEPS | PROBLEM | SOLUTION |
|---|---|---|
| 7, 8, 9 | Cell culture, transfection | Problems with cell culture and transfection will prevent detection of protein interactions. Use plasmids encoding intact fluorescent proteins to establish the transfection efficiency and expression of heterologous proteins in the cells. |
| 10, 13 | Protein expression | Problems with protein expression can be generic or specific to the fusion proteins to be tested. Establish that the chosen expression vectors are appropriate for the cells that are used. Determine the expression of the fusion proteins by western blot analysis. If the fusion proteins cannot be detected, construct new expression vectors with a different arrangement of fluorescent protein fragments or change the linker sequences. |
| 2, 13 | Protein localization | If the fusion proteins have a different subcellular distribution than the endogenous proteins, it is possible that overexpression of the proteins causes their mislocalization. Reduce the level of fusion protein expression by using regulated expression vectors, ideally as stably transfected integrants. If the proteins are transiently expressed, shorten the time allowed for protein expression or reduce the amount of plasmid transfected into the cells. It is also possible that the fusions or the interaction between them affect the distributions of the proteins. Test different arrangements of the fluorescent protein fragments and compare the distributions of the fusion proteins when expressed singly or in combination. |
| 2, 3, 12 | Complementation | If the proteins are expressed but no fluorescence complementation is detected, it is possible that the steric arrangement of fluorescent protein fragments does not allow their association. Test other arrangements of fluorescent protein fragments or change the linker sequences. If the cells are grown at 37°C, transfer them to 30°C for a few hours before imaging to promote fluorophore maturation. It is also possible that the proteins do not interact under the conditions tested. |
Acknowledgments
I thank Dr. Changdeng Hu for his participation in the design and implementation of the BiFC assay in mammalian cells and all members of the Kerppola laboratory for their contributions to the improvement and adaptation of the BiFC approach.
References
- 1.Förster T. 10th Spiers Memorial Lecture. Transfer mechanisms of electronic excitation. Discussions of the Faraday Society. 1959;27:7–17. [Google Scholar]
- 2.Hu CD, Chinenov Y, Kerppola TK. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol Cell. 2002;9:789–98. doi: 10.1016/s1097-2765(02)00496-3. [DOI] [PubMed] [Google Scholar]
- 3.Brock R, Jovin TM. Fluorescence correlation microscopy (FCM)-fluorescence correlation spectroscopy (FCS) taken into the cell. Cell Mol Biol (Noisy-le-grand) 1998;44:847–56. [PubMed] [Google Scholar]
- 4.Baudendistel N, Muller G, Waldeck W, Angel P, Langowski J. Two-hybrid fluorescence cross-correlation spectroscopy detects protein-protein interactions in vivo. Chemphyschem. 2005;6:984–90. doi: 10.1002/cphc.200400639. [DOI] [PubMed] [Google Scholar]
- 5.Petersen NO, Hoddelius PL, Wiseman PW, Seger O, Magnusson KE. Quantitation of membrane receptor distributions by image correlation spectroscopy: concept and application. Biophys J. 1993;65:1135–1146. doi: 10.1016/S0006-3495(93)81173-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnsson N, Varshavsky A. Split ubiquitin as a sensor of protein interactions in vivo. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:10340–4. doi: 10.1073/pnas.91.22.10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rossi F, Charlton CA, Blau HM. Monitoring protein-protein interactions in intact eukaryotic cells by beta-galactosidase complementation. Proc Natl Acad Sci U S A. 1997;94:8405–10. doi: 10.1073/pnas.94.16.8405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pelletier JN, Campbell-Valois FX, Michnick SW. Oligomerization domain-directed reassembly of active dihydrofolate reductase from rationally designed fragments. Proc Natl Acad Sci U S A. 1998;95:12141–6. doi: 10.1073/pnas.95.21.12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kerppola TK. Visualization of molecular interactions by fluorescence complementation. Nature Reviews Molecular Cell Biology. 