

Abstract
Stress dramatically exacerbates pain in diseases such as fibromyalgia and rheumatoid arthritis, but the underlying mechanisms are unknown. We tested the hypothesis that stress causes generalized hyperalgesia by enhancing pronociceptive effects of immune mediators. Rats exposed to nonhabituating sound stress exhibited no change in mechanical nociceptive threshold, but showed a marked increase in hyperalgesia evoked by local injections of prostaglandin E2 or epinephrine. This enhancement, which developed more than a week after exposure to stress, required concerted action of glucocorticoids and catecholamines at receptors located in the periphery on sensory afferents. The altered response to pronociceptive mediators involved a switch in coupling of their receptors from predominantly stimulatory to inhibitory G-proteins (Gs to Gi), and for prostaglandin E2, emergence of novel dependence on protein kinase Cε. Thus, an important mechanism in generalized pain syndromes may be stress-induced coactivation of the hypothalamo-pituitary-adrenal and sympathoadrenal axes, causing a long-lasting alteration in intracellular signaling pathways, enabling normally innocuous levels of immune mediators to produce chronic hyperalgesia.
Keywords: fibromyalgia, G-protein, hyperalgesia, protein kinase Cε, catecholamine, glucocorticoid
Introduction
Stress exacerbates generalized pain syndromes such as fibromyalgia (Nilsen et al., 2007), irritable bowel syndrome (Bach et al., 2006), and interstitial cystitis (Temml et al., 2007), and painful inflammatory diseases like rheumatoid arthritis (Zautra et al., 2007). However, mechanisms mediating the impact of stress on pain are poorly understood.
Clinical studies of the role of stress in the pathogenesis of chronic pain syndromes have implicated the hypothalamo-pituitary-adrenal (HPA) axis in, for example, fibromyalgia and chronic fatigue syndrome (Crofford et al., 1996; Neeck and Crofford, 2000). Although better known for their antiinflammatory actions, glucocorticoids (principal stress mediators released by the hypothalamo-pituitary-adrenal axis) have recently been reported to enhance pain, both in animal models of peripheral neuropathy (Wang et al., 2004; Takasaki et al., 2005) and when used for the treatment of osteoarthritis in humans (Wollstein et al., 2007). In addition, we previously reported that stress enhances the hyperalgesic effect of the cytokine bradykinin in the rat (Khasar et al., 2005), an effect mediated by the sympathoadrenal stress axis mediator, epinephrine.
The present study tested the hypothesis that stress-induced activation of the hypothalamo-pituitary-adrenal and sympathoadrenal axes exacerbates pain by enhancing the pronociceptive effects of immune mediators produced in peripheral tissues. Specifically, we examined the effect of stress on hyperalgesia evoked by prostaglandin E2 (PGE2) or by a mediator of neurogenic inflammation, epinephrine.
Materials and Methods
Animals.
Experiments were performed on adult male Sprague Dawley rats (250–350 g; Charles River). Rats were housed in the Laboratory Animal Resource Center of the University of California, San Francisco, under a 12 h light/dark cycle. Animal care and use conformed to National Institutes of Health guidelines. The University of California, San Francisco, Institutional Animal Care and Use Committee approved experimental protocols. Concerted effort was made to reduce the suffering and number of animals used.
Drugs.
Stock solutions of PGE2 (Sigma-Aldrich) were 4 mg/ml in 10% ethanol, with subsequent dilutions made in 0.9% saline. Stock solutions of epinephrine [(−)-epinephrine bitartrate; Sigma-Aldrich] were prepared daily as 4 mg/ml in distilled water, with 4 mg/ml ascorbic acid to minimize oxidation. This stock solution was further diluted in 0.9% saline for use. Working solutions of epinephrine and PGE2 were kept on ice and in subdued light during experiments. Pertussis toxin (Bordetella pertussis; Calbiochem), dissolved in 0.9% saline, was injected intradermally, on the dorsum of the hindpaw. Nociceptive thresholds were measured before and 30 min after its injection. PGE2 or epinephrine was injected at the same site, 30 min after pertussis toxin, and nociceptive thresholds were again measured 10, 15, and 20 min later. Corticosterone (Cort) (HBC complex; Sigma-Aldrich) was also dissolved in 0.9% saline and injected intradermally 24 h before nociceptive testing or implanted as subdermal pellet. The protein kinase Cε translocation inhibitor (PKCεI) peptide (Calbiochem) was injected intradermally at the site of nociceptive testing at a concentration of 1 μg/2.5 μl in distilled water. Nociceptive thresholds were measured before and 30 min after its injection. PGE2 was then injected at the same site and nociceptive thresholds measured 10, 15, and 20 min later. Injections of PKCεI were immediately preceded by injection of distilled water (2.5 μl), in the same syringe, which produces hypoosmotic shock, facilitating cell penetrance by peptides (Taiwo and Levine, 1989; Khasar et al., 1995).
Mechanical threshold in the skin.
Nociceptive thresholds were quantified in lightly restrained rats using an Ugo Basile Algesymeter (Stoelting), which applies linearly increasing mechanical pressure to the dorsum of the hindpaw. To decrease variability of measurements, the rats were trained for the paw withdrawal reflex test at 5 min intervals for 1 h each day for a period of 3 d (Taiwo et al., 1989). Mechanical nociceptive thresholds were determined both before and 10, 15, and 20 min after the intradermal injection of epinephrine or PGE2, in a volume of 2.5 μl. For each dose of an agent, the mean of the nociceptive threshold at the three time points was determined and the percentage change in nociceptive threshold calculated as follows: (postinjection nociceptive threshold) − (preinjection nociceptive threshold)/(preinjection nociceptive threshold) × 100.
Mechanical threshold in the gastrocnemius muscle.
Because fibromyalgia syndrome affects the muscles, we tested whether there is increased sensitivity to immune mediator, PGE2 in muscle in the sound-stressed rats compared with nonstressed controls. Mechanical nociceptive threshold in the gastrocnemius muscle was quantified using a digital force transducer (Chatillon; model DFI2; Amtek) (Dina et al., 2008). Rats were restrained in a vented tubular Plexiglas holder with slated openings on the side that allow easy access to the hindlimb. A 6-mm-diameter probe attached to the transducer was applied to the gastrocnemius muscle to deliver an increasing compression force, and the nociceptive threshold defined as the force, in newtons, at which the rat withdrew its hindleg. Baseline withdrawal threshold was defined as the mean of two readings taken at 5 min intervals before administration of test agent. Each experimental manipulation was performed on a separate group of rats.
Intramuscular injection of agents.
Increasing doses of PGE2 (0.1–1000 ng) were administered cumulatively, at 25 min intervals, in a volume of 10 μl, into the belly of the gastrocnemius muscle. Injection sites were marked on the skin, using an indelible pen, so that the same site on the muscle could be tested for mechanical nociceptive threshold. Mechanical nociceptive thresholds were measured again 15 and 20 min after injection of PGE2.
Stress.
Exposure to sound stress occurred over 4 d as initially described by Singh et al. (1990) and previously used in our laboratory (Strausbaugh et al., 2003; Khasar et al., 2005). Animals were placed three per cage 25 cm from a speaker that emitted a 105 dB tone of mixed frequencies (11–19 kHz). Over a period of 30 min, rats were exposed to 5 or 10 s sound epochs each minute at random intervals during the minute. Sham-stressed animals were placed in the sound chamber for 30 min, but without exposure to the sound stimulus. After sound stress, rats were returned to the animal care facility in their home cages. Animals were exposed to the stressor on days 1, 3, and 4. This 4 d sound stress protocol is schematically illustrated in Figure 1A. Rats were used for nociceptive studies 24 h, 7 d, or 14 d after the last exposure to sound stress.
Figure 1.

Sound stress enhances hyperalgesia induced by prostaglandin E2 and epinephrine. A, A 4 d cycle of sound stress consists of 30 min of intermittent exposure to sound stress (s) administered on days 1, 3, and 4. Pain threshold measurements (M) were taken on poststress days 1, 7, 14, and 21. B, C, Dose–response relationships for PGE2- and epinephrine-induced hyperalgesia were determined after exposure to sound stress or sham (exposure to the sound stress device without sound). Logarithmically increasing doses of PGE2 or epinephrine were administered cumulatively as indicated. B, Changes in PGE2 hyperalgesia produced by sound stress exposure. There was a significant main effect of group for sound stress on PGE2 hyperalgesia (F(4,261) = 29.70; p < 0.001). Post hoc analysis showed that PGE2 hyperalgesia in the group of rats that was tested 14 d (n = 24) or 21 d (n = 12) after sound stress was significantly enhanced (p < 0.001 each) compared with the sham sound-stressed group (n = 20). PGE2 hyperalgesia in the rest of the groups was not significantly different from the sham-stressed group (p > 0.05). C, Effect of stress on epinephrine hyperalgesia. There was a significant main effect of sound stress on epinephrine hyperalgesia compared with the sham sound stress group (F(4,207) = 110.70; p < 0.001). Post hoc analysis showed that epinephrine hyperalgesia in the group of rats that was tested 24 h after sound stress (n = 22) was significantly less (p < 0.001) than in the sham-stressed group (n = 12). However, 14 d (n = 12) or 21 d (n = 12) after sound stress, epinephrine hyperalgesia was significantly enhanced compared with the sham sound-stressed group (p < 0.001). D, Dose–response relationship for PGE2 in the gastrocnemius muscle in naive rats and 14 d after sound stress. Repeated-measures ANOVA showed a significant difference in the response of gastrocnemius muscle to PGE2 between naive and sound-stressed rats (F(1,55) = 53.2; p < 0.001; n = 12 each). Error bars indicate SEM.
