

Abstract
Reductive alkylation of 5-methoxy-1-tetralone (6) with 2,3-dibromopropene gave an equilibrium mixture of bicyclic diones 7 (51%) and 8 (35%). Radical cyclization of 7 afforded tricyclic dione 5 (84%), which was reduced, cyclized and dehydrated to give tetracyclic alkene 13 in 63% yield. Allylic oxidation of 13 with SeO2 and activated MnO2 afforded enone 2 in 85% yield, thereby completing a short formal synthesis of (±)-platensimycin.
The broad spectrum antibiotic platensimycin (1) (see Scheme 1) was recently isolated by a Merck group from Streptomyces platensis as part of a screening program designed to isolate inhibitors of bacterial fatty acid biosynthesis by the highly conserved condensing enzyme FabF.1 Only the weak antibiotics cerulenin and thiolactomycin were known to act by this mechanism. Potent inhibitors of this enzyme are expected to be antibiotics with no cross-resistance to existing drugs. Platensimycin acts by specific binding with the acyl-enzyme intermediate of FabF. The structure and absolute stereochemistry of platensimycin were determined by a combination of spectroscopic methods and X-ray crystallography of a bromo derivative.1
Scheme 1.
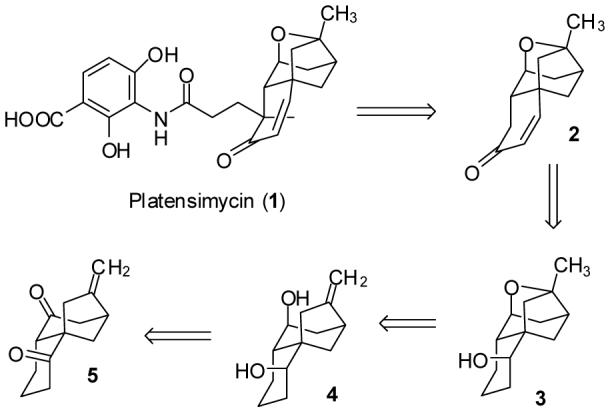
Retrosynthesis of Platensimycin
We thought that the acyl portion of platensimycin should be readily accessible by introduction of a methyl group and a propanoic acid side chain onto enone 2. Nicolaou recently reported the first synthesis of platensimycin (1) in which he prepared 2 in 10 steps and elaborated it to (±)-platensimycin (1).2 We planned to prepare 2 by dehydration of the alcohol of 3 and allylic oxidation. Acid-catalyzed cyclization of unsaturated diol 4 should afford the ether linkage of 3. L-Selectride reduction of dione 5 should provide the bis axial alcohol 4 (see Scheme 1).
This approach was attractive because Marinovic reported a two-step synthesis of dione 5 in 1983.3 Reductive alkylation of 5-methoxy-1-tetralone (6) with 2,3-dibromopropene by Narisada’s procedure4 afforded bicyclic diones 7 and 8 in 68% yield with unspecified stereochemistry (see Scheme 2). Radical cyclization of this mixture of 7 and 8 with n-Bu3SnH in benzene at reflux afforded the tricyclic diones 5 and 9 in 85% yield, again with unspecified stereochemistry.
Scheme 2.
