

Abstract
Pertussis toxin (PT) is an AB-type protein toxin that consists of a catalytic A subunit (PT S1) and an oligomeric, cell-binding B subunit. It belongs to a subset of AB toxins that move from the cell surface to the endoplasmic reticulum (ER) before A chain passage into the cytosol. Toxin translocation is thought to involve A chain unfolding in the ER and the quality control mechanism of ER-associated degradation (ERAD). The absence of lysine residues in PT S1 may allow the translocated toxin to avoid ubiquitin-dependent degradation by the 26S proteasome, which is the usual fate of exported ERAD substrates. As the conformation of PT S1 appears to play an important role in toxin translocation, we used biophysical and biochemical methods to examine the structural properties of PT S1. Our in vitro studies found that the isolated PT S1 subunit is a thermally unstable protein that can be degraded in a ubiquitin-independent fashion by the core 20S proteasome. The thermal denaturation of PT S1 was inhibited by its interaction with NAD, a donor molecule used by PT S1 for the ADP-ribosylation of target G proteins. These observations support a model of intoxication in which toxin translocation, degradation, and activity are all influenced by the heat-labile nature of the isolated toxin A chain.
Pertussis toxin (PT) is an AB-type protein toxin that consists of an enzymatic A moiety and a cell-binding B moiety (reviewed in (1, 2)). PT A (the S1 subunit) activates certain Gα proteins by an ADP-ribosylation reaction that utilizes NAD as a donor molecule. PT B is composed of an S2 subunit, an S3 subunit, two S4 subunits, and an S5 subunit. The oligomeric PT B complex forms a ring-like structure that is stable for temperatures up to 60ºC–70ºC (3). Non-covalent interactions position the catalytic S1 subunit within and on top of the B ring to form the PT holotoxin.
PT B binds to glycoproteins or glycolipids on the plasma membrane of a target cell (4–6). The surface-bound toxin then travels by vesicular transport to the Golgi apparatus and most likely to the endoplasmic reticulum (ER) as well (7–12). In the ER, ATP can bind to the central pore of the B oligomer and destabilize the holotoxin (9, 13, 14). Subsequent reduction of the PT S1 intramolecular disulfide bond may further destabilize the holotoxin and allow PT S1 to dissociate from PT B (15). The isolated S1 subunit can then cross the ER membrane and enter the cytosol to interact with its Gα targets (16, 17).
The quality control system of ER-associated degradation (ERAD) may be involved with the ER-to-cytosol translocation of PT S1 (10). ERAD recognizes misfolded proteins (often identified by surface-exposed hydrophobic residues) in the ER and exports them to the cytosol for degradation by the ubiquitin-proteasome pathway (18). Export occurs through the Sec61p translocon, a gated pore in the ER membrane that allows bi-directional movement of partially or fully unfolded proteins between the ER and the cytosol (19). The C-terminal hydrophobic region of PT S1 is thought to trigger ERAD activity and stimulate PT S1 translocation into the cytosol; proteasome-mediated degradation in the cytosol is presumably avoided because PT S1 has no lysine residues for ubiquitin attachment (10). Similar predictions have been made for cholera toxin (CT) and ricin, two other AB-type toxins that move from the cell surface to the ER and exploit ERAD for A chain entry into the cytosol (10, 20).
Recent work has shown that the C-terminal region of PT S1 is not required for passage into the cytosol and has suggested that PT S1 may be degraded in the cytosol (16, 17). Likewise, the CT A1 polypeptide does not require its C-terminal hydrophobic domain to move from the ER to the cytosol (21). Furthermore, CT A1 and ricin A chain are both degraded in the cytosol by ubiquitin-independent proteasomal mechanisms (22–25) (Teter et. al., unpublished experiments). These observations call for a modification to the current ERAD models of toxin translocation.
We have proposed a revised model of toxin-ERAD interactions in which both toxin translocation and toxin degradation are linked to the heat-labile nature of the isolated toxin A moiety (21). With this model, thermal instability in the dissociated toxin A chain generates an unfolded conformational state at 37ºC that triggers ERAD activity and renders the cytosolic pool of toxin susceptible to degradation by the 20S proteasome (in contrast to the usual route of ubiquitin-dependent degradation by the 26S proteasome). Both CT A1 and ricin A chain are heat-labile proteins, and in vitro degradation of CT A1 by the 20S proteasome has been observed by our group (26) (Teter et. al., unpublished experiments). Here, structural studies and biochemical assays were used to examine the putative heat-labile nature of PT S1 and its possible degradation by the 20S proteasome. Analysis of the physical properties of the isolated PT A moiety support our revised model of toxin-ERAD interactions. Furthermore, we provide a mechanism by which the PT S1 subunit can retain significant enzymatic activity in the cytosol despite its heat-labile state.
EXPERIMENTAL PROCEDURES
Materials
Chemicals and thermolysin were purchased from Sigma-Aldrich (St. Louis, MO). PT, PT S1, and PT B were from List Biological Laboratories, Inc. (Campbell, CA). ATP and the CT A1/A2 heterodimer were purchased from Calbiochem (La Jolla, CA). The purified 20S proteasome was from Boston Biochem (Cambridge, MA).
Circular Dichroism Measurements
Temperature-dependent unfolding of the PT S1 subunit was studied by circular dichroism (CD) experiments, using a J-810 spectrofluoropolarimeter equipped with a PFD-425S Peltier temperature controller (Jasco Corp., Tokyo, Japan). Protein concentration was 37.4 μg in 0.225 ml of 20 mM Na-phosphate buffer (pH 7.4) containing 150 mM NaCl and 10 mM β-mercaptoethanol (β-ME). Measurements were taken with a 4-mm optical path length rectangular quartz cuvette as the temperature increased stepwise from 18ºC–50ºC. As shown in the thermal unfolding profiles, measurements were taken in 2ºC increments from 18–30ºC; 1ºC increments from 30–33ºC; 0.5ºC increments from 33–37ºC; a 1ºC increment from 37–38ºC; and 2ºC increments from 40–50ºC. Samples were allowed to equilibrate at each temperature for 4 min before readings. CD spectra were recorded from 200–315 nm, which covers both near-UV and far-UV range and thus allowed us to detect thermal changes in both the tertiary and secondary structures of PT S1. The same sample was used for near-UV and far-UV measurements in order to eliminate possible errors resulting from sample-to-sample variability. In all cases the spectral resolution was 1 nm. CD spectra were averaged from 5 scans. The observed ellipticity was converted to mean residue molar ellipticity, [θ], in units of degrees × cm2 × dmol−1 using
| (1) |
where thetas;obs is the measured ellipticity in millidegrees, c is the molar concentration of the protein, nres is the number of amino acid residues in the protein, and l is the optical path-length in millimeters.