2006;7:449–456. doi: 10.1038/nrm1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grinberg AV, Hu CD, Kerppola TK. Visualization of Myc/Max/Mad family dimers and the competition for dimerization in living cells. Molecular and Cellular Biology. 2004;24:4294–4308. doi: 10.1128/MCB.24.10.4294-4308.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu CD, Kerppola TK. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat Biotechnol. 2003;21:539–45. doi: 10.1038/nbt816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fang DY, Kerppola TK. Ubiquitin-mediated fluorescence complementation reveals that Jun ubiquitinated by Itch/AIP4 is localized to lysosomes. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:14782–14787. doi: 10.1073/pnas.0404445101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zamyatnin AA, et al. Assessment of the integral membrane protein topology in living cells. Plant Journal. 2006;46:145–154. doi: 10.1111/j.1365-313X.2006.02674.x. [DOI] [PubMed] [Google Scholar]
- 14.MacDonald ML, et al. Identifying off-target effects and hidden phenotypes of drugs in human cells. Nature Chemical Biology. 2006;2:329–337. doi: 10.1038/nchembio790. [DOI] [PubMed] [Google Scholar]
- 15.Demidov VV, et al. Fast complementation of split fluorescent protein triggered by DNA hybridization. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:2052–2056. doi: 10.1073/pnas.0511078103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh I, Hamilton AD, Regan L. Antiparallel leucine zipper-directed protein reassembly: application to the green fluorescent protein. J Am Chem Soc. 2000;122:5658–5659. [Google Scholar]
- 17.Magliery TJ, et al. Detecting protein-protein interactions with a green fluorescent protein fragment reassembly trap: Scope and mechanism. Journal of the American Chemical Society. 2005;127:146–157. doi: 10.1021/ja046699g. [DOI] [PubMed] [Google Scholar]
- 18.Guo YJ, Rebecchi M, Scarlata S. Phospholipase C beta(2) binds to and inhibits phospholipase C delta(1) Journal of Biological Chemistry. 2005;280:1438–1447. doi: 10.1074/jbc.M407593200. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt C, et al. Mechanisms of proinflammatory cytokine-induced biphasic NF-kappa B activation. Molecular Cell. 2003;12:1287–1300. doi: 10.1016/s1097-2765(03)00390-3. [DOI] [PubMed] [Google Scholar]
- 20.Shyu YJ, Liu H, Deng X, Hu CD. Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions. Biotechniques. 2006;40:61–66. doi: 10.2144/000112036. [DOI] [PubMed] [Google Scholar]
- 21.Zhang J, Campbell RE, Ting AY, Tsien RY. Creating new fluorescent probes for cell biology. Nature Reviews Molecular Cell Biology. 2002;3:906–918. doi: 10.1038/nrm976. [DOI] [PubMed] [Google Scholar]
- 22.Nagai T, et al. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nature Biotechnology. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- 23.von der Lehr N, et al. The F-Box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Molecular Cell. 2003;11:1189–1200. doi: 10.1016/s1097-2765(03)00193-x. [DOI] [PubMed] [Google Scholar]
- 24.Deppmann CD, Thornton TM, Utama FE, Taparowsky EJ. Phosphorylation of BATF regulates DNA binding: a novel mechanism for AP-1 (activator protein-1) regulation. Biochemical Journal. 2003;374:423–431. doi: 10.1042/BJ20030455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rajaram N, Kerppola TK. Synergistic transcription activation by Maf and Sox and their subnuclear localization are disrupted by a mutation in Maf that causes cataract. Molecular and Cellular Biology. 2004;24:5694–5709. doi: 10.1128/MCB.24.13.5694-5709.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu LQ, et al. Inhibition of Mist1 homodimer formation induces pancreatic acinar-to-ductal metaplasia. Molecular and Cellular Biology. 2004;24:2673–2681. doi: 10.1128/MCB.24.7.2673-2681.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanno T, et al. Selective recognition of acetylated histones by bromodomain proteins visualized in living cells. Molecular Cell. 2004;13:33–43. doi: 10.1016/s1097-2765(03)00482-9. [DOI] [PubMed] [Google Scholar]