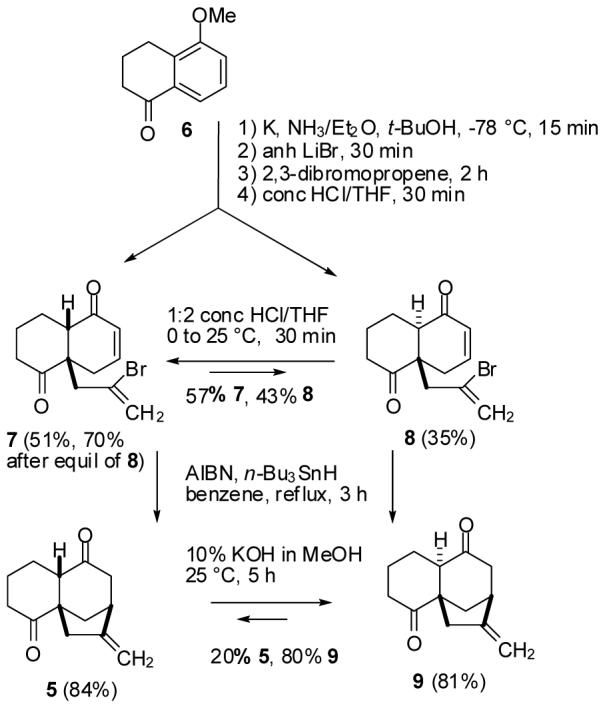
Synthesis of Tricyclic Diones 5 and 9
Although this route is very short, it is only attractive if the desired tricyclic dione 5 can be prepared cleanly and in good yield. Unfortunately, molecular mechanics calculations5 suggested that the desired tricyclic dione 5 is 1.6 kcal/mol less stable than epimeric dione 9. However, calculations also suggested that the desired bicyclic dione 7 is 0.1 kcal/mol more stable than epimeric dione 8. Therefore, it might be possible to isolate 7 in acceptable yield and convert it to 5 if the radical cyclization can be carried out without epimerization.
In our hands, the reduction of 6 was best carried out with potassium in NH3/Et2O at -78 °C.6 Addition of LiBr and then 2,3-dibromopropene effected alkylation. Hydrolysis of the enol ether with conc HCl in THF for 30 min afforded a readily separable mixture from which the desired bicyclic dione 7 was isolated 51% yield and the epimer 8 was obtained in 35% yield. The structures of 7 and 8 could not be assigned at this point so both compounds were carried on. The HCl hydrolysis step produced close to an equilibrium mixture. Acid catalyzed equilibration of either 7 or 8 provided a 4:3 mixture of 7 and 8. Equilibration of 8 afforded additional 7 (19%), which was therefore isolated in 70% overall yield from 5-methoxy-1-tetralone (6).
Radical cyclization of 7 with n-Bu3SnH and catalytic AIBN in benzene at reflux afforded the desired tricyclic dione 5 in 84% yield without any epimerization. This practical two-step route to 5 proceeds in 59% overall yield. A similar sequence converted the undesired bicyclic dione 8 to tricyclic dione 9 in 81% yield. Equilibration of either 5 or 9 with KOH in MeOH gave a 1:4 mixture of 5 and the more stable tricyclic dione 9. The equilibration of both bicyclic diones 7 and 8 and tricyclic diones 5 and 9 thus gave results close to those expected from molecular mechanics calculations.
The 1H NMR spectra of 5 and 7-9 were hard to analyze because of extensive overlap. Fortunately, all the hydrogens of 5 could be resolved in C6D6 at 800 MHz and the stereochemistry of 5 was tentatively assigned based on an NOE between the ring fusion hydrogen and one of the allylic methylene hydrogens (see Figure 1). The stereochemical assignments of 5 and 7-9 were unam-biguously established by X-ray crystal structure determination of both tricyclic diones 5 and 9 (see Figure 1).
Figure 1.
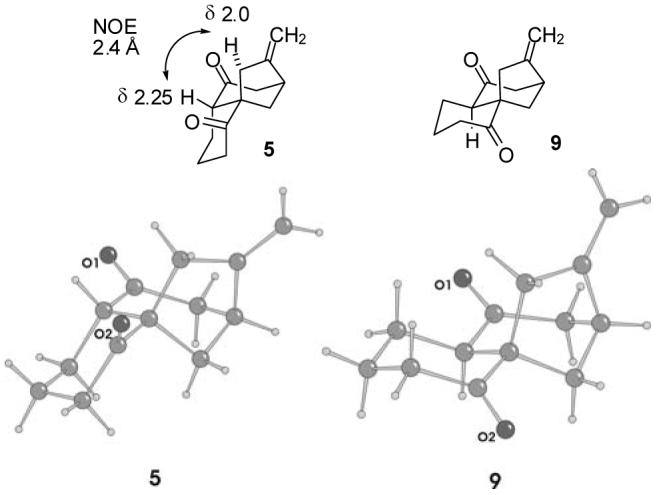
Three Dimensional Representations and Molecular Structures of 5 and 9 Established by X-ray Structure Determination.
L-Selectride reduction of the unhindered ketone of 5 occurred readily at -78 °C, but gave a 1:1 mixture of equatorial and axial alcohols. The other ketone was reduced at 25 °C affording a 12:1 mixture favoring the desired axial alcohol. This resulted in the formation of an inseparable 1:1 mixture of 4 and 11 in 90% yield (see Scheme 3).7 This mixture was treated with TFA and CH2Cl2 to effect formation of the ether linkage as described by Nicolaou2 for a related substrate with different functionality in the isolated ring. This two-step sequence afforded axial alcohol 3 in 39% yield and the equatorial alcohol 12 in 42% yield from the mixture of diols. Treatment of the axial alcohol 3 with Tf2O and pyridine in CH2Cl2 afforded the triflate, which eliminated readily to give alkene 13 in 90% yield. Similar treatment of the equatorial alcohol 12 afforded triflate 14, which did not undergo E2 elimination readily because there is no β-hydrogen anti to the triflate.8 Eventually, we found that treatment of crude triflate 14 with either silica gel or hydrochloric acid resulted in clean elimination, possibly by an E1 mechanism, to form alkene 13 in 84% yield from 12.9 This three-step sequence converts tricyclic dione 5 to tetracyclic alkene 13 in 63% overall yield, but diols 4 and 11 cannot be characterized.
Scheme 3.
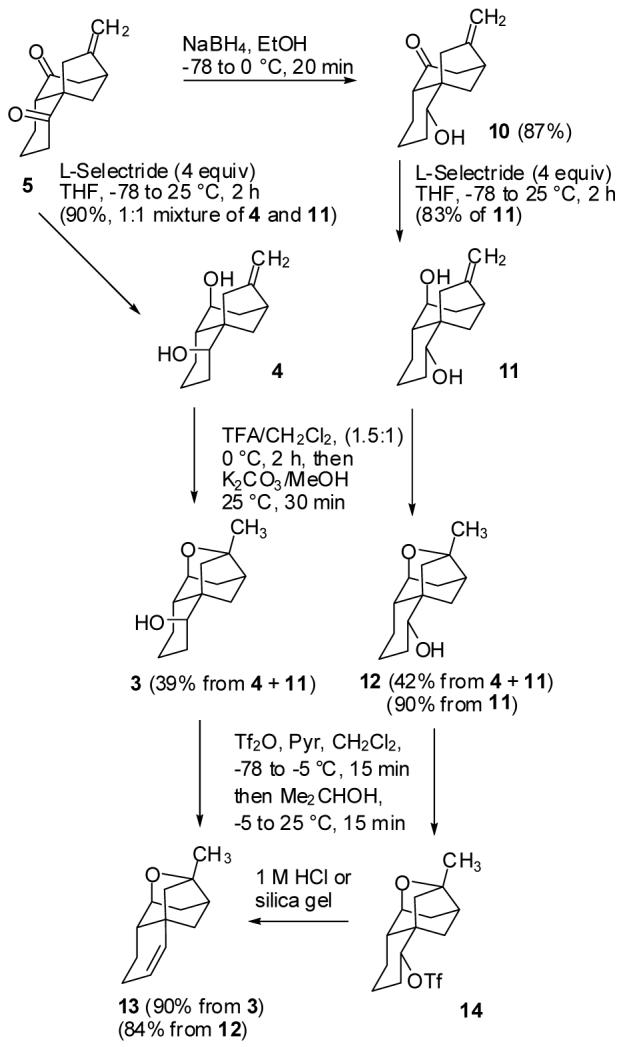
Reduction, Cyclization and Dehydration of 5
Alternatively, reduction of dione 5 with NaBH4 in EtOH at -78 to 0 °C afforded equatorial keto alcohol 10. Reduction of 10 with L-Selectride afforded diol 11 contaminated with a few percent of the equatorial alcohol7 in 83% yield. Acid catalyzed cyclization afforded 12 (90%), which was elaborated to 13 as previously described. This four-step sequence converts tricyclic dione 5 to tetracyclic alkene 13 in 55% overall yield.