Adrenal adrenalectomy and medullectomy.
Rats were anesthetized with isoflurane (2.5% in 97.5% O2). The adrenal glands were located through bilateral incisions in the abdominal wall, and either removed (for adrenalectomy) or the capsules incised and the medullas removed (for adrenal medullectomy) (Wilkinson et al., 1981; Miao et al., 1992). Fascia were closed with suture, and the skin was closed with wound clips. Rats were provided with 0.45% saline to drink for the first 7 d after surgery. Adrenal medullectomy was performed at least 5 weeks before additional experimental procedures to allow maximum recovery of HPA axis function (Wilkinson et al., 1981).
Measurement of epinephrine.
One day before initiation of sound stress and 1, 7, or 14 d after its completion, whole-blood samples (0.5 ml) were collected from the tail veins of anesthetized rats, and immediately placed on ice. Plasma was isolated by centrifugation and stored at −80°C until analysis. Catecholamines were extracted from plasma by alumina adsorption, and levels were determined using HPLC with electrochemical detection (5200 electrochemical detector; ESA) (Khasar et al., 2003a).
Measurement of plasma corticosterone.
Rats were anesthetized in 2–3% isoflurane for at least 15 min before sample collection and all efforts were made to minimize stress before blood collection. Peripheral blood was collected via the tail vein in heparinized tubes, centrifuged, and plasma was stored at −80°C until analysis. Plasma corticosterone was measured (in duplicate) per manufacturer's instructions using a commercially available radioimmunoassay kit (Siemens Medical Solutions Diagnostics). Briefly, 50 μl of standard or plasma and 1 ml of 125I rat corticosterone were added to corticosterone Ab-coated tubes and incubated for 2 h. Tubes were decanted and each radiolabeled tube was read using a gamma counter for 1 min. Corticosterone concentration is expressed as nanograms per milliliter.
Tissue preparation and immunohistochemistry.
Naive rats were deeply anesthetized with sodium pentobarbital (100 mg/kg, i.p.) and perfused through the left ventricle with ∼35 ml of PBS containing heparin (100 U/ml), followed by 300 ml of PBS, pH 7.3, containing 4% paraformaldehyde (Sigma-Aldrich). L5 dorsal root ganglia (DRGs) were immediately dissected out and sequentially transferred and stored in PBS containing 10, 20, and 30% sucrose at 4°C for 12–24 h each. The DRGs were embedded in Tissue Tek (OCT compound; Electron Microscopy Sciences) sectioned at 20 μm in a Hacker/Bright microtome cryostat (Hacker Instruments) and mounted on microscope slides (Fisher Scientific).
Before antigen detection, tissue sections were rinsed with PBS, blocked, and permeabilized for 1 h at room temperature with PBS containing 10% normal goat serum (Jackson ImmunoResearch Laboratories) and 0.3% Triton X-100. Glucocorticoid receptor (GCR) expression was revealed by a 12 h incubation with a 1:500 dilution of the anti-GCR antibody (Santa Cruz Biotechnology) with PBS containing 10% normal goat serum (antibody dilution buffer) followed by three washing steps with PBS containing 0.3% Triton X-100 (for 10 min each) and a 2 h incubation with an 1:500 dilution of an Alexa Fluor 594 conjugated anti-rabbit antibody (Invitrogen) in antibody dilution buffer. Tissue sections were washed three times with PBS containing 0.3% Triton X-100 and mounted with Fluoromount G (Southern Biotechnology Associates).
Oligodeoxynucleotide antisense.
The glucocorticoid receptor antisense oligodeoxynucleotide (ODN) sequence: 5′-TGG AGT CCA TTG GCA AAT-3′ was directed against the translation initiation site of the rat mRNA (Engelmann et al., 1998) (GenBank accession number Y12264). The mismatch ODN sequence was designed by mismatching five bases (Engelmann et al., 1998) (denoted here by boldface type) of the antisense sequence, 5′-TGA AGT TCA GTG TCA ACT-3′. A BLAST search was performed to check for the theoretical specificity and activity of the sequences, to confirm that they were not homologous to other sequences in the rat. ODNs were reconstituted in nuclease-free 0.9% NaCl. ODN was injected at a dose of 2 μg/μl in 20 μl. As described previously (Alessandri-Haber et al., 2003, 2004; Dina et al., 2005; Malik-Hall et al., 2005), rats were anesthetized with 2.5% isoflurane (97.5% O2), a 30-gauge needle was inserted into the subarachnoid space on the midline between the L4 and L5 vertebrae, and the ODN injected at 1 μl/s by microsyringe. It has been shown that intrathecal administration of ODNs provides an effective mode of delivery of these molecules to the DRG neurons in vivo (Lai et al., 2002). Rats received injections of corticosterone (1 μg/2.5 μl, intradermally) into one hindpaw on the sixth day of glucocorticoid receptor ODN treatment. In stressed animals, ODN treatment was initiated on the same day as sound stress and continued for 13 d after sound stress. In either case, nociceptive testing was performed the day after the last ODN treatment.
Chronic administration of epinephrine and corticosterone.
Chronic administration of stress levels of epinephrine was performed by implanting Alzet miniosmotic pumps (model 2004; Durect) filled with epinephrine, subcutaneously in the interscapular region to deliver epinephrine at the rate of 5.4 μg · 0.25 μl−1 · h−1 (Khasar et al., 2005). Rats were anesthetized with isoflurane to insert implants. To produce levels of circulating corticosterone that mimic the time course of corticosterone levels induced by the sound stress protocol, 100 mg corticosterone pellets (Akana et al., 1985) were implanted into adrenalectomized rats (to produce the normal baseline corticosterone level) and corticosterone (25 mg/kg, i.p.) (Kavushansky and Richter-Levin, 2006) injected on days 1, 3, and 4 (to mimic the sound-stress-induced transient increases in corticosterone). Nociceptive threshold was measured in rats with corticosterone and epinephrine implants 14 d after the last injection of corticosterone or vehicle.
Western blots.
In anesthetized rat, the saphenous nerve was ligated with silk surgical suture (4-0) 1 cm above its knee-level bifurcation. On the fourth day after initiation of ODN treatment, a 5 mm section of saphenous nerve proximal to the ligation was harvested. Nerves were homogenized in radioimmunoprecipitation assay buffer with 2× protease inhibitor by probe sonication. Cell lysates were then run on 7.5% polyacrylamide gels and transferred to the membrane. The membrane was probed with M-20 anti-glucocorticoid receptor antibody (raised in the rabbit; 1:200; Santa Cruz Biotechnology) followed by incubation with horseradish peroxidase-conjugated goat anti-mouse IgG (1:5000; Pierce Biotechnology). The specificity of the anti-glucocorticoid receptor antibody was controlled for by using the corresponding blocking peptide. The antibody only detected three bands, one intense band at the expected size of 90 kDa, and two much fainter bands around 65 and 120 kDa. The band at the expected size was used for our statistical quantification. To normalize the amount of sample loaded, affinity-purified mouse monoclonal anti-GAPDH antibody (1:5000; Abcam) was used, followed by horseradish peroxidase-conjugated goat anti-mouse IgG (1:5000; Pierce). Membranes were incubated with enhanced chemiluminescence reagents (SuperSignal West solution; Pierce), and images of the membrane were acquired with CHEMILMAGER chemiluminescence imaging system and analyzed with AlphaEaseFC software (Alpha Innotech).
Statistical analysis.
Group data are presented as mean ± SEM and analyzed using repeated-measures ANOVA, two-way or one-way ANOVA, or Student's t test, as appropriate. Where the overall ANOVA showed significant differences between the groups, Scheffé's post hoc test was used to determine the pairs of groups that were different. The accepted level of significance was p < 0.05. The p values for main effects are from ANOVA, and all subsequent p values are from Scheffé's post hoc tests, unless otherwise stated.
Results
Stress causes a delayed-onset, long-lasting increase in hyperalgesia evoked by PGE2 or epinephrine
As we reported previously (Khasar et al., 2005), 24 h after exposure to a 4 d protocol of unpredictable sound stress (Fig. 1A), the magnitude of hyperalgesia induced by an intradermal injection of PGE2 into the hindpaw was unchanged (Fig. 1B). However, in the present study we tested for effects over a much longer time period, and found that 14 d after sound stress, a marked enhancement of PGE2 hyperalgesia became apparent (40% greater decrease in the threshold for mechanically evoked paw withdrawal) (Fig. 1B). This state of greater susceptibility to PGE2-induced hyperalgesia persisted without diminution for at least 21 d (Fig. 1B). The 4 d sound stress protocol alone did not significantly affect nociceptive threshold, measured 24 h [sham sound stress, 107.9 ± 1.1 g (n = 30); sound stress, 107.9 ± 1.4 g (n = 30)] or 14 d [sham sound, 105.9 ± 1.7 g (n = 42); sound stress, 106.7 ± 1.1 g (n = 40)] after the last sound stress.
To determine whether stress-induced enhancement is restricted to hyperalgesia elicited via prostaglandin receptors, we tested the effect of sound stress on hyperalgesia elicited by the pronociceptive mediator, epinephrine. Epinephrine produces mechanical hyperalgesia via G-protein-coupled β2-adrenergic receptors on nociceptors (Khasar et al., 1999). Twenty-four hours after the end of sound stress, epinephrine-induced hyperalgesia was attenuated (Fig. 1C). However, similar to the observed enhancement of PGE2-induced hyperalgesia, 14 d after stress, epinephrine hyperalgesia was significantly enhanced (Fig. 1C). This indicates that the effect of stress is not limited to prostaglandin receptor signaling, but may be generalized to other G-protein-coupled receptors. Importantly, the concentration of immune mediator needed to induce hyperalgesia after stress is 1–1.5 orders of magnitude less than that required in the nonstressed control condition (Fig. 1B,C).
In the gastrocnemius muscle, 14 d after sound stress, PGE2 hyperalgesia was significantly enhanced compared with that in the nonstressed control group (Fig. 1D) (p < 0.001).
Plasma glucocorticoids contribute to the effect of stress
We previously showed that plasma levels of glucocorticoids are increased by our unpredictable sound stress protocol (Strausbaugh et al., 2003), and others have shown that sensory neurons express glucocorticoid receptors (DeLeon et al., 1994). Therefore, we assessed the role of glucocorticoids in the stress-induced enhancement of hyperalgesia evoked by immune mediators, using a glucocorticoid receptor antagonist and intrathecal administration of ODN antisense to the glucocorticoid receptor. As shown in Figure 2A, daily subcutaneous administration of the glucocorticoid receptor antagonist, mifepristone, during the period of sound stress exposure, and 13 d thereafter, prevented the stress-induced enhancement of epinephrine hyperalgesia compared with mifepristone vehicle-treated group (p < 0.001), while not affecting baseline nociceptive thresholds [mifepristone vehicle, 107.5 ± 4.4 g (n = 4); mifepristone, 107.8 ± 2.7 (n = 12)].
Figure 2.

The effect of stress is mediated by glucocorticoid receptors in sensory neurons. A, Dose–response relationship for epinephrine hyperalgesia shows that systemic administration of the glucocorticoid receptor antagonist mifepristone (RU 38486), starting at the same time as initiation of sound stress and continuing for 13 d after, prevented the stress-induced enhancement of epinephrine hyperalgesia. All measurements were taken 14 d after the final exposure to sound stress. There was a significant main effect of mifepristone treatment on epinephrine hyperalgesia between the groups (F(3,132) = 23.5; p < 0.001). Post hoc analysis showed that subcutaneous injection of mifepristone (n = 12), but not its vehicle (n = 4), prevented sound-stress-induced enhancement of epinephrine hyperalgesia (p = 0.001). Epinephrine hyperalgesia in the mifepristone-treated group was not significantly different from that in the sham sound-stressed group (p > 0.05). B, Immunohistochemical analysis of the GCR expression in rat lumbar dorsal root ganglia. Anti-GCR immunoreactivity (ir) was detected in sensory neurons. As expected, anti-GCR-ir shows a predominantly nuclear distribution. A, Negative control, immunohistochemistry in the absence of the anti-GCR antibody. B, Immunohistochemistry in the presence of the anti-GCR antibody. Scale bars, 100 μm. C, Expression of glucocorticoid receptor was decreased in peripheral nerves after intrathecal antisense treatment. A 90 kDa band corresponding to the glucocorticoid receptor was detected by Western blotting of saphenous nerve. There was a 33 ± 8% decrease in protein expression in antisense compared with mismatch ODN-treated rats (p = 0.004, unpaired Student's t test; n = 11 for both antisense- and mismatch-treated rats). D, Glucocorticoid receptor antisense inhibits sound-stress-induced enhancement of epinephrine hyperalgesia. Rats were injected intrathecally daily with antisense (AS) or mismatch control (MM) oligodeoxynucleotides to the GCR. These rats were exposed to sound stress, and epinephrine hyperalgesia was measured at 14 d postexposure. The antisense, but not mismatch, ODN eliminated the enhancement of epinephrine hyperalgesia seen 14 d after sound stress. Overall ANOVA showed significant main effect of ODN treatment on epinephrine hyperalgesia between the groups (F(3,156) = 30.3; p < 0.001). Glucocorticoid receptor antisense treatment (n = 12) significantly inhibited stress-induced enhancement (n = 12) of epinephrine hyperalgesia [GCR AS compared with GCR MM or sound stress (14 d); p = 0.001]. Error bars indicate SEM.
To determine whether glucocorticoids act directly on sensory nerve fibers to enhance epinephrine- and PGE2-induced hyperalgesia, we tested whether the stress-induced enhancement of epinephrine hyperalgesia is blocked by intrathecally injected ODN antisense to the glucocorticoid receptor. As shown in Figure 2B, sensory neurons express glucocorticoid receptors. As shown in Figure 2C, expression of glucocorticoid receptor protein was reduced by 33 ± 8% in the saphenous nerve in antisense- versus mismatch-treated rats. As shown in Figure 2D, glucocorticoid receptor antisense treatment eliminated the stress-induced enhancement of epinephrine hyperalgesia (tested 14 d after the last exposure to sound stress; p = 0.001; n = 12).
We determined whether elevated glucocorticoids alone, acting locally at the site of nociceptive testing, are sufficient to mimic the pronociceptive effect of stress. As shown in Figure 3A, intradermal injection of Cort (1 μg in 2.5 μl) at the site of mechanical nociceptive testing on the dorsum of the hindpaw, increased epinephrine-induced hyperalgesia (p = 0.004 at 24 h; p < 0.005 at 7 d). Although the glucocorticoid-induced enhancement of epinephrine hyperalgesia was of more rapid onset (detectable within 24 h) than that induced by stress, it was similarly long-lasting. Corticosterone injection had no effect on the baseline nociceptive threshold after 24 h [Cort, 110.4 ± 1.7; vehicle, 111.3 ± 2.1 (n = 22 each)] or 7 d [Cort, 107.3 ± 1.8; vehicle, 111.0 ± 1.7 (n = 12 each)], and epinephrine hyperalgesia in the contralateral, vehicle-treated paw was not enhanced (Fig. 3A). Local intradermal corticosterone treatment in sound-stressed rats did not further enhance epinephrine-evoked hyperalgesia (Fig. 3B). The increase in the hyperalgesic response produced by local administration of glucocorticoids was not limited to epinephrine-evoked hyperalgesia; it also enhanced PGE2 hyperalgesia (Fig. 4A).
Figure 3.
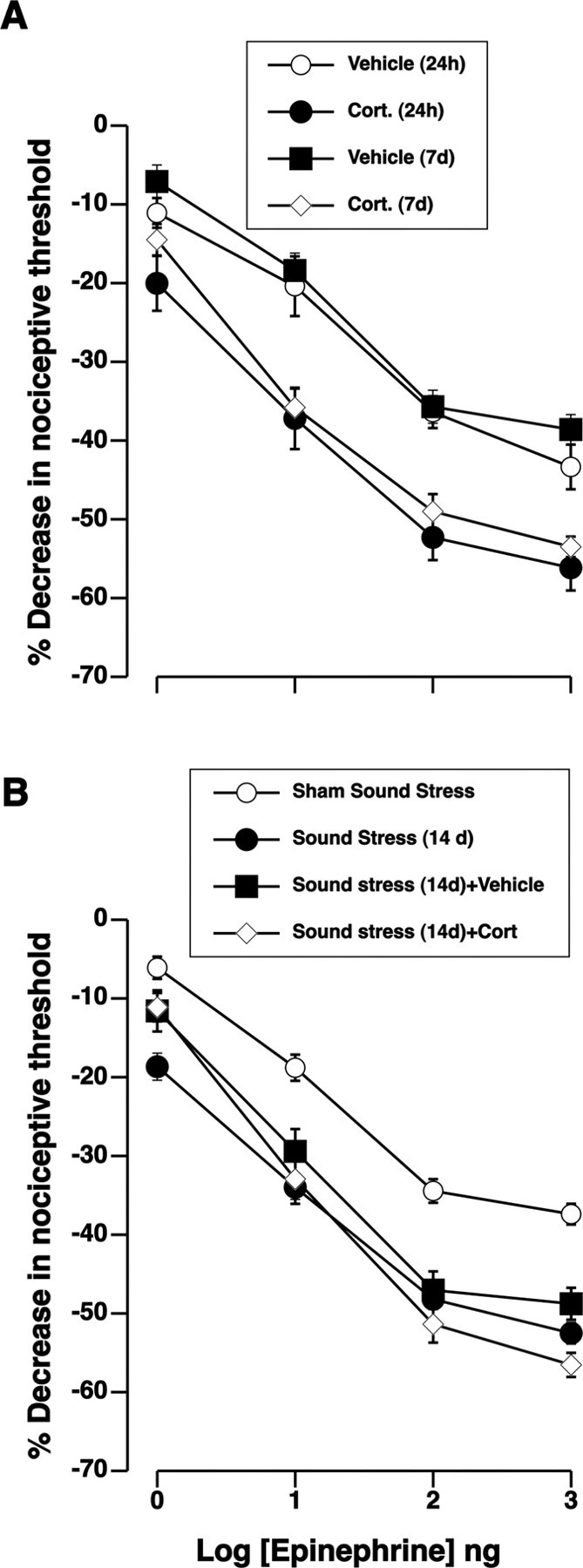
Corticosterone mimics the ability of stress to enhance epinephrine hyperalgesia. A, Dose–response relationships for hyperalgesia induced by intradermal injection of epinephrine show that pretreatment with intradermal corticosterone (1 μg/2.5 μl) enhanced responses to epinephrine, measured 24 h later. Corticosterone was injected in one paw (Cort), while vehicle was injected in the contralateral paw (vehicle). No effect was observed in untreated contralateral paws. There was a significant main effect of corticosterone-treatment on epinephrine hyperalgesia between the groups (F(3,84) = 8.32; p < 0.001). Post hoc analysis showed significant enhancement of epinephrine hyperalgesia 24 h (n = 10) after corticosterone treatment compared with the vehicle-treated paws (n = 10) during the same time period (p = 0.004). Epinephrine hyperalgesia remained significantly enhanced in the corticosterone-treated paws 7 d after corticosterone injection (n = 6), whereas the vehicle-treated paws (n = 6) remained unaffected (p < 0.005). B, Intradermal corticosterone did not significantly increase sound stress enhancement of epinephrine hyperalgesia compatible with an occlusion of overlapping mechanisms. There was a significant main effect of corticosterone treatment on epinephrine hyperalgesia (F(3,144) = 32.4; p < 0.001). Post hoc analysis showed that corticosterone treatment did not further enhance epinephrine hyperalgesia 14 d after sound stress (n = 10) compared with sound stress with (p > 0.05; n = 10) or without (p > 0.05; n = 20) corticosterone vehicle. Error bars indicate SEM.
Figure 4.
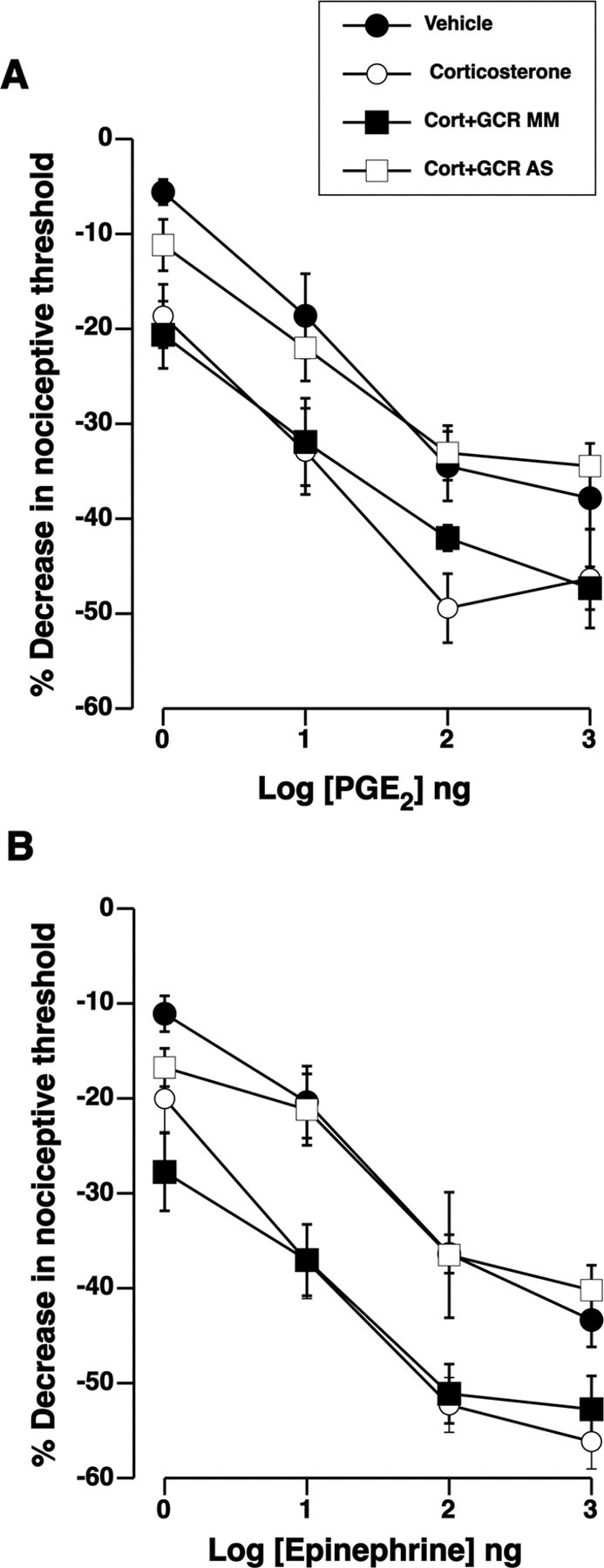
Corticosterone-enhanced hyperalgesia is mediated by glucocorticoid receptors on sensory neurons. A, Intrathecally administered glucocorticoid receptor antisense (AS), but not mismatch (MM), ODN treatment abolished enhancement of PGE2 hyperalgesia after intradermal glucocorticoid treatment. Overall ANOVA showed significant main effect of ODN treatment group on PGE2 hyperalgesia (F(3,60) = 7.2; p = 0.002). Post hoc analysis showed that glucocorticoid receptor antisense ODN treatment (n = 6) significantly reversed corticosterone-induced enhancement (n = 6) of PGE2 hyperalgesia (p = 0.030, GCR AS compared with GCR MM or corticosterone). B, Rats received intrathecal injections of glucocorticoid receptor antisense and mismatch ODN and were then treated with intradermal corticosterone 24 h before measurement of epinephrine hyperalgesia. Antisense but not mismatch glucocorticoid receptor treatment abolished enhancement of epinephrine hyperalgesia that is normally seen after intradermal glucocorticoid treatment. There were significant main effects of ODN treatment on epinephrine hyperalgesia between the antisense and mismatch groups (F(3,84) = 8.0; p < 0.001). Post hoc analysis showed that glucocorticoid antisense treatment (n = 6) significantly reversed corticosterone-induced enhancement (n = 6) of epinephrine hyperalgesia (p = 0.035, GCR AS compared with GCR MM or corticosterone). Epinephrine-induced hyperalgesia in the vehicle-treated paw of control rats was not different from that of the paws in the antisense ODN-treated group (p > 0.05). Error bars indicate SEM.
To address the possibility of an indirect effect of locally injected glucocorticoids via non-neuronal cells in the skin, antisense ODN was intrathecally administered to reduce expression of glucocorticoid receptor in primary afferents selectively among all other cells in the skin. Intrathecal administration of ODN antisense to glucocorticoid receptor blocked the enhancement of PGE2-evoked hyperalgesia induced by intradermal administration of corticosterone, compared with the effect of corticosterone in the control group (Fig. 4A) (p = 0.03). As shown in Figure 4B, ODN antisense, but not mismatch, to the glucocorticoid receptor also eliminated the enhancement of epinephrine-induced hyperalgesia after intradermal corticosterone treatment (p = 0.035), but baseline nociceptive threshold was unaltered [mismatch, 109.0 ± 2.0 g; vs antisense, 107.2 ± 1.8 g (p > 0.05; n = 12 each)]. Epinephrine-induced hyperalgesia in the vehicle-treated (contralateral) paw of control rats was not different from that of the antisense ODN-treated group (Fig. 4B) (p > 0.05). Thus, we conclude that the action of corticosterone in the skin to enhance immune mediator-evoked hyperalgesia occurs directly on the peripheral nerve endings of primary afferents, presumably enhancing sensitization of nociceptors by the immune mediator.
Plasma epinephrine augments the glucocorticoid-mediated effect
To investigate the contribution of circulating epinephrine in stress-induced hyperalgesia, we first tested whether sound stress does, in fact, elevate the serum level of epinephrine. Plasma levels of epinephrine were significantly increased 24 h after the final sound stress exposure compared with those in sham-stressed rats (Table 1), and plasma epinephrine levels were still significantly elevated 21 d after sound stress (Table 1).
Table 1.
Plasma epinephrine and corticosterone levels in sound-stressed male Sprague Dawley rats
| Epinephrine (pg/ml) | Corticosterone (ng/ml) | ||
|---|---|---|---|
| Prestress | 273.17 ± 51.01 (12) | 34.4 ± 8.0 (12) | |
| 1 h | Sham sound stress | 338.6 ± 106.1 (9) | 37.8 ± 17.5 (12) |
| Sound stress | 1034.0 ± 87.5 (12) | 268.3 ± 25.9 (12) | |
| 24 h | Sham sound stress | 289.1 ± 88.0 (8) | 17.0 ± 8.8 (12) |
| Sound stress | 801.5 ± 29.5 (11) | 76.8 ± 21.4 (11) | |
| 7 d | Sham sound stress | 348.9 ± 103.8 (7) | 27.7 ± 14.1 (12) |
| Sound stress | 754.7 ± 38.3 (10) | 86.9 ± 23.0 (11) | |
| 14 d | Sham sound stress | 142.6 ± 15.9 (10) | 65.9 ± 16.7 (12) |
| Sound stress | 910.0 ± 106.3 (20) | 133.1 ± 34.4 (11) | |
| 21 d | Sham sound stress | 230.3 ± 62.4 (10) | 41.0 ± 11.5 (11) |
| Sound stress | 609.7 ± 95.0 (12) | 124.0 ± 35.0 (11) |
As shown in Figure 5A, adrenal medullectomy was performed in two groups of rats and one group stressed, to test whether release of epinephrine from the adrenal medulla is necessary for stress-induced enhancement of hyperalgesia evoked by immune mediators. In adrenal-medullectomized rats, hyperalgesia evoked by intradermal injection of epinephrine was not significantly different in stressed compared with nonstressed animals (p > 0.05), suggesting that both glucocorticoids and epinephrine are necessary for the initiation of the process leading to enhancement of hyperalgesia later on.
Figure 5.
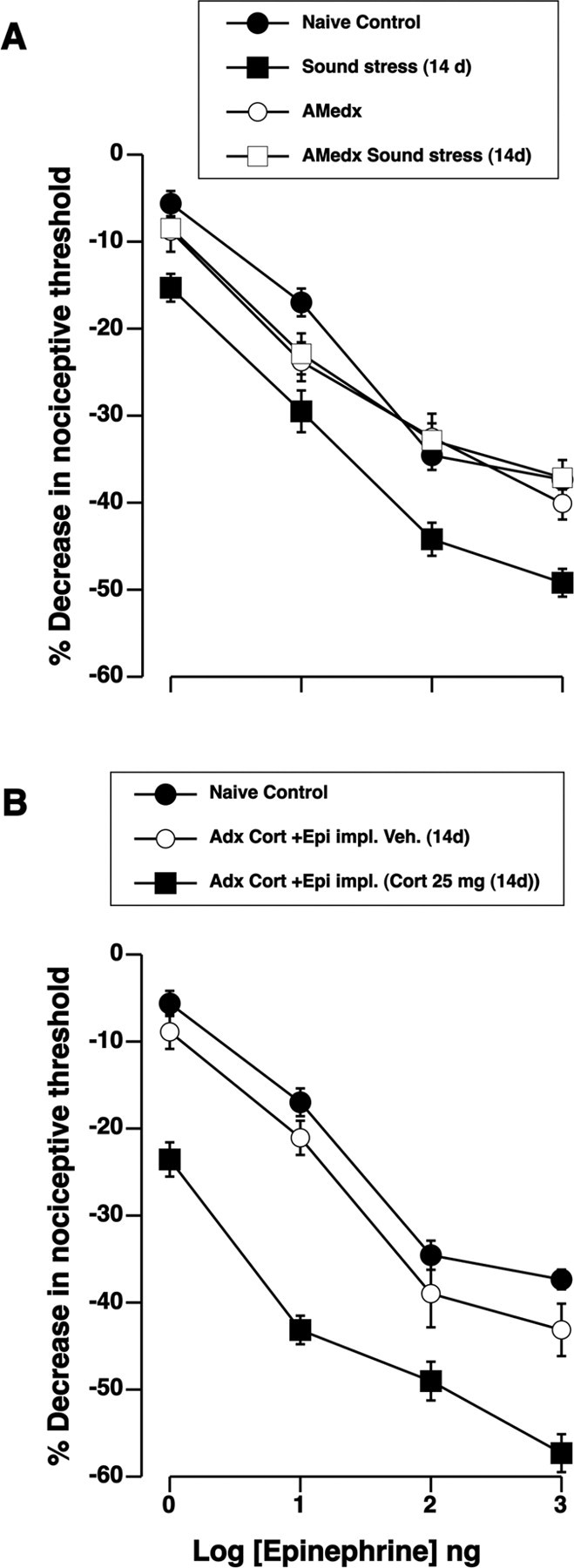
Epinephrine augments the glucocorticoid-mediated enhancement of epinephrine hyperalgesia. A, Dose–response curves for epinephrine show that adrenal medullectomy prevented sound-stress-induced enhancement of epinephrine hyperalgesia. There were differences between the groups (F(3,89) = 10.2; p < 0.001). Post hoc analysis showed that, whereas epinephrine hyperalgesia in naive control (n = 30), AMedx (n = 12), and AMedx sound stress (n = 20) groups was not significantly different (p > 0.05), it was significantly enhanced in the sound stress (n = 20) group (p < 0.001). B, Adrenalectomized rats (n = 6) were reconstituted with both corticosterone (100 mg) pellets and epinephrine (5.4 μg/h) on the day of surgery. Three days after the implants, three rats were given bolus injection of corticosterone (25 mg/kg, i.p.) on days 1, 3, and 4 (similar to a sound stress protocol), the other three rats received injection of vehicle. Fourteen days after the last injection, intradermal injection of epinephrine (1–1000 ng) produced enhanced hyperalgesia compared with the naive control or vehicle-treated groups. Overall ANOVA showed significant main effect of reconstitution group on epinephrine hyperalgesia (F(3,189) = 20.3; p < 0.001). Post hoc analysis showed a significant enhancement of epinephrine hyperalgesia (p < 0.001; n = 8) in adrenalectomized rats that were given a bolus injection of corticosterone, compared with naive controls (n = 31). Error bars indicate SEM.
We performed additional experiments to examine the influence of sympathoadrenal-released epinephrine on the glucocorticoid-mediated enhancement of immune mediator hyperalgesia. First, rats were adrenalectomized to eliminate the release of both epinephrine and glucocorticoids by the sympathoadrenal and HPA axes, respectively. Corticosterone (100 mg) fused pellets as well as epinephrine-containing osmotic minipumps were implanted in adrenalectomized rats. In addition to the implants, a subgroup of the rats was given three injections of corticosterone (25 mg/kg body weight, i.p.) (Kavushansky and Richter-Levin, 2006) over a 4 d period, on days 1, 3, and 4, to produce stress levels of corticosterone, similar to the sound stress protocol. One hour after the last injection of corticosterone, plasma levels were [640.5 ± 103.5 ng/ml (n = 6)]. Plasma levels for corticosterone 1 h after sound stress in the current study were [268.3 ± 25.9 ng/ml (n = 12)] (Table 1). As shown in Figure 5B, rats exposed to stress plasma levels of corticosterone for 3 d and epinephrine for 14 d exhibited an enhanced epinephrine-evoked compared with either naive or rats injected with corticosterone vehicle (p < 0.001).
Stress alters intracellular signaling pathways in nociceptors
We hypothesized that the initial decrease of sensitivity to intradermal epinephrine (measured 24 h after stress) reflects desensitization of β2-adrenergic receptors in response to the high plasma levels of epinephrine. However, the enhancement of epinephrine hyperalgesia, first observed 14 d after sound stress, which also occurs in the presence of sustained elevation of plasma epinephrine, would seem to require some other mechanistic change in the signaling pathways underlying PGE2- and epinephrine-induced hyperalgesia. Therefore, we tested the hypothesis that the enhancement of epinephrine-induced hyperalgesia involves a change in intracellular signaling pathways by which epinephrine sensitizes nociceptors.
In cardiac cells, chronic exposure to catecholamines can induce a switch in the a subunit of the G-protein complex associated with β2-adrenergic receptors, from αs to αi (Gs to Gi/o) (Lamba and Abraham, 2000). To test for a similar change in intracellular signaling pathways in cutaneous nociceptors caused by stress, we assayed the sensitivity of epinephrine-evoked hyperalgesia to pertussis toxin, a Gi/o protein inhibitor. As shown in Figure 6A, in nonstressed rats, ∼50% of epinephrine-induced decrease in paw withdrawal threshold was blocked by pertussis toxin (100 ng in 2.5 ml intradermal); after sound stress, however, ∼80% of the epinephrine-induced hyperalgesia became sensitive to this dose of pertussis toxin (p < 0.001).
Figure 6.

Adrenal medulla is required for stress-induced enhancement of hyperalgesia and pertussis toxin sensitivity. A, The Gi/o inhibitor pertussis toxin (PTx) (100 ng/2.5 μl) partially inhibited epinephrine hyperalgesia (shown here as a decrease in the nociceptive threshold). PTx sensitivity was enhanced after sound stress. One-way ANOVA showed a significant main effect of sound stress group on epinephrine hyperalgesia (F(3,40) = 95.8; p < 0.001). Post hoc analysis showed epinephrine hyperalgesia after sound stress (n = 16) was significantly more inhibited by PTx than epinephrine hyperalgesia in the nonstressed group (sham sound; p = 0.013; n = 8). B, Adrenal medullectomy prevented the inhibition of epinephrine hyperalgesia by PTx in sham sound-stressed rats. Adrenal medullectomy also prevented sound-stress-induced enhancement as well as inhibition of epinephrine hyperalgesia by PTx. One-way ANOVA showed no significant main effect of adrenal medullectomy on epinephrine hyperalgesia between the sham sound-stressed and sound-stressed groups, whether PTx-treated or not (F(3,44) = 0.26; p > 0.05). C, Whereas PTx treatment enhanced PGE2 hyperalgesia in sham sound-stressed rats, it inhibited it in sound-stress-induced enhancement of PGE2 hyperalgesia. One-way ANOVA showed a significant main effect of PTx treatment on PGE2 hyperalgesia between the sham sound-stressed and sound-stressed groups (F(3,24) = 8.2; p < 0.001). Post hoc analysis showed that, whereas PTx significantly enhanced PGE2 hyperalgesia in sham sound-stressed rats [p = 0.002, sham sound stress (n = 6) compared with sham sound stress plus PTx (n = 6)], PTx inhibited sound-stress-induced enhancement of PGE2 hyperalgesia [p = 0.002, sound stress (n = 8) compared with sound stress plus PTx (n = 8)]. D, PKCεI (1 μg/2.5 μl) peptide had no effect on PGE2-evoked hyperalgesia in sham-stressed rats, but it abolished the enhancement of PGE2 hyperalgesia caused by sound stress. The scrambled peptide [PKCεI (-ve)] had no effect on stress-induced enhancement of PGE2 hyperalgesia. The dose of epinephrine used in A and B was 100 ng/2.5 μl, whereas the dose of PGE2 used in both C and D was 1 ng/2.5 μl. Error bars indicate SEM.
To determine whether the stress-induced increase in signaling via Gi/o is attributable to release of epinephrine by the sympathoadrenal stress axis, we performed adrenal medullectomy to eliminate the effect of neuroendocrine epinephrine. Hyperalgesia evoked by intradermal injection of epinephrine was not inhibited by pertussis toxin in both stressed as well as nonstressed adrenal-medullectomized animals (Fig. 6B). Thus, even levels of adrenal-derived epinephrine in the systemic circulation affect the G-protein dependence of signaling pathways by which epinephrine produces hyperalgesia, with basal levels of epinephrine maintaining ∼50% dependence on Gi/o and stress levels markedly increasing this dependence to 80%.
To determine whether the stress-induced enhancement of PGE2 hyperalgesia also involves a similar switch to Gi/o-dependent mechanisms, we assayed the effect of pertussis toxin on PGE2 hyperalgesia. In sham-stressed control rats, pertussis toxin actually enhances PGE2-evoked hyperalgesia (Fig. 6C) [a result similar to that previously observed in naive rats, presumably because of the toxin attenuating some baseline inhibition of adenylyl cyclase by Gi/o (Taiwo and Levine, 1991)]. After exposure to sound stress, however, pertussis toxin no longer enhanced PGE2 hyperalgesia, rather it significantly inhibited it (Fig. 6C) (p = 0.002). This suggests that, as for β2-adrenergic receptors, sound stress induces a switch in G-protein signaling, enhancing a hyperalgesic, pertussis toxin-sensitive pathway linked to PGE2 receptors. It also suggests that signaling downstream from Gi/o switches from predominantly inhibiting adenylyl cyclase to an alternative pathway that sensitizes primary afferent nociceptors.
We have shown previously that in hyperalgesic priming, a state induced by previous inflammatory lesion, PGE2 hyperalgesia switches to PKCε-dependent mechanisms (Aley et al., 2000). Therefore we investigated whether stress-enhanced PGE2 hyperalgesia, in addition to switching to a Gi/o-dependent pathway, also gains a new linkage to PKCε. An inhibitor of PKCε was administered intradermally before administration of PGE2 at the same site. The PKCε translocation inhibitor peptide had no effect on PGE2-evoked hyperalgesia in sham-stressed rats, but it abolished the enhancement of PGE2 hyperalgesia caused by sound stress (Fig. 6D). This reduction in stress-induced enhancement of PGE2 hyperalgesia was similar in magnitude to the reduction caused by the Gi/o inhibitor, pertussis toxin.
Discussion
We have identified a stress-induced prolonged enhancement of mechanical hyperalgesia mediated by neuroendocrine release of both glucocorticoids and catecholamines, which act by modifying signaling pathways in primary afferent nociceptors. This phenomenon may explain the prominent influence of stress in chronic generalized pain syndromes, such as fibromyalgia (Nilsen et al., 2007) and irritable bowel syndrome (Bach et al., 2006), as well as in other illnesses, like posttraumatic stress disorder (Otis et al., 2003) and depression (Blackburn-Munro and Blackburn-Munro, 2001).
The pronociceptive effect of stress that we observed differs markedly from previously reported models of “stress-induced hyperalgesia” (Imbe et al., 2006). Most notably, our protocol of unpredictable sound stress by itself did not cause hyperalgesia (there was no change in the baseline nociceptive threshold after stress). Instead, the effect of stress was only manifest as an enhancement of the hyperalgesia evoked by immune mediators. Specifically, stress caused a leftward shift in the dose–response relationship for immune mediator hyperalgesia by 1.5 orders of magnitude. Therefore, we speculate that in disease states associated with increased cytokines, even at low levels normally insufficient to produce hyperalgesia, stress (also at levels normally insufficient to produce hyperalgesia) could precipitate a long-lasting hyperalgesic condition.
Our model also differs from other models of stress-induced hyperalgesia in the very long delay (7–14 d) between exposure to stress and the expression of enhanced immune mediator hyperalgesia. The present results do not implicate a mechanism for the delay, but it may be instructive to note that a similar delay in enhancement of hyperalgesia occurs in rats after subdiaphragmatic vagotomy (Khasar et al., 1998a), an effect also dependent on neuroendocrine stress axes (Khasar et al., 1998b, 2003b). Furthermore, whereas hyperalgesia is a component of inflammation, in our previous studies on the effect of nonhabituating sound stress in a model of inflammation, bradykinin-induced plasma extravasation from the knee joint, we found no evidence of the involvement of the HPA axis in sound-stress-induced suppression of plasma extravasation (Strausbaugh et al., 2003). This lack of involvement of the HPA axis suggests differences in the mechanisms mediating the effect of nonhabituating sound stress in the inflammation model (Strausbaugh et al., 2003) and enhancement of immune mediator hyperalgesia in the current study.
Previous investigations have implicated changes in CNS circuitry as underlying mechanisms of stress-induced hyperalgesia (Imbe et al., 2006). In contrast, although the stress-induced hypersensitivity to immune mediator hyperalgesia that we observed could potentially also involve changes in the CNS, it clearly depends on changes within the primary afferent nociceptor. Thus, the effect of stress could be mimicked by local administration of corticosterone in the skin, an effect attenuated by intrathecal administration of antisense to the glucocorticoid receptor. These results suggest that glucocorticoids act at receptors located on sensory nerve fibers to participate in stress-enhancement of epinephrine hyperalgesia.
Within nociceptors, stress caused changes in second messenger signaling pathways that are activated by hyperalgesic mediators. This was indicated by a shift toward greater dependence on Gi/o signaling in nociceptors of stressed rats. This shift seems related to a previously reported shift in β2-adrenergic receptor signaling from Gs to Gi/o caused by chronic exposure to epinephrine (Lamba and Abraham, 2000). However, that shift involves an increased role for Gi/o inhibition of adenylyl cyclase, which would be expected to attenuate hyperalgesia (because elevation of cAMP causes hyperalgesia). In contrast, our finding of enhanced hyperalgesia associated with a Gs-to-Gi/o switch suggests that such inhibition of adenylyl cyclase is not the dominant effect of the stress-induced signaling switch in nociceptors. In this case, chronic exposure to elevated levels of epinephrine in peripheral tissue appears to induce novel coupling via Gi/o to second messenger pathways that produce hyperalgesia. Thus, stress caused a switch in downstream signaling pathways by which PGE2 produces hyperalgesia, from its usual dependence on a cAMP/protein kinase A pathway (Levine and Reichling, 2005), to a novel additional dependence on PKCε. The emergence of PKCε dependence may be particularly significant in view of our previous finding that PKCε plays a crucial role in “hyperalgesic priming,” a model of inflammation-induced chronic enhancement of hyperalgesia (Aley et al., 2000).
Although the switch in G-protein-coupled receptor signaling, from Gs to Gi, and the signaling from Gi to PKCε have not been reported previously in neurons, such mechanisms are well established in another excitable tissue, the myocardium. Thus, in heart muscle cells, β2-adrenegic receptor signaling can switch from Gs to Gi (Daaka et al., 1997; Hasseldine et al., 2003; Hill and Baker, 2003; Magocsi et al., 2007), and signaling from Gi to mediators known to be upstream of PKCε (Herrlich et al., 1996; Daaka et al., 1997; Steinberg, 1999; Mackay and Mochly-Rosen, 2001; Pavoine et al., 2003; Pavoine and Defer, 2005) or to PKCε (Fraser et al., 2000; Otani et al., 2003; Paruchuri and Sjolander, 2003) have been reported.
Conceivably, the interaction between the hypothalamo-pituitary-adrenal and sympathoadrenal axes that we observed occurs within the primary afferent nociceptor, potentially involving known interactions between glucocorticoid and β2-adrenergic receptor signaling pathways. Thus, epinephrine can increase the efficacy of glucocorticoid receptors, via the βγ phosphokinase pathway (Schmidt et al., 2001), which can contribute to immune mediator hyperalgesia (Khasar et al., 1995). Conversely, glucocorticoids can increase the expression of β2-adrenergic receptors (Fraser and Venter, 1980) as well as enhance their downstream second messenger signaling (Gs to adenylate cyclase to cAMP) (Aksoy et al., 2002).
Our findings may also provide an explanation for why generalized pain syndromes such as secondary fibromyalgia and irritable bowel syndrome occur at higher frequencies in diseases associated with the production of immune mediators [for example, interleukin-1β, which signals through production of PGE2 (Hori et al., 2000)]. Such cytokine production has been noted in several chronic conditions comorbid with generalized pain, including rheumatoid arthritis (Petrovic-Rackov and Pejnovic, 2006) and inflammatory bowel disease (Raddatz et al., 2005), as well as in depression and in self-reported poor health (Lekander et al., 2004; Calcagni and Elenkov, 2006).
Our demonstration of enhanced hyperalgesia after administration of glucocorticoids may further explain why fibromyalgia patients treated with glucocorticoids have been reported to show a trend toward worsened outcomes (Clark et al., 1985), and why patients receiving steroid injections for arthritis sometimes report a period of increased pain (Wollstein et al., 2007). Furthermore, it is known that genes that modulate β-adrenergic receptor-mediated signaling, such as catechol-O-methyltransferase (COMT), affect pain sensitivity in vivo (Nackley et al., 2007), and genetic variants of COMT are predictive of development of temporamandibular disorder, a generalized pain syndrome (Diatchenko et al., 2005). Also consistent with this idea, the β-adrenergic receptor antagonist pindolol has shown promise in a preliminary trial for fibromyalgia (Wood et al., 2005).
In summary, our work provides a potential mechanistic explanation for the pathophysiology of enhanced pain and disease flairs in stress-sensitive pain syndromes such as fibromyalgia, irritable bowel syndrome, interstitial cystitis, and rheumatoid arthritis. Importantly, our findings provide testable hypotheses for interventions in people with generalized pain syndromes. Specifically, elucidating the complex time-dependent interactions between the HPA and sympathoadrenal axes including the switch in second messenger signaling for pronociceptive immune mediators in the primary afferent nociceptor may identify specific targets for the development of novel therapeutic strategies.
Footnotes
This work was supported by National Institutes of Health Grant AR048821.
References
- Akana SF, Cascio CS, Shinsako J, Dallman MF. Corticosterone: narrow range required for normal body and thymus weight and ACTH. Am J Physiol. 1985;249:R527–R532. doi: 10.1152/ajpregu.1985.249.5.R527. [DOI] [PubMed] [Google Scholar]
- Aksoy MO, Mardini IA, Yang Y, Bin W, Zhou S, Kelsen SG. Glucocorticoid effects on the beta-adrenergic receptor-adenylyl cyclase system of human airway epithelium. J Allergy Clin Immunol. 2002;109:491–497. doi: 10.1067/mai.2002.122154. [DOI] [PubMed] [Google Scholar]
- Alessandri-Haber N, Yeh JJ, Boyd AE, Parada CA, Chen X, Reichling DB, Levine JD. Hypotonicity induces TRPV4-mediated nociception in rat. Neuron. 2003;39:497–511. doi: 10.1016/s0896-6273(03)00462-8. [DOI] [PubMed] [Google Scholar]
- Alessandri-Haber N, Dina OA, Yeh JJ, Parada CA, Reichling DB, Levine JD. Transient receptor potential vanilloid 4 is essential in chemotherapy-induced neuropathic pain in the rat. J Neurosci. 2004;24:4444–4452. doi: 10.1523/JNEUROSCI.0242-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley KO, Messing RO, Mochly-Rosen D, Levine JD. Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase C. J Neurosci. 2000;20:4680–4685. doi: 10.1523/JNEUROSCI.20-12-04680.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach DR, Erdmann G, Schmidtmann M, Monnikes H. Emotional stress reactivity in irritable bowel syndrome. Eur J Gastroenterol Hepatol. 2006;18:629–636. doi: 10.1097/00042737-200606000-00009. [DOI] [PubMed] [Google Scholar]
- Blackburn-Munro G, Blackburn-Munro RE. Chronic pain, chronic stress and depression: coincidence or consequence? J Neuroendocrinol. 2001;13:1009–1023. doi: 10.1046/j.0007-1331.2001.00727.x. [DOI] [PubMed] [Google Scholar]
- Calcagni E, Elenkov I. Stress system activity, innate and T helper cytokines, and susceptibility to immune-related diseases. Ann NY Acad Sci. 2006;1069:62–76. doi: 10.1196/annals.1351.006. [DOI] [PubMed] [Google Scholar]
- Clark S, Tindall E, Bennett RM. A double blind crossover trial of prednisone versus placebo in the treatment of fibrositis. J Rheumatol. 1985;12:980–983. [PubMed] [Google Scholar]
- Crofford LJ, Engleberg NC, Demitrack MA. Neurohormonal perturbations in fibromyalgia. Baillieres Clin Rheumatol. 1996;10:365–378. doi: 10.1016/s0950-3579(96)80022-7. [DOI] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- DeLeon M, Covenas R, Chadi G, Narvaez JA, Fuxe K, Cintra A. Subpopulations of primary sensory neurons show coexistence of neuropeptides and glucocorticoid receptors in the rat spinal and trigeminal ganglia. Brain Res. 1994;636:338–342. doi: 10.1016/0006-8993(94)91034-0. [DOI] [PubMed] [Google Scholar]
- Diatchenko L, Slade GD, Nackley AG, Bhalang K, Sigurdsson A, Belfer I, Goldman D, Xu K, Shabalina SA, Shagin D, Max MB, Makarov SS, Maixner W. Genetic basis for individual variations in pain perception and the development of a chronic pain condition. Hum Mol Genet. 2005;14:135–143. doi: 10.1093/hmg/ddi013. [DOI] [PubMed] [Google Scholar]
- Dina OA, Hucho T, Yeh J, Malik-Hall M, Reichling DB, Levine JD. Primary afferent second messenger cascades interact with specific integrin subunits in producing inflammatory hyperalgesia. Pain. 2005;115:191–203. doi: 10.1016/j.pain.2005.02.028. [DOI] [PubMed] [Google Scholar]
- Dina OA, Green PG, Levine JD. Role of interleukin-6 in chronic muscle hyperalgesic priming. Neuroscience. 2008;152:521–525. doi: 10.1016/j.neuroscience.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelmann M, Landgraf R, Lorscher P, Conzelmann C, Probst JC, Holsboer F, Reul JM. Downregulation of brain mineralocorticoid and glucocorticoid receptor by antisense oligodeoxynucleotide treatment fails to alter spatial navigation in rats. Eur J Pharmacol. 1998;361:17–26. doi: 10.1016/s0014-2999(98)00702-x. [DOI] [PubMed] [Google Scholar]
- Fraser CM, Venter JC. The synthesis of beta-adrenergic receptors in cultured human lung cells: induction by glucocorticoids. Biochem Biophys Res Commun. 1980;94:390–397. doi: 10.1016/s0006-291x(80)80233-6. [DOI] [PubMed] [Google Scholar]
- Fraser ID, Cong M, Kim J, Rollins EN, Daaka Y, Lefkowitz RJ, Scott JD. Assembly of an A kinase-anchoring protein-beta(2)-adrenergic receptor complex facilitates receptor phosphorylation and signaling. Curr Biol. 2000;10:409–412. doi: 10.1016/s0960-9822(00)00419-x. [DOI] [PubMed] [Google Scholar]
- Hasseldine AR, Harper EA, Black JW. Cardiac-specific overexpression of human beta2 adrenoceptors in mice exposes coupling to both Gs and Gi proteins. Br J Pharmacol. 2003;138:1358–1366. doi: 10.1038/sj.bjp.0705191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrlich A, Kuhn B, Grosse R, Schmid A, Schultz G, Gudermann T. Involvement of Gs and Gi proteins in dual coupling of the luteinizing hormone receptor to adenylyl cyclase and phospholipase C. J Biol Chem. 1996;271:16764–16772. doi: 10.1074/jbc.271.28.16764. [DOI] [PubMed] [Google Scholar]
- Hill SJ, Baker JG. The ups and downs of Gs- to Gi-protein switching. Br J Pharmacol. 2003;138:1188–1189. doi: 10.1038/sj.bjp.0705192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori T, Oka T, Hosoi M, Abe M, Oka K. Hypothalamic mechanisms of pain modulatory actions of cytokines and prostaglandin E2. Ann NY Acad Sci. 2000;917:106–120. doi: 10.1111/j.1749-6632.2000.tb05375.x. [DOI] [PubMed] [Google Scholar]
- Imbe H, Iwai-Liao Y, Senba E. Stress-induced hyperalgesia: animal models and putative mechanisms. Front Biosci. 2006;11:2179–2192. doi: 10.2741/1960. [DOI] [PubMed] [Google Scholar]
- Kavushansky A, Richter-Levin G. Effects of stress and corticosterone on activity and plasticity in the amygdala. J Neurosci Res. 2006;84:1580–1587. doi: 10.1002/jnr.21058. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Wang JF, Taiwo YO, Heller PH, Green PG, Levine JD. Mu-opioid agonist enhancement of prostaglandin-induced hyperalgesia in the rat: a G-protein beta gamma subunit-mediated effect? Neuroscience. 1995;67:189–195. doi: 10.1016/0306-4522(94)00632-f. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Miao JP, Janig W, Levine JD. Modulation of bradykinin-induced mechanical hyperalgesia in the rat by activity in abdominal vagal afferents. Eur J Neurosci. 1998a;10:435–444. doi: 10.1046/j.1460-9568.1998.00030.x. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Miao FJ, Janig W, Levine JD. Vagotomy-induced enhancement of mechanical hyperalgesia in the rat is sympathoadrenal-mediated. J Neurosci. 1998b;18:3043–3049. doi: 10.1523/JNEUROSCI.18-08-03043.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasar SG, McCarter G, Levine JD. Epinephrine produces a beta-adrenergic receptor-mediated mechanical hyperalgesia and in vitro sensitization of rat nociceptors. J Neurophysiol. 1999;81:1104–1112. doi: 10.1152/jn.1999.81.3.1104. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Green PG, Miao FJ, Levine JD. Vagal modulation of nociception is mediated by adrenomedullary epinephrine in the rat. Eur J Neurosci. 2003a;17:909–915. doi: 10.1046/j.1460-9568.2003.02503.x. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Miao FJ, Gear RW, Green PG, Levine JD. Vagal modulation of bradykinin-induced mechanical hyperalgesia in the female rat. J Pain. 2003b;4:278–283. doi: 10.1016/s1526-5900(03)00631-x. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Green PG, Levine JD. Repeated sound stress enhances inflammatory pain in the rat. Pain. 2005;116:79–86. doi: 10.1016/j.pain.2005.03.040. [DOI] [PubMed] [Google Scholar]
- Lai J, Gold MS, Kim CS, Bian D, Ossipov MH, Hunter JC, Porreca F. Inhibition of neuropathic pain by decreased expression of the tetrodotoxin-resistant sodium channel, NaV1.8. Pain. 2002;95:143–152. doi: 10.1016/s0304-3959(01)00391-8. [DOI] [PubMed] [Google Scholar]
- Lamba S, Abraham WT. Alterations in adrenergic receptor signaling in heart failure. Heart Fail Rev. 2000;5:7–16. doi: 10.1023/A:1009885822076. [DOI] [PubMed] [Google Scholar]
- Lekander M, Elofsson S, Neve IM, Hansson LO, Unden AL. Self-rated health is related to levels of circulating cytokines. Psychosom Med. 2004;66:559–563. doi: 10.1097/01.psy.0000130491.95823.94. [DOI] [PubMed] [Google Scholar]
- Levine JD, Reichling DB. Peripheral mechanisms of inflammatory pain. In: Wall PD, Melzack R, editors. Textbook of pain. Edinburgh: Churchill Livingstone; 2005. [Google Scholar]
- Mackay K, Mochly-Rosen D. Arachidonic acid protects neonatal rat cardiac myocytes from ischaemic injury through epsilon protein kinase C. Cardiovasc Res. 2001;50:65–74. doi: 10.1016/s0008-6363(00)00322-9. [DOI] [PubMed] [Google Scholar]
- Magocsi M, Vizi ES, Selmeczy Z, Brozik A, Szelenyi J. Multiple G-protein-coupling specificity of beta-adrenoceptor in macrophages. Immunology. 2007;122:503–513. doi: 10.1111/j.1365-2567.2007.02658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik-Hall M, Dina OA, Levine JD. Primary afferent nociceptor mechanisms mediating NGF-induced mechanical hyperalgesia. Eur J Neurosci. 2005;21:3387–3394. doi: 10.1111/j.1460-9568.2005.04173.x. [DOI] [PubMed] [Google Scholar]
- Miao FJ, Benowitz NL, Basbaum AI, Levine JD. Sympathoadrenal contribution to nicotinic and muscarinic modulation of bradykinin-induced plasma extravasation in the knee joint of the rat. J Pharmacol Exp Ther. 1992;262:889–895. [PubMed] [Google Scholar]
- Nackley AG, Tan KS, Fecho K, Flood P, Diatchenko L, Maixner W. Catechol-O-methyltransferase inhibition increases pain sensitivity through activation of both beta2- and beta3-adrenergic receptors. Pain. 2007;128:199–208. doi: 10.1016/j.pain.2006.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neeck G, Crofford LJ. Neuroendocrine perturbations in fibromyalgia and chronic fatigue syndrome. Rheum Dis Clin North Am. 2000;26:989–1002. doi: 10.1016/s0889-857x(05)70180-0. [DOI] [PubMed] [Google Scholar]
- Nilsen KB, Sand T, Westgaard RH, Stovner LJ, White LR, Bang Leistad R, Helde G, Ro M. Autonomic activation and pain in response to low-grade mental stress in fibromyalgia and shoulder/neck pain patients. Eur J Pain. 2007;11:743–755. doi: 10.1016/j.ejpain.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Otani H, Okada T, Fujiwara H, Uchiyama T, Sumida T, Kido M, Imamura H. Combined pharmacological preconditioning with a G-protein-coupled receptor agonist, a mitochondrial KATP channel opener and a nitric oxide donor mimics ischaemic preconditioning. Clin Exp Pharmacol Physiol. 2003;30:684–693. doi: 10.1046/j.1440-1681.2003.03896.x. [DOI] [PubMed] [Google Scholar]
- Otis JD, Keane TM, Kerns RD. An examination of the relationship between chronic pain and post-traumatic stress disorder. J Rehabil Res Dev. 2003;40:397–405. doi: 10.1682/jrrd.2003.09.0397. [DOI] [PubMed] [Google Scholar]
- Paruchuri S, Sjolander A. Leukotriene D4 mediates survival and proliferation via separate but parallel pathways in the human intestinal epithelial cell line Int 407. J Biol Chem. 2003;278:45577–45585. doi: 10.1074/jbc.M302881200. [DOI] [PubMed] [Google Scholar]
- Pavoine C, Defer N. The cardiac beta2-adrenergic signalling a new role for the cPLA2. Cell Signal. 2005;17:141–152. doi: 10.1016/j.cellsig.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Pavoine C, Behforouz N, Gauthier C, Le Gouvello S, Roudot-Thoraval F, Martin CR, Pawlak A, Feral C, Defer N, Houel R, Magne S, Amadou A, Loisance D, Duvaldestin P, Pecker F. beta2-Adrenergic signaling in human heart: shift from the cyclic AMP to the arachidonic acid pathway. Mol Pharmacol. 2003;64:1117–1125. doi: 10.1124/mol.64.5.1117. [DOI] [PubMed] [Google Scholar]
- Petrovic-Rackov L, Pejnovic N. Clinical significance of IL-18, IL-15, IL-12 and TNF-alpha measurement in rheumatoid arthritis. Clin Rheumatol. 2006;25:448–452. doi: 10.1007/s10067-005-0106-0. [DOI] [PubMed] [Google Scholar]
- Raddatz D, Bockemuhl M, Ramadori G. Quantitative measurement of cytokine mRNA in inflammatory bowel disease: relation to clinical and endoscopic activity and outcome. Eur J Gastroenterol Hepatol. 2005;17:547–557. doi: 10.1097/00042737-200505000-00012. [DOI] [PubMed] [Google Scholar]
- Schmidt P, Holsboer F, Spengler D. Beta(2)-adrenergic receptors potentiate glucocorticoid receptor transactivation via G protein beta gamma-subunits and the phosphoinositide 3-kinase pathway. Mol Endocrinol. 2001;15:553–564. doi: 10.1210/mend.15.4.0613. [DOI] [PubMed] [Google Scholar]
- Singh VB, Corley KC, Phan TH, Boadle-Biber MC. Increases in the activity of tryptophan hydroxylase from rat cortex and midbrain in response to acute or repeated sound stress are blocked by adrenalectomy and restored by dexamethasone treatment. Brain Res. 1990;516:66–76. doi: 10.1016/0006-8993(90)90898-l. [DOI] [PubMed] [Google Scholar]
- Steinberg SF. The molecular basis for distinct beta-adrenergic receptor subtype actions in cardiomyocytes. Circ Res. 1999;85:1101–1111. doi: 10.1161/01.res.85.11.1101. [DOI] [PubMed] [Google Scholar]
- Strausbaugh HJ, Green PG, Dallman MF, Levine JD. Repeated, non-habituating stress suppresses inflammatory plasma extravasation by a novel, sympathoadrenal dependent mechanism. Eur J Neurosci. 2003;17:805–812. doi: 10.1046/j.1460-9568.2003.02493.x. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Levine JD. Contribution of guanine nucleotide regulatory proteins to prostaglandin hyperalgesia in the rat. Brain Res. 1989;492:400–403. doi: 10.1016/0006-8993(89)90929-3. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Levine JD. Kappa- and delta-opioids block sympathetically dependent hyperalgesia. J Neurosci. 1991;11:928–932. doi: 10.1523/JNEUROSCI.11-04-00928.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taiwo YO, Coderre TJ, Levine JD. The contribution of training to sensitivity in the nociceptive paw-withdrawal test. Brain Res. 1989;487:148–151. doi: 10.1016/0006-8993(89)90950-5. [DOI] [PubMed] [Google Scholar]
- Takasaki I, Kurihara T, Saegusa H, Zong S, Tanabe T. Effects of glucocorticoid receptor antagonists on allodynia and hyperalgesia in mouse model of neuropathic pain. Eur J Pharmacol. 2005;524:80–83. doi: 10.1016/j.ejphar.2005.09.045. [DOI] [PubMed] [Google Scholar]
- Temml C, Wehrberger C, Riedl C, Ponholzer A, Marszalek M, Madersbacher S. Prevalence and correlates for interstitial cystitis symptoms in women participating in a health screening project. Eur Urol. 2007;51:803–809. doi: 10.1016/j.eururo.2006.08.028. [DOI] [PubMed] [Google Scholar]
- Wang S, Lim G, Zeng Q, Sung B, Ai Y, Guo G, Yang L, Mao J. Expression of central glucocorticoid receptors after peripheral nerve injury contributes to neuropathic pain behaviors in rats. J Neurosci. 2004;24:8595–8605. doi: 10.1523/JNEUROSCI.3058-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson CW, Shinsako J, Dallman MF. Return of pituitary-adrenal function after adrenal enucleation or transplantation: diurnal rhythms and responses to ether. Endocrinology. 1981;109:162–169. doi: 10.1210/endo-109-1-162. [DOI] [PubMed] [Google Scholar]
- Wollstein R, Chaimsky G, Carlson L, Watson HK, Wollstein G, Saleh J. Evaluating short-term pain after steroid injection. Am J Orthop. 2007;36:128–131. [PubMed] [Google Scholar]
- Wood PB, Kablinger AS, Caldito GS. Open trial of pindolol in the treatment of fibromyalgia. Ann Pharmacother. 2005;39:1812–1816. doi: 10.1345/aph.1G014. [DOI] [PubMed] [Google Scholar]
- Zautra AJ, Parrish BP, Van Puymbroeck CM, Tennen H, Davis MC, Reich JW, Irwin M. Depression history, stress, and pain in rheumatoid arthritis patients. J Behav Med. 2007;30:187–197. doi: 10.1007/s10865-007-9097-4. [DOI] [PubMed] [Google Scholar]