- 28.Farina A, et al. Bromodomain protein Brd4 binds to GTPase-activating SPA-1, modulating its activity and subcellular localization. Molecular and Cellular Biology. 2004;24:9059–9069. doi: 10.1128/MCB.24.20.9059-9069.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jang MK, et al. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Molecular Cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 30.Remy I, Montmarquette A, Michnick SW. PKB/Akt modulates TGF-beta signalling through a direct interaction with Smad3. Nature Cell Biology. 2004;6:358–365. doi: 10.1038/ncb1113. [DOI] [PubMed] [Google Scholar]
- 31.Remy I, Michnick SW. Regulation of apoptosis by the Ft1 protein, a new modulator of protein kinase B/Akt. Molecular and Cellular Biology. 2004;24:1493–1504. doi: 10.1128/MCB.24.4.1493-1504.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laricchia-Robbio L, et al. Partner-regulated interaction of IFN regulatory factor 8 with chromatin visualized in live macrophages. PNAS. 2005;102:14368–14373. doi: 10.1073/pnas.0504014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoff B, Kuck U. Use of bimolecular fluorescence complementation to demonstrate transcription factor interaction in nuclei of living cells from the filamentous fungus Acremonium chrysogenum. Current Genetics. 2005;47:132–138. doi: 10.1007/s00294-004-0546-0. [DOI] [PubMed] [Google Scholar]
- 34.Blondel M, et al. Degradation of Hof1 by SCFGrr1 is important for actomyosin contraction during cytokinesis in yeast. Embo Journal. 2005;24:1440–1452. doi: 10.1038/sj.emboj.7600627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stolpe T, et al. In planta analysis of protein-protein interactions related to light signaling by bimolecular fluorescence complementation. Protoplasma. 2005;226:137–146. doi: 10.1007/s00709-005-0122-6. [DOI] [PubMed] [Google Scholar]
- 36.Marrocco K, et al. Functional analysis of EID1, an F-box protein involved in phytochrome A-dependent light signal transduction. Plant Journal. 2006;45:423–438. doi: 10.1111/j.1365-313X.2005.02635.x. [DOI] [PubMed] [Google Scholar]
- 37.Atmakuri K, Ding ZY, Christie PJ. VirE2, a type IV secretion substrate, interacts with the VirD4 transfer protein at cell poles of Agrobacterium tumefaciens. Molecular Microbiology. 2003;49:1699–1713. doi: 10.1046/j.1365-2958.2003.03669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tzfira T, Vaidya M, Citovsky V. Involvement of targeted proteolysis in plant genetic transformation by Agrobacterium. Nature. 2004;431:87–92. doi: 10.1038/nature02857. [DOI] [PubMed] [Google Scholar]
- 39.Loyter A, et al. The plant VirE2 interacting protein 1. A molecular link between the Agrobacterium T-complex and the host cell chromatin? Plant Physiology. 2005;138:1318–1321. doi: 10.1104/pp.105.062547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li JX, Krichevsky A, Vaidya M, Tzfira T, Citovsky V. Uncoupling of the functions of the Arabidopsis VIN protein in transient and stable plant genetic transformation by Agrobacterium. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:5733–5738. doi: 10.1073/pnas.0404118102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lacroix B, Vaidya M, Tzfira T, Citovsky V. The VirE3 protein of Agrobacterium mimics a host cell function required for plant genetic transformation. Embo Journal. 2005;24:428–437. doi: 10.1038/sj.emboj.7600524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hynes TR, Mervine SM, Yost EA, Sabo JL, Berlot CH. Live cell imaging of G(s) and the beta(2)-adrenergic receptor demonstrates that both alpha(s) and beta(1)gamma(7) internalize upon stimulation and exhibit similar trafficking patterns that differ from that of the beta(2)-adrenergic receptor. Journal of Biological Chemistry. 2004;279:44101–44112. doi: 10.1074/jbc.M405151200. [DOI] [PubMed] [Google Scholar]
- 43.Hynes TR, et al. Visualization of g protein beta gamma dimers using bimolecular fluorescence complementation demonstrates roles for both beta and gamma in subcellular targeting. Journal of Biological Chemistry. 2004;279:30279–30286. doi: 10.1074/jbc.M401432200. [DOI] [PubMed] [Google Scholar]
- 44.Takahashi Y, et al. Loss of Bif-1 suppresses Bax/Bak conformational change and mitochondrial apoptosis. Molecular and Cellular Biology. 2005;25:9369–9382. doi: 10.1128/MCB.25.21.9369-9382.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blumenstein A, et al. The Aspergillus nidulans phytochrome FphA represses sexual development in red light. Current Biology. 2005;15:1833–1838. doi: 10.1016/j.cub.2005.08.061. [DOI] [PubMed] [Google Scholar]
- 46.Tsuchisaka A, Theologis A. Heterodimeric interactions among the 1-amino-cyclopropane-1-carboxylate synthase polypeptides encoded by the Arabidopsis gene family. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:2275–2280. doi: 10.1073/pnas.0308515101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ozalp C, Szczesna-Skorupa E, Kemper B. Bimolecular fluorescence complementation analysis of cytochrome P4502C2, 2E1, and NADPH-cytochrome P450 reductase molecular interactions in living cells. Drug Metabolism and Disposition. 2005;33:1382–1390. doi: 10.1124/dmd.105.005538. [DOI] [PubMed] [Google Scholar]
- 48.Szczesna-Skorupa E, Kemper B. BAP31 is involved in the retention of cytochrome P4502C2 in the endoplasmic reticulum. Journal of Biological Chemistry. 2006;281:4142–4148. doi: 10.1074/jbc.M509522200. [DOI] [PubMed] [Google Scholar]
- 49.de Virgilio M, Kiosses WB, Shattil SJ. Proximal, selective, and dynamic interactions between integrin alpha II beta 3 and protein tyrosine kinases in living cells. Journal of Cell Biology. 2004;165:305–311. doi: 10.1083/jcb.200402064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Niu TK, Pfeifer AC, Lippincott-Schwartz J, Jackson CL. Dynamics of GBF1, a brefeldin A-sensitive Arf1 exchange factor at the Golgi. Molecular Biology of the Cell. 2005;16:1213–1222. doi: 10.1091/mbc.E04-07-0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nyfeler B, Michnick SW, Hauri HP. Capturing protein interactions in the secretory pathway of living cells. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:6350–6355. doi: 10.1073/pnas.0501976102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Giese B, et al. Dimerization of the cytokine receptors gp130 and LIFR analysed in single cells. Journal of Cell Science. 2005;118:5129–5140. doi: 10.1242/jcs.02628. [DOI] [PubMed] [Google Scholar]
- 53.Rackham O, Brown CM. Visualization of RNA-protein interactions in living cells: FMRP and IMP1 interact on mRNAs. Embo Journal. 2004;23:3346–3355. doi: 10.1038/sj.emboj.7600341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ye HH, Choi HJ, Poe J, Smithgall TE. Oligomerization is required for HIV-1 nef-induced activation of the Src family protein-tyrosine kinase, Hck. Biochemistry. 2004;43:15775–15784. doi: 10.1021/bi048712f. [DOI] [PubMed] [Google Scholar]
- 55.Schmidt U, Richter K, Berger AB, Lichter P. In vivo BiFC analysis of Y14 and NXF1 mRNA export complexes: preferential localization within and around SC35 domains. Journal of Cell Biology. 2006;172:373–381. doi: 10.1083/jcb.200503061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bracha-Drori K, et al. Detection of protein-protein interactions in plants using bimolecular fluorescence complementation. Plant Journal. 2004;40:419–427. doi: 10.1111/j.1365-313X.2004.02206.x. [DOI] [PubMed] [Google Scholar]
- 57.Walter M, et al. Visualization of protein interactions in living plant cells using bimolecular fluorescence complementation. Plant Journal. 2004;40:428–438. doi: 10.1111/j.1365-313X.2004.02219.x. [DOI] [PubMed] [Google Scholar]
- 58.Hackbusch J, Richter K, Muller J, Salamini F, Uhrig JF. A central role of Arabidopsis thaliana ovate family proteins in networking and subcellular localization of 3-aa loop extension homeodomain proteins. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:4908–4912. doi: 10.1073/pnas.0501181102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Diaz I, Martinez M, Isabel-LaMoneda I, Rubio-Somoza I, Carbonero P. The DOF protein, SAD, interacts with GAMYB in plant nuclei and activates transcription of endosperm-specific genes during barley seed development. Plant Journal. 2005;42:652–662. doi: 10.1111/j.1365-313X.2005.02402.x. [DOI] [PubMed] [Google Scholar]
- 60.Shimizu H, et al. LIP19, a basic region leucine zipper protein, is a fos-like molecular switch in the cold signaling of rice plants. Plant and Cell Physiology. 2005;46:1623–1634. doi: 10.1093/pcp/pci178. [DOI] [PubMed] [Google Scholar]
- 61.Abe M, et al. FD, a bZIP protein mediating signals from the floral pathway integrator FT at the shoot apex. Science. 2005;309:1052–1056. doi: 10.1126/science.1115983. [DOI] [PubMed] [Google Scholar]
- 62.Maple J, Aldridge C, Moller SG. Plastid division is mediated by combinatorial assembly of plastid division proteins. Plant Journal. 2005;43:811–823. doi: 10.1111/j.1365-313X.2005.02493.x. [DOI] [PubMed] [Google Scholar]
- 63.MacDonald ML, et al. Identifying off-target effects and hidden phenotypes of drugs in human cells. Nat Chem Biol. 2006;2:329–337. doi: 10.1038/nchembio790. [DOI] [PubMed] [Google Scholar]
- 64.Ding YH, Liu NY, Tang ZS, Liu J, Yang WC. Arabidopsis GLUTAMINE-RICH PROTEIN23 is essential for early embryogenesis and encodes a novel nuclear PPR motif protein that interacts with RNA polymerase II subunit III. Plant Cell. 2006;18:815–830. doi: 10.1105/tpc.105.039495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang KZQ, et al. TRAF6 activation of PI 3-kinase-dependent cytoskeletal changes is cooperative with Ras and is mediated by an interaction with cytoplasmic Src. Journal of Cell Science. 2006;119:1579–1591. doi: 10.1242/jcs.02889. [DOI] [PubMed] [Google Scholar]
- 66.Xu XM, Moller SG. AtSufE is an essential activator of plastidic and mitochondrial desulfurases in Arabidopsis. Embo Journal. 2006;25:900–909. doi: 10.1038/sj.emboj.7600968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen CD, Oh SY, Hinman JD, Abraham CR. Visualization of APP dimerization and APP-Notch2 heterodimerization in living cells using bimolecular fluorescence complementation. Journal of Neurochemistry. 2006;97:30–43. doi: 10.1111/j.1471-4159.2006.03705.x. [DOI] [PubMed] [Google Scholar]
- 68.Hu CD, Kerppola TK. In: Protein-Protein Interactions. Adams P, Golemis E, editors. Cold Spring Harbor Laboratory Press; 2005. pp. 673–693. [Google Scholar]
- 69.Liu H, et al. Mutual regulation of c-Jun and ATF2 by transcriptional activation and subcellular localization. EMBO Journal. 2006;25:1058–1069. doi: 10.1038/sj.emboj.7601020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pazhouhandeh M, et al. F-box-like domain in the polerovirus protein P0 is required for silencing suppressor function. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:1994–1999. doi: 10.1073/pnas.0510784103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cascales E, Atmakuri K, Liu Z, Binns AN, Christie PJ. Agrobacterium tumefaciens oncogenic suppressors inhibit T-DNA and VirE2 protein substrate binding to the VirD4 coupling protein. Molecular Microbiology. 2005;58:565–579. doi: 10.1111/j.1365-2958.2005.04852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stains CI, Porter JR, Ooi AT, Segal DJ, Ghosh I. DNA sequence-enabled reassembly of the green fluorescent protein. Journal of the American Chemical Society. 2005;127:10782–10783. doi: 10.1021/ja051969w. [DOI] [PubMed] [Google Scholar]