Allylic oxidation of alkene 13 with CrO3·3,5-dimethylpyrazole10 in CH2Cl2 at -25 °C provided an inseparable 4:1 mixture of the desired enone 2 and the regioisomer 15 in 75% yield (see Scheme 4). Oxidation of 13 with CrO3·pyridine was slower, but gave the same ratio of products. The formation of mixtures of products was expected11 because both ends of the intermediate allylic cation are secondary.
Scheme 4.
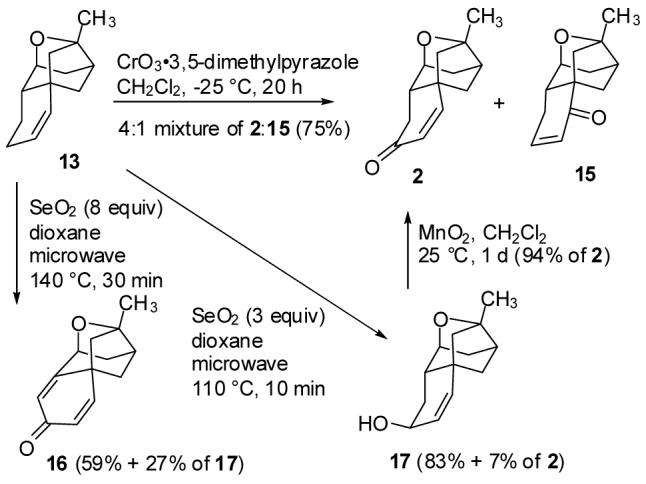
Allylic Oxidation of 13 to Give Enone 2
We therefore turned to SeO2 oxidation, which should be regiospecific because the oxygen is introduced by an ene reaction followed by a [2,3]-sigmatropic rearrangement.12 Oxidation of 13 with 8 equiv of SeO2 in dioxane at 140 °C in a microwave reactor for 30 min afforded allylic alcohol 17 in only 27% yield. The major product was dienone 16 (59%). Oxidation of alkene 13 with SeO2 should give allylic alcohol 17 and enone 2, which is apparently oxidized further to dienone 16 as has been observed in related systems.13 Oxidation of alkene 13 with only 3 equiv of SeO2 in dioxane at 110 °C in a microwave reactor for only 10 min afforded allylic alcohol 17 in 83% yield and enone 2 in 7% yield. Oxidation of alcohol 17 with activated MnO2 provided enone 2 in 94% yield. This two-step sequence converts alkene 13 to enone 2 in 85% yield. The 1H and 13C NMR spectra of 2 are identical to those reported by Nicolaou.
In conclusion, we have developed an efficient route (seven steps, 32% overall yield) to tetracyclic enone 2, a late intermediate in Nicolaou’s (±)-platensimycin (1) synthesis.
Supplementary Material
Acknowledgment
We are grateful to the National Institutes of Health (GM-50151) for support of this work. We thank the National Science Foundation for the partial support of this work through grant CHE-0521047 for the purchase of an X-ray diffractometer. We thank Prof. Susan S. Pochapsky, Brandeis University, for assistance in obtaining 800 MHz NOESY data for 5. The 800 MHz spectrometer was purchased under NIH RR High-End Instrumentation program, 1S10RR017269-01. We thank Prof. K. C. Nicolaou, The Scripps Research Institute, for copies of the 1H and 13C NMR spectra of 2.
Footnotes
snider@brandeis.edu
References
- 1(a).Wang J, Soisson SM, Young K, Shoop W, Kodali S, Galgoci A, Painter R, Parthasarathy G, Tang YS, Cummings R, Ha S, Dorso K, Motyl M, Jayasuriya H, Ondeyka J, Herath K, Zhang C, Hernandez L, Allocco J, Basilio Á, Tormo JR, Genilloud O, Vicente F, Pelaez F, Colwell L, Lee SH, Michael B, Felcetto T, Gill C, Silver LL, Hermes JD, Bartizal K, Barrett J, Schmatz D, Becker JW, Cully D, Singh SB. Nature. 2006;441:358–361. doi: 10.1038/nature04784. [DOI] [PubMed] [Google Scholar]; (b) Singh SB, Jayasuriya H, Ondeyka JG, Herath KB, Zhang C, Zink DL, Tsou NN, Ball RG, Basilio A, Genilloud O, Diez MT, Vicente F, Pelaez F, Young K, Wang J. J. Am. Chem. Soc. 2006;128:11916–11920. doi: 10.1021/ja062232p. [DOI] [PubMed] [Google Scholar]; (c) Häbich D, von Nussbaum F. ChemMedChem. 2006;1:951–954. doi: 10.1002/cmdc.200600145. [DOI] [PubMed] [Google Scholar]
- 2.Nicolaou KC, Li A, Edmonds DJ. Angew. Chem., Int. Ed. 2006;45:7086–7090. doi: 10.1002/anie.200603892. [DOI] [PubMed] [Google Scholar]
- 3.Marinovic NN, Ramanathan H. Tetrahedron Lett. 1983;24:1871–1874. [Google Scholar]
- 4(a).Narisada M, Watanabe F. J. Org. Chem. 1973;38:3887–3892. [Google Scholar]; (b) Brown JM, Cresp TM, Mander LN. J. Org. Chem. 1977;42:3984–3986. [Google Scholar]
- 5.PCMODEL version 8.0 from Serena Software was used with MMX.
- 6(a).For more recent studies of this reductive alkylation see:Labadie GR, Estiú GL, Cravero RM, Gonzalez Sierra M. THEOCHEM. 2003;635:173–182.Marcinow Z, Rabideau PW. J. Org. Chem. 1988;53:2117–2119.Labadie GR, Cravero RM, Gonzalez-Sierra M. Synth. Commun. 2000;30:4065–4079.
- 7.This mixture contained a few percent of the epimers of 4 and 11 with an equatorial alcohol in the bicyclic moiety. These isomers cannot form an ether on treatment with TFA and are easily separated from 3 and 12.
- 8.Atempted dehydration of 12 with Burgess’ reagent, Martin sulfurane, or via the mesylate failed.
- 9.LiCl in THF has been reported to facilitate elimination of triflates:Finch H, Harwood LM, Highcock R, Jackson B, Prout K, Robertson G, Sewell RC. Synlett. 1990;7:384–386.
- 10.Salmond WG, Barta MA, Havens JL. J. Org. Chem. 1978;43:2057–2059. [Google Scholar]
- 11(a).Dauben WG, Lorber M, Fullerton DS. J. Org. Chem. 1969;34:3587–3592. [Google Scholar]; (b) Gulge R, Shaligram AM. Indian J. Chem., Sect. B. 1985;24B:815–819. [Google Scholar]; (c) Engler TA, Sampath U, Velde DV, Takusagawa F. Tetrahedron. 1992;48:9399–9416. [Google Scholar]; (d) Zhao J, Zhao F, Wang Y, Li H, Zhang Q, Guénard D, Ge Q, Wei E, Jiang H, Wu Y, Wang L, Jiang H, Guéritte F, Wu X, Cheng CHK, Lee S-S, Zhao Y. Helv. Chim. Acta. 2004;87:1832–1853. [Google Scholar]
- 12(a).Rabjohn N. Org. React. 1976;24:261–415. [Google Scholar]; (b) Bullman Page PC, McCarthy TJ. In: Comprehensive Organic Synthesis. Ley SV, editor. Vol. 7. Pergamon Press; New York: 1991. pp. 83–117. [Google Scholar]
- 13.Kocór M, Tuszy-Maczka M. Bull. Acad. Pol. Sci., Ser. Sci. Chim. 1961;9:405–409. Chem. Abstr. 1964, 60, 6910e or 38978. [Google Scholar]; (b) Moreno-Dorado FJ, Guerra FM, Aladro FJ, Bustamante JM, Jorge ZD, Massanet GM. Tetrahedron. 1999;55:6997–7010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.