The temperature-dependent protein unfolding data were analyzed as described by Lavigne et al. (27) using the following equation:
| (2) |
where X is the ellipticity measured at a given temperature, fL is the fraction of amino acids representing the native conformation at low temperature, XL and XH are limiting values of X at low and high temperatures, respectively. The parameter, fL, is given by
| (3) |
where the temperature dependence of the free energy of unfolding (ΔG) is described by
| (4) |
Here T is the absolute temperature, Tm is the transition temperature, ΔH is the apparent enthalpy of unfolding and ΔC is the heat capacity of unfolding.
Thermolysin Degradation Assay
6 μg of PT S1, 6 μg of the CT A1/A2 heterodimer, 12 μg of PT B, or 18 μg of PT was added to 0.12 ml of 20 mM Na-phosphate buffer (pH 7.0) containing 10 mM β-ME. Where indicated, 1 mM NAD, or 1% CHAPS, or both 1 mM NAD and 1% CHAPS together were present in the Na-phosphate buffer. 20 μl sample aliquots were transferred to fresh microcentrifuge tubes and incubated at 4°C, 25°C, 33°C, 37°C, or 41°C for 45 min. All samples were then shifted to 4°C for 10 minutes. Thermolysin (prepared as a 10x stock in 50 mM CaCl2 and 100 mM Hepes pH 8.0) was added to a final concentration of 0.04 mg/ml for a 45 min incubation at 4°C. Digests were halted by the addition of 10 mM EDTA (final concentration) and SDS-PAGE sample buffer to the reaction mix. Toxin samples were visualized by SDS-PAGE and Coomassie staining. The NAD glycohydrolase activity of PT S1 at 37°C is approximately 0.6 pmol/min/μg toxin (28). From this value, we estimate an inconsequential 0.14% of the NAD present in our assay buffer was hydrolyzed during the course of the experiment.
20S Proteasome Assay
5 μg of PT S1, 5 μg of the CT A1/A2 heterodimer, 10 μg of PT B, or 15 μg of PT was mixed with 100 nM of the purified 20S proteasome in 0.1 ml assay buffer (100 mM KCl, 10 mM MgCl2, 0.1 mM CaCl2, 10 mM β-ME, 3 mM ATP, and 50 mM Hepes pH 7.5) and placed at 37°C. 20 μl aliquots taken at the stated intervals were mixed with SDS-PAGE sample buffer visualized by SDS-PAGE with Coomassie staining.
RESULTS
Effect of temperature on PT S1 structure
The structure of PT has been determined by X-ray crystallography, which identified the S1 subunit as a 26 kDa protein with 28% α-helix content and 14% β-sheet content (29). To examine the thermal stability of PT S1, temperature-induced changes in the structure of PT S1 were monitored by near-UV and far-UV CD (Fig. 1). The disruption of PT S1 tertiary structure was detected by a decrease in the near-UV CD signal from PT S1 aromatic side chains at ~280 nm (Fig. 1A), while the loss of PT S1 secondary structure was detected by a decrease in the ellipticity in the far-UV region at ~ 225 nm (Fig. 1B). The mean residue molar ellipticities at 280 nm and 225 nm then were plotted as a function of temperature in order to generate thermal unfolding profiles for the tertiary and secondary structures of PT S1. With these studies, we found thermotropic conformational changes occurred in a sigmodial manner for both the tertiary (Fig. 1C) and secondary (Fig. 1D) structures of PT S1. The transition temperature (Tm; the midpoint of transition) for the tertiary structure of PT S1 was 28.5ºC. The secondary structure of PT S1 exhibited a slightly higher Tm of 31.0ºC. Both values demonstrated that PT S1 is a heat-labile protein, with perturbed tertiary and secondary structures at the physiological temperature of 37ºC (see also Fig. 2).
Figure 1.
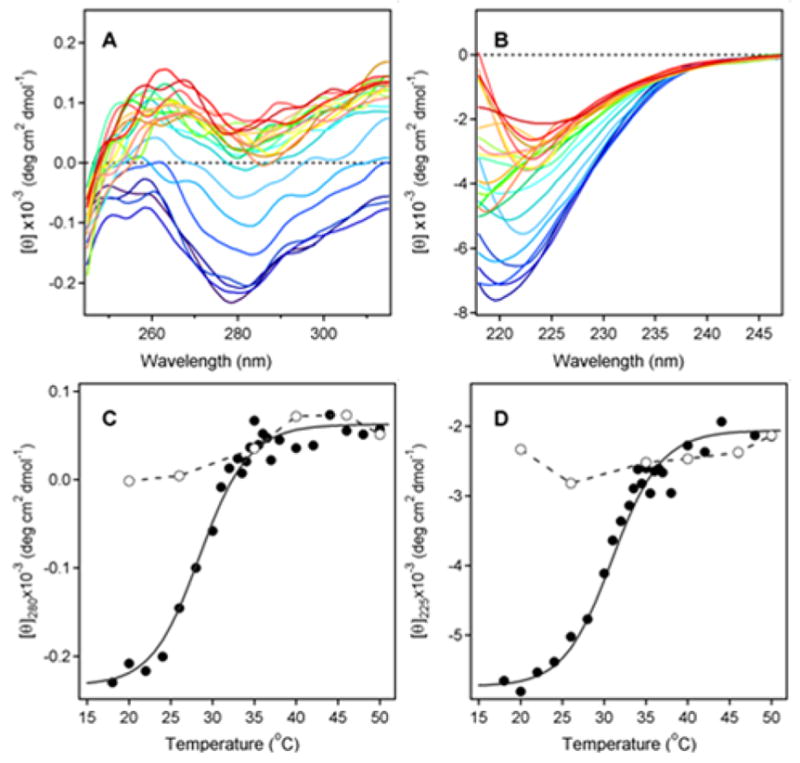
Temperature-Induced Unfolding of the PT S1 Subunit. (A–B): 37.4 μg of PT S1 was dissolved in 0.225 ml of 20 mM Na-phosphate buffer (pH 7.4) containing 150 mM NaCl and 10 mM β-ME. Thermotropic conformational changes to the structure of PT S1 were then monitored by near-UV CD (A) and far-UV CD (B). Both measurements were conducted on the same sample with the use of a 4-mm optical path-length rectangular quartz cuvette. Samples were equilibrated for 4 min at each temperature before measurements were taken. CD spectra were recorded from 200–315 nm, which covers both the near-UV and far-UV range. The change in color from blue to red corresponds to a change in temperature from 18°C to 50°C, as shown in panels C & D. (C–D): Thermal unfolding profiles for PT S1 tertiary structure (C) and PT S1 secondary structure (D) were derived from the data in panels A & B. The mean residue molar ellipticities at 280 nm (near-UV CD; tertiary structure) and 225 nm (far-UV CD; secondary structure) were plotted as a function of temperature. As described in Experimental Procedures, curves were simulated according to a two-state phase transition approach using the following best-fit parameters: heat capacities were ΔC = 0.39 kcal mol−1 K−1, and the values of ΔH were 55.0 kcal mol−1. The open circles represent measurements taken at the indicated temperatures during sample cooling from 50°C to 18°C.
Figure 2.
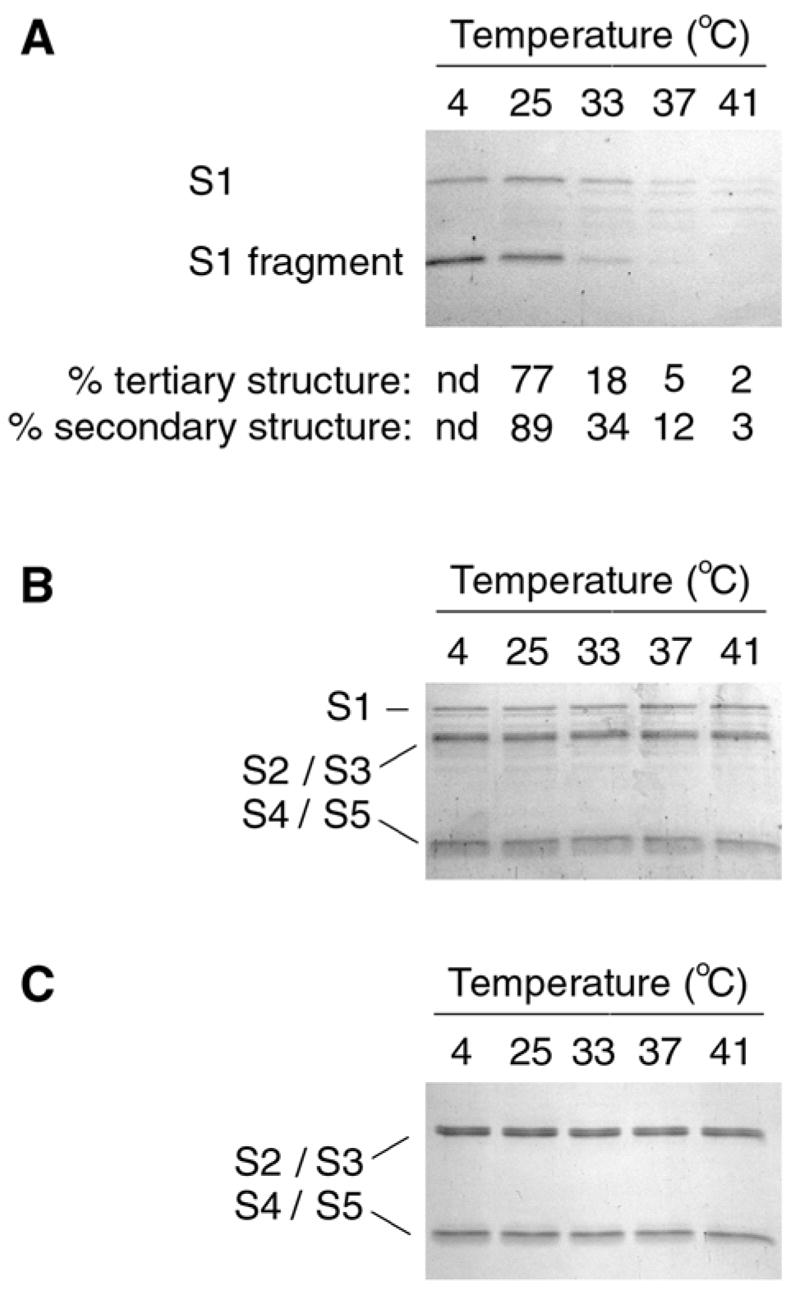
PT S1 Protease Sensitivity Assay. (A): PT S1 was placed in 20 mM Na-phosphate buffer (pH 7.0) containing 10 mM β-ME. Toxin samples were incubated for 45 min at the indicated temperatures and then shifted to 4°C for 10 min. Thermolysin was subsequently added for another 45 min at 4°C. Samples were visualized by SDS-PAGE and Coomassie staining. The percentage of native tertiary or secondary structure remaining in PT S1 at the indicated temperatures was calculated from the thermal unfolding profiles presented in Figs. 1C & 1D, respectively. nd; not determined. (B–C): PT (B) and PT B (C) were processed as described above for PT S1. The subunits of PT are identified: S1 (26 kDa), S2/S3 (~22 kDa each), and S4/S5 (11–12 kDa).
The thermal denaturation of PT S1 was irreversible (Figs. 1C & 1D). After heating PT S1 to 50ºC, the toxin was cooled in stepwise increments to 18ºC. Measurements of the near- and far-UV CD spectra taken at various temperatures during sample cooling are represented by the open circles in panels 1C and 1D. The data clearly indicates that PT S1 was unable to assume a native conformation following its temperature-induced unfolding at 50ºC.
Effect of temperature, NAD, and CHAPS on PT S1 protease sensitivity
The temperature-dependent folding state of PT S1 was also monitored with a protease sensitivity assay (Fig. 2). Samples of the purified and reduced PT S1 subunit were placed in 20 mM Na-Phosphate buffer (pH 7.0) and incubated at 4ºC, 25ºC, 33ºC, 37ºC, or 41ºC for 45 minutes. All samples were then shifted to 4ºC and exposed to the metalloprotease thermolysin for another 45 minutes. Since all protease treatments were conducted at 4ºC, differential degradation of the PT S1 samples could only result from temperature-induced changes to the structure of PT S1. Thermolysin-treated samples were resolved by SDS-PAGE and visualized with Coomassie staining. Similar experiments were performed for PT and PT B.
As shown in Fig. 2A, PT S1 samples pre-incubated at 37ºC or 41ºC were degraded by thermolysin. Substantial degradation of the PT S1 sample pre-incubated at 33ºC was also observed. PT S1 samples pre-incubated at 4ºC or 25ºC were nicked but not degraded by thermolysin. The ~18 kDa S1 fragment generated by thermolysin nicking of PT S1 most likely represents the N-terminal catalytic core of PT S1; similar N-terminal S1 fragments have also been detected in intoxicated cells (30, 31) and after in vitro exposure of PT S1 to trypsin or chymotrypsin (32, 33). For our assay, the S1 fragment did not appear without thermolysin treatment and was not generated from PT S1 samples co-incubated with EDTA and thermolysin (data not shown).
The extent of PT S1 degradation correlated well with its conformational state (Fig. 2A, bottom panel): the protease-resistant PT S1 sample that was incubated at 25ºC retained a significant amount of native tertiary and secondary structure, whereas the protease-sensitive PT S1 samples that were incubated at temperatures ≥37ºC retained little of the native toxin structure. When PT S1 was incubated at 33ºC, it retained an intermediate amount of native structure and was partially resistant to thermolysin-mediated degradation. These observations validated the use of a protease sensitivity assay to probe the folding state of PT S1 and demonstrated that PT S1 is in an unfolded, protease-sensitive conformation at 37ºC.
PT and PT B samples pre-incubated at 4ºC, 25ºC, 33ºC, 37ºC, and 41ºC were uniformly resistant thermolysin-mediated degradation (Figs. 2B & 2C). This demonstrated the specificity of thermolysin activity against the isolated PT S1 subunit and was consistent with the heat-stable properties of both PT and PT B (3, 34). It also indicated that the association of PT S1 with PT B protected PT S1 from thermolysin-mediated degradation. Likewise, PT S1 is resistant to processing by trypsin and chymotrypsin when incorporated into the PT holotoxin (32, 33). Thus, protease sensitivity (and thermal instability) in the PT S1 subunit is only apparent after it dissociates from PT B.
The heat-labile nature of the isolated PT S1 subunit suggests that the translocated, cytosolic pool of toxin would not function at 37ºC. Yet intoxication occurs in vivo at 37ºC. In vitro NAD-glycohydrolase and ADP-ribosylation assays have also documented PT S1 activity at 37ºC (28). Since both of these in vitro assays included NAD as a donor molecule, we hypothesized that an interaction between PT S1 and NAD could stabilize the structure of PT S1 and allow the toxin to function at 37ºC despite its heat-labile state. A similar relationship has been established for the heat-labile ricin A chain and its ribosome target (26).
To determine whether NAD has a stabilizing effect on the structure of PT S1, we performed our protease sensitivity assay on toxin samples that had been incubated with 1 mM NAD (Fig. 3). In comparison to toxin samples incubated without NAD, PT S1 samples incubated with NAD exhibited increased resistance to thermolysin-mediated degradation (Fig. 3A). Co-incubation with NAD increased the susceptibility of PT S1 to thermolysin nicking, but in the presence of NAD a substantial amount of this nicked S1 fragment (as well as the remaining full-length PT S1 polypeptide) was detected after incubations at 37ºC and 41ºC. Generation of the nicked PT S1 fragment indicated that thermolysin was active in the presence of NAD. Furthermore, NAD did not inhibit the thermolysin-mediated degradation of CT A1 (Fig. 3B). Thus, NAD specifically inhibited the temperature-induced structural shift of PT S1 to a protease-sensitive conformation.
Figure 3.
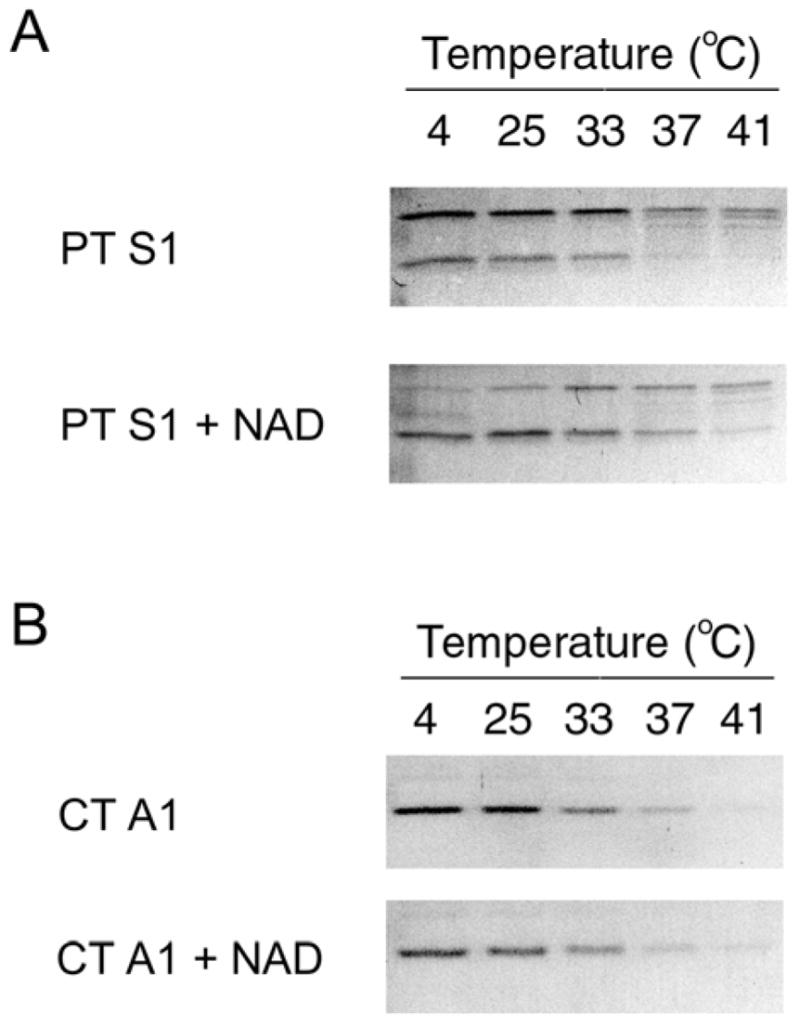
Effect of NAD on PT S1 Protease Sensitivity. PT S1 (A) and CT A1/CT A2 (B) were placed in 20 mM Na-phosphate buffer (pH 7.0) containing 10 mM β-ME. 1 mM NAD was present in the Na-phosphate buffer as indicated. Toxin samples were incubated at the stated temperatures for 45 min and then shifted to 4°C for 10 min. Thermolysin was added for another 45 min at 4°C. Samples were visualized by SDS-PAGE with Coomassie staining. In panel A, the upper band is full-length PT S1 and the lower band is the S1 fragment. In panel B, the 5 kDa CT A2 polypeptide was not visible after Coomassie staining.
We also examined PT S1 protease sensitivity in the presence of CHAPS, a zwitterionic detergent that stimulates the in vitro enzymatic activity of PT and PT S1 (28, 35, 36). This stimulatory effect could conceivably result from CHAPS-mediated stabilization of the heat-labile PT S1 structure. Yet CHAPS did not inhibit the temperature-induced structural shift of PT S1 to a protease-sensitive conformation (Fig. 4). Instead, PT S1 proteolysis was slightly more efficient in the presence of CHAPS: toxin samples pre-incubated at 33ºC in the absence of CHAPS were partially degraded by thermolysin, whereas PT S1 samples pre-incubated at 33ºC in the presence of CHAPS were completely degraded by thermolysin. Furthermore, in the presence of CHAPS the conversion of full-length PT S1 to the S1 fragment was complete. These observations indicated that CHAPS had some effect on the structural state of PT S1, but this effect did not inhibit the temperature-induced shift of PT S1 to a protease-sensitive conformation.
Figure 4.
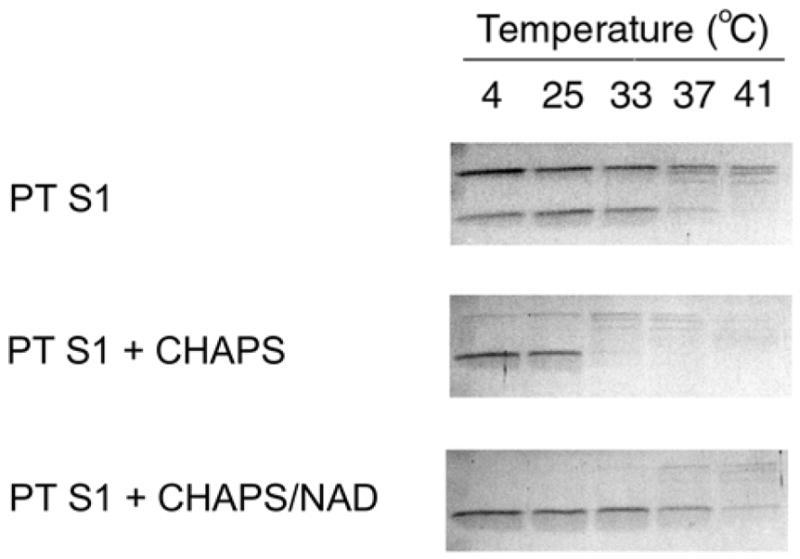
Effect of CHAPS on PT S1 Protease Sensitivity. PT S1 was placed in 20 mM Na-phosphate buffer (pH 7.0) containing 10 mM β-ME. 1% CHAPS or 1% CHAPS with 1 mM NAD were present in the Na-phosphate buffer as indicated. Toxin samples were incubated at the stated temperatures for 45 min and then shifted to 4°C for 10 min. Thermolysin was added for another 45 min at 4°C. Samples were visualized by SDS-PAGE with Coomassie staining. For the untreated PT S1 sample, the upper band is full-length PT S1 and the lower band is the S1 fragment. CHAPS-treated samples only produced the S1 fragment.
CHAPS and NAD both exerted structural effects on PT S1 when the two agents were mixed together with PT S1 (Fig. 4). In the presence of CHAPS and NAD, there was a full conversion of PT S1 to the S1 fragment. However, the PT S1 fragment also exhibited substantial resistance to degradation by thermolysin. CHAPS therefore stimulated the conversion of PT S1 to the S1 fragment, but it did not override the NAD-induced structural shift of the S1 fragment to a protease-resistant conformation. As CHAPS is often used to stimulate the in vitro enzymatic activity of PT and PT S1 (28, 35, 36), our results allow a direct comparison between these published studies and the structural state of PT S1.
PT S1 degradation by the 20S proteasome
The unfolded, protease-sensitive conformation of PT S1 at 37ºC could facilitate its degradation by the 20S proteasome. This barrel-shaped structure comprises the catalytic core of the 26S proteasome, which is generated by the attachment of 19S regulatory particles to one or both ends of the 20S proteasome. Most proteasomal substrates are degraded in a ubiquitin-dependent process by the 26S proteasome, but a few proteins are instead degraded by a ubiquitin-independent mechanism involving only the core 20S proteasome (37, 38). Proteins degraded by the 20S proteasome must be unfolded in order to pass through the central catalytic pore of the proteasome, and this unfolding event represents the rate-limiting step in proteolysis by the 20S proteasome (38, 39). The unfolded structural state of PT S1 at 37ºC would therefore make it a suitable substrate for degradation by the 20S proteasome.
To monitor toxin degradation by the 20S proteasome, we incubated reduced CT A1 or reduced PT S1 with the 20S proteasome at 37ºC for 0, 3, 9, and 20 hours (Fig. 5A). CT A1 is a substrate for the 20S proteasome (Teter et. al., unpublished experiments), and it was used here as a positive control for proteasomal activity. Toxin samples were resolved by SDS-PAGE and visualized with Commaisse staining. Both CT A1 and PT S1 were degraded by the 20S proteasome, even though ubiquitin and the ubiquitin conjugation machinery were absent from our assay conditions. CT A1 was degraded more efficiently than PT S1: whereas significant degradation of CT A1 was observed after 3 hours of incubation, more than 9 hours of incubation were required to detect the degradation of PT S1. This slow rate of in vitro proteolysis is common for 20S proteasome substrates (40–42), and may be accelerated in vivo by proteasome accessory factors such as hsp90 (43).
Figure 5.

PT S1 Degradation by the 20S Proteasome. (A) Reduced PT S1 and the reduced CT A1/CT A2 heterodimer were incubated at 37°C with the 20S proteasome. Toxin samples taken at the indicated time points were visualized by SDS-PAGE and Coomassie staining. (B) PT S1, PT, and PT B were incubated at 37°C with the 20S proteasome under reducing conditions. Toxin samples taken after 0 or 20 hrs of incubation were visualized by SDS-PAGE and Coomassie staining.
To further evaluate the specificity of our proteasome assay, we incubated PT S1, PT, or PT B with the 20S proteasome for 20 hours at 37ºC (Fig. 5B). The isolated PT S1 subunit was effectively degraded by the 20S proteasome. However, PT S1 degradation did not occur when the A moiety was incorporated into the PT holotoxin. This again indicated that the structural arrangement of the holotoxin prevented PT S1 from assuming a protease-sensitive (i.e., unfolded) conformation. It also demonstrated that indiscriminate proteasomal degradation was not responsible for proteolysis of the isolated PT S1 subunit. The specificity of proteasomal activity was further confirmed by the stability of the PT B oligomer in the presence of the 20S proteasome.
As the core 20S proteasome can only degrade unfolded substrates, the stabilizing effect of NAD on PT S1 structure could protect the toxin from ubiquitin-independent degradation by the 20S proteasome. However, addition of 1 mM NAD to the assay buffer did not prevent PT S1 degradation by the 20S proteasome (data not shown). Thus, NAD-induced stabilization of the PT S1 catalytic core (see Figures 3 & 4) is not sufficient to protect the PT S1 polypeptide from processing by the 20S proteasome.
DISCUSSION
Contrary to current models of ERAD-mediated toxin translocation, the C-terminal region of PT S1 is not required for toxin passage into the cytosol (16, 17). PT S1 may also be degraded in the cytosol (16). Based on these observations and our recent work with the CT A1 polypeptide (21), we hypothesized ER-translocating toxins such as PT contain thermally unstable A subunits that are susceptible to degradation by the 20S proteasome. The present work provides direct experimental evidence to support this hypothesis and suggests a new model for toxin-ERAD interactions.
The tertiary structure of PT S1 unfolds with a Tm of 28.5ºC, and the secondary structure of PT S1 becomes disordered with a Tm of 31.0ºC. Thus, the isolated and reduced PT S1 subunit is structurally unstable at physiological temperature. PT S1 is also in a protease-sensitive state at 37ºC. These observations stand in marked contrast to the heat-stable and protease-resistant properties of both PT and PT B, which only unfold at temperatures greater than 60ºC (3, 34). The structure of the PT holotoxin thus maintains PT S1 in a stable conformation until the toxin is exposed to an environment (i.e., the ER lumen) that allows PT S1 to dissociate from PT B. A similar structural relationship exists for the subunits of CT (44, 45) (Teter et. al., unpublished experiments), so the stabilizing effect of A/B interactions on the heat-labile structure of the toxin A moiety may be a general phenomenon for AB-type, ER-translocating toxins. The proposed role of PT B as a scaffold to stabilize the heat-labile structure of PT S1 complements the apparent role of PT B as a targeting vehicle for PT S1 delivery to the ER (12, 16, 17).
Dissociation of PT S1 from PT B in the ER of an intoxicated cell would apparently result in the spontaneous unfolding of PT S1. This unfolding event, which is likely required for passage through the Sec61p translocon, could stimulate the ERAD-mediated translocation of PT S1 into the cytosol. By this model, the unfolded conformation of PT S1 at 37ºC allows it to masquerade as a misfolded protein. ERAD-mediated toxin translocation was originally thought to depend upon a hydrophobic domain in the C-terminal region of the toxin A chain (10), but neither CT A1 nor PT S1 require their C-terminal domains for passage into the cytosol (16, 17, 21). Our structural studies provide an alternative trigger for the ERAD mechanism that is based upon the heat-labile state of the isolated toxin A chain. This property results in chaperone-independent unfolding of PT S1 at 37ºC, although in vivo unfolding of the toxin A chain may be initiated or aided by ER-localized chaperones and oxidoreductases.
The translocated and cytosolic pool of PT S1 might be stabilized by its association with NAD, the donor molecule for the ADP-ribosylation reaction that modifies the G protein targets of PT. We could not obtain a thermal unfolding profile for PT S1 in the presence of NAD because of the interfering signal produced by NAD in CD measurements. Structural measurements with fluorescence spectroscopy were also hampered by the quenching effect NAD has on PT S1 fluorescence (46, 47). However, a protease sensitivity assay demonstrated the inhibitory effect of NAD on the temperature-induced structural shift of PT S1 to a protease-sensitive (i.e., unfolded) conformation. Although full-length PT S1 was still susceptible to thermolysin nicking in the presence of NAD, the resulting S1 fragment (which represents the catalytic core of PT S1) exhibited increased stability in the presence of NAD. This result was consistent with the reported ability of NAD to protect the enzymatic activity of reduced PT S1 during incubations at 30ºC (48). Collectively, these observations provide an explanation for how PT S1 can regain an active conformation (via NAD binding) after entering the cytosol in an unfolded state.
The cellular concentration of NAD is estimated at 3 mM (49, 50). Up to 80% of cellular NAD is found in the cytosol; the bulk of the remaining NAD is located in the mitochondria (49). As the ER does not contain a substantial pool of NAD, the thermal unfolding of PT S1 can occur at this site without interference from NAD. Furthermore, the cytosolic concentration of NAD is well above the 25 μM Kd for NAD with PT S1 (47). It is therefore feasible for the cytosolic pool of NAD to readily interact with, and stabilize, the translocated PT S1 subunit. The subcellular distribution of NAD, which is absent from the ER and present in the cytosol at millimolar levels, thus conforms to our model of PT S1 translocation.
CT A1 and other toxins also use NAD as a donor molecule for the ADP-ribosylation of target substrates (1). However, as assessed with our protease sensitivity assay, NAD did not inhibit the thermal denaturation of CT A1. This indicated that (i) NAD does not directly inhibit thermolysin activity; and (ii) the stabilizing effect of NAD on PT S1 is not a common phenomenon for all ADP-ribosylating toxins. In the case of CT, an interaction with ADP-ribosylation factors is likely to stabilize and/or renature the heat-labile CT A1 subunit (51).
CHAPS, a zwitterionic detergent that stimulates PT and PT S1 activity in vitro (28, 35, 36), also affected the structural state of PT S1. It did not, however, inhibit the thermal denaturation of PT S1: nearly identical near- and far-UV CD thermal unfolding profiles were obtained for PT S1 samples incubated in either the presence or absence of 0.5% CHAPS (data not shown). Instead, CHAPS induced a structural shift that stimulated the thermolysin-mediated conversion of PT S1 to the S1 fragment. This structural shift may enhance the enzymatic activity of PT S1, as CHAPS provides a 2-fold stimulation of PT S1 NAD glycohydrolase activity at 37ºC and a greater level of stimulation at 30ºC (28, 35). Interestingly, the S1 fragment generated by exposure to CHAPS and thermolysin was held in a protease-resistant conformation when NAD was also present in the sample buffer. This again emphasized the stabilizing effect of NAD on the catalytic core of PT S1.
PT S1 may be degraded in the eukaryotic cytosol despite the absence of lysine residues for ubiquitin attachment (16). A ubiquitin-independent mechanism for toxin degradation could involve processing by the core 20S proteasome, which acts upon CT A1 and other unfolded or partially unfolded proteins (37, 38). The unfolded, protease-sensitive state of PT S1 at 37ºC would therefore place it in the proper conformation for degradation by the 20S proteasome. This prediction was confirmed with an in vitro assay demonstrating the susceptibility of PT S1 to ubiquitin-independent degradation by the core 20S proteasome. Thus, the heat-labile nature of PT S1 could possibly influence both its translocation into the cytosol and its degradation in the cytosol.
The 20S proteasome degraded CT A1 more rapidly than PT S1, even though CT A1 retains more of its native structure at 37ºC than PT S1. Thus, the efficiency of substrate processing by the 20S proteasome is not always directly proportional to the extent of substrate unfolding. Additional factors such as substrate recognition/binding and structural constraints on substrate passage into the central pore of the proteasome are likely to affect the efficiency of substrate processing by the 20S proteasome as well. The differential processing of CT A1 and PT S1 by the 20S proteasome suggests that these toxins could serve as useful probes in future work to dissect the mechanics of degradation by the core 20S proteasome.
The in vitro studies of this work have demonstrated that the isolated and reduced PT S1 subunit is a thermally unstable protein susceptible to degradation by the 20S proteasome. This provides experimental evidence for a revised model of toxin-ERAD interactions in which both toxin translocation and toxin degradation are linked to an inherent physical property (i.e., thermal instability) of the toxin A moiety.
Footnotes
This work was supported by National Institutes of Health grant K22 AI054568 to K. Teter and by start-up funds provided to K. Teter from the University of Central Florida Department of Molecular Biology and Microbiology.
Abbreviations: β-ME, β-mercaptoethanol; CHAPS, 3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate; CT, cholera toxin; CD, circular dichroism; ER, endoplasmic reticulum; ERAD, endoplasmic reticulum-associated degradation; PT, pertussis toxin; SDS-PAGE; sodium dodecyl sulfate polyacrylamide gel electrophoresis; Tm, transition temperature.
References
- 1.Krueger KM, Barbieri JT. The family of bacterial ADP-ribosylating exotoxins. Clin Microbiol Rev. 1995;8:34–47. doi: 10.1128/cmr.8.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaslow HR, Burns DL. Pertussis toxin and target eukaryotic cells: binding, entry, and activation. FASEB J. 1992;6:2684–2690. doi: 10.1096/fasebj.6.9.1612292. [DOI] [PubMed] [Google Scholar]
- 3.Yang J, Mou J, Shao Z. Structure and stability of pertussis toxin studied by in situ atomic force microscopy. FEBS Lett. 1994;338:89–77792. doi: 10.1016/0014-5793(94)80122-3. [DOI] [PubMed] [Google Scholar]
- 4.Brennan MJ, David JL, Kenimer JG, Manclark CR. Lectin-like binding of pertussis toxin to a 165-kilodalton Chinese hamster ovary cell glycoprotein. J Biol Chem. 1988;263:4895–4899. [PubMed] [Google Scholar]
- 5.Hausman SZ, Burns DL. Binding of pertussis toxin to lipid vesicles containing glycolipids. Infect Immun. 1993;61:335–337. doi: 10.1128/iai.61.1.335-337.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Witvliet MH, Burns DL, Brennan MJ, Poolman JT, Manclark CR. Binding of pertussis toxin to eucaryotic cells and glycoproteins. Infect Immun. 1989;57:3324–3330. doi: 10.1128/iai.57.11.3324-3330.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu Y, Barbieri JT. Pertussis toxin-mediated ADP-ribosylation of target proteins in Chinese hamster ovary cells involves a vesicle trafficking mechanism. Infect Immun. 1995;63:825–832. doi: 10.1128/iai.63.3.825-832.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu Y, Barbieri JT. Pertussis toxin-catalyzed ADP-ribosylation of Gi-2 and Gi-3 in CHO cells is modulated by inhibitors of intracellular trafficking. Infect Immun. 1996;64:593–599. doi: 10.1128/iai.64.2.593-599.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hazes B, Boodhoo A, Cockle SA, Read RJ. Crystal structure of the pertussis toxin-ATP complex: a molecular sensor. J Mol Biol. 1996;258:661–671. doi: 10.1006/jmbi.1996.0277. [DOI] [PubMed] [Google Scholar]
- 10.Hazes B, Read RJ. Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry. 1997;36:11051–11054. doi: 10.1021/bi971383p. [DOI] [PubMed] [Google Scholar]
- 11.el Baya A, Linnemann R, von Olleschik-Elbheim L, Robenek H, Schmidt MA. Endocytosis and retrograde transport of pertussis toxin to the Golgi complex as a prerequisite for cellular intoxication. Eur J Cell Biol. 1997;73:40–48. [PubMed] [Google Scholar]
- 12.Carbonetti NH, Irish TJ, Chen CH, O’Connell CB, Hadley GA, McNamara U, Tuskan RG, Lewis GK. Intracellular delivery of a cytolytic T-lymphocyte epitope peptide by pertussis toxin to major histocompatibility complex class I without involvement of the cytosolic class I antigen processing pathway. Infect Immun. 1999;67:602–607. doi: 10.1128/iai.67.2.602-607.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burns DL, Manclark CR. Adenine nucleotides promote dissociation of pertussis toxin subunits. J Biol Chem. 1986;261:4324–4327. [PubMed] [Google Scholar]
- 14.Krueger KM, Barbieri JT. Molecular characterization of the in vitro activation of pertussis toxin by ATP. J Biol Chem. 1993;268:12570–12578. [PubMed] [Google Scholar]
- 15.Burns DL, Manclark CR. Role of cysteine 41 of the A subunit of pertussis toxin. J Biol Chem. 1989;264:564–568. [PubMed] [Google Scholar]
- 16.Castro MG, McNamara U, Carbonetti NH. Expression, activity and cytotoxicity of pertussis toxin S1 subunit in transfected mammalian cells. Cell Microbiol. 2001;3:45–54. doi: 10.1046/j.1462-5822.2001.00092.x. [DOI] [PubMed] [Google Scholar]
- 17.Veithen A, Raze D, Locht C. Intracellular trafficking and membrane translocation of pertussis toxin into host cells. Int J Med Microbiol. 2000;290:409–413. doi: 10.1016/S1438-4221(00)80053-3. [DOI] [PubMed] [Google Scholar]
- 18.McCracken AA, Brodsky JL. Evolving questions and paradigm shifts in endoplasmic-reticulum-associated degradation (ERAD) Bioessays. 2003;25:868–877. doi: 10.1002/bies.10320. [DOI] [PubMed] [Google Scholar]
- 19.Romisch K. Surfing the Sec61 channel: bidirectional protein translocation across the ER membrane. J Cell Sci. 1999;112(Pt 23):4185–4191. doi: 10.1242/jcs.112.23.4185. [DOI] [PubMed] [Google Scholar]
- 20.Lord JM, Roberts LM. Toxin entry: retrograde transport through the secretory pathway. J Cell Biol. 1998;140:733–736. doi: 10.1083/jcb.140.4.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teter K, Jobling MG, Stentz D, Holmes RK. The Cholera Toxin A13 Subdomain Is Essential for Interaction with ADP-Ribosylation Factor 6 and Full Toxic Activity but Is Not Required for Translocation from the Endoplasmic Recticulum to the Cytosol. Infect Immun. 2006;74:2259–2267. doi: 10.1128/IAI.74.4.2259-2267.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simpson JC, Roberts LM, Romisch K, Davey J, Wolf DH, Lord JM. Ricin A chain utilises the endoplasmic reticulum-associated protein degradation pathway to enter the cytosol of yeast. FEBS Lett. 1999;459:80–84. doi: 10.1016/s0014-5793(99)01222-3. [DOI] [PubMed] [Google Scholar]
- 23.Deeks ED, Cook JP, Day PJ, Smith DC, Roberts LM, Lord JM. The low lysine content of ricin A chain reduces the risk of proteolytic degradation after translocation from the endoplasmic reticulum to the cytosol. Biochemistry. 2002;41:3405–3413. doi: 10.1021/bi011580v. [DOI] [PubMed] [Google Scholar]
- 24.Di Cola A, Frigerio L, Lord JM, Ceriotti A, Roberts LM. Ricin A chain without its partner B chain is degraded after retrotranslocation from the endoplasmic reticulum to the cytosol in plant cells. Proc Natl Acad Sci U S A. 2001;98:14726–14731. doi: 10.1073/pnas.251386098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teter K, Allyn RL, Jobling MG, Holmes RK. Transfer of the cholera toxin A1 polypeptide from the endoplasmic reticulum to the cytosol is a rapid process facilitated by the endoplasmic reticulum-associated degradation pathway. Infect Immun. 2002;70:6166–6171. doi: 10.1128/IAI.70.11.6166-6171.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Argent RH, Parrott AM, Day PJ, Roberts LM, Stockley PG, Lord JM, Radford SE. Ribosome-mediated folding of partially unfolded ricin A-chain. J Biol Chem. 2000;275:9263–9269. doi: 10.1074/jbc.275.13.9263. [DOI] [PubMed] [Google Scholar]
- 27.Lavigne P, Crump MP, Gagne SM, Hodges RS, Kay CM, Sykes BD. Insights into the mechanism of heterodimerization from the 1H-NMR solution structure of the c-Myc-Max heterodimeric leucine zipper. J Mol Biol. 1998;281:165–181. doi: 10.1006/jmbi.1998.1914. [DOI] [PubMed] [Google Scholar]
- 28.Murayama T, Hewlett EL, Maloney NJ, Justice JM, Moss J. Effect of temperature and host factors on the activities of pertussis toxin and Bordetella adenylate cyclase. Biochemistry. 1994;33:15293–15297. doi: 10.1021/bi00255a010. [DOI] [PubMed] [Google Scholar]
- 29.Stein PE, Boodhoo A, Armstrong GD, Cockle SA, Klein MH, Read RJ. The crystal structure of pertussis toxin. Structure. 1994;2:45–57. doi: 10.1016/s0969-2126(00)00007-1. [DOI] [PubMed] [Google Scholar]
- 30.Finck-Barbancon V, Barbieri JT. Preferential processing of the S1 subunit of pertussis toxin that is bound to eukaryotic cells. Mol Microbiol. 1996;22:87–95. doi: 10.1111/j.1365-2958.1996.tb02658.x. [DOI] [PubMed] [Google Scholar]
- 31.Carbonetti NH, Mays RM, Artamonova GV, Plaut RD, Worthington ZE. Proteolytic cleavage of pertussis toxin S1 subunit is not essential for its activity in mammalian cells. BMC Microbiol. 2005;5:7. doi: 10.1186/1471-2180-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burns DL, Hausman SZ, Lindner W, Robey FA, Manclark CR. Structural characterization of pertussis toxin A subunit. J Biol Chem. 1987;262:17677–17682. [PubMed] [Google Scholar]
- 33.Krueger KM, Mende-Mueller LM, Barbieri JT. Protease treatment of pertussis toxin identifies the preferential cleavage of the S1 subunit. J Biol Chem. 1991;266:8122–8128. [PubMed] [Google Scholar]
- 34.Krell T, Greco F, Nicolai MC, Dubayle J, Renauld-Mongenie G, Poisson N, Bernard I. The use of microcalorimetry to characterize tetanus neurotoxin, pertussis toxin and filamentous haemagglutinin. Biotechnol Appl Biochem. 2003;38:241–251. doi: 10.1042/BA20030089. [DOI] [PubMed] [Google Scholar]
- 35.Moss J, Stanley SJ, Watkins PA, Burns DL, Manclark CR, Kaslow HR, Hewlett EL. Stimulation of the thiol-dependent ADP-ribosyltransferase and NAD glycohydrolase activities of Bordetella pertussis toxin by adenine nucleotides, phospholipids, and detergents. Biochemistry. 1986;25:2720–2725. doi: 10.1021/bi00357a066. [DOI] [PubMed] [Google Scholar]
- 36.Kaslow HR, Lim LK, Moss J, Lesikar DD. Structure-activity analysis of the activation of pertussis toxin. Biochemistry. 1987;26:123–127. doi: 10.1021/bi00375a018. [DOI] [PubMed] [Google Scholar]
- 37.Orlowski M, Wilk S. Ubiquitin-independent proteolytic functions of the proteasome. Arch Biochem Biophys. 2003;415:1–5. doi: 10.1016/s0003-9861(03)00197-8. [DOI] [PubMed] [Google Scholar]
- 38.Shringarpure R, Grune T, Davies KJ. Protein oxidation and 20S proteasome-dependent proteolysis in mammalian cells. Cell Mol Life Sci. 2001;58:1442–1450. doi: 10.1007/PL00000787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferrington DA, Sun H, Murray KK, Costa J, Williams TD, Bigelow DJ, Squier TC. Selective degradation of oxidized calmodulin by the 20 S proteasome. J Biol Chem. 2001;276:937–943. doi: 10.1074/jbc.M005356200. [DOI] [PubMed] [Google Scholar]
- 40.Pacifici RE, Kono Y, Davies KJ. Hydrophobicity as the signal for selective degradation of hydroxyl radical-modified hemoglobin by the multicatalytic proteinase complex, proteasome. J Biol Chem. 1993;268:15405–15411. [PubMed] [Google Scholar]
- 41.Reinheckel T, Sitte N, Ullrich O, Kuckelkorn U, Davies KJ, Grune T. Comparative resistance of the 20S and 26S proteasome to oxidative stress. Biochem J. 1998;335(Pt 3):637–642. doi: 10.1042/bj3350637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tanaka K, Waxman L, Goldberg AL. ATP serves two distinct roles in protein degradation in reticulocytes, one requiring and one independent of ubiquitin. J Cell Biol. 1983;96:1580–1585. doi: 10.1083/jcb.96.6.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whittier JE, Xiong Y, Rechsteiner MC, Squier TC. Hsp90 enhances degradation of oxidized calmodulin by the 20 S proteasome. J Biol Chem. 2004;279:46135–46142. doi: 10.1074/jbc.M406048200. [DOI] [PubMed] [Google Scholar]
- 44.Surewicz WK, Leddy JJ, Mantsch HH. Structure, stability, and receptor interaction of cholera toxin as studied by Fourier-transform infrared spectroscopy. Biochemistry. 1990;29:8106–1811. doi: 10.1021/bi00487a017. [DOI] [PubMed] [Google Scholar]
- 45.Goins B, Freire E. Thermal stability and intersubunit interactions of cholera toxin in solution and in association with its cell-surface receptor ganglioside GM1. Biochemistry. 1988;27:2046–5202. doi: 10.1021/bi00406a035. [DOI] [PubMed] [Google Scholar]
- 46.Antoine R, Tallett A, van Heyningen S, Locht C. Evidence for a catalytic role of glutamic acid 129 in the NAD-glycohydrolase activity of the pertussis toxin S1 subunit. J Biol Chem. 1993;268:24149–24155. [PubMed] [Google Scholar]
- 47.Lobban MD, Irons LI, van Heyningen S. Binding of NAD+ to pertussis toxin. Biochim Biophys Acta. 1991;1078:155–160. doi: 10.1016/0167-4838(91)99004-c. [DOI] [PubMed] [Google Scholar]
- 48.Kaslow HR, Schlotterbeck JD, Mar VL, Burnette WN. Alkylation of cysteine 41, but not cysteine 200, decreases the ADP-ribosyltransferase activity of the S1 subunit of pertussis toxin. J Biol Chem. 1989;264:6386–6390. [PubMed] [Google Scholar]
- 49.Devin A, Nogueira V, Leverve X, Guerin B, Rigoulet M. Allosteric activation of pyruvate kinase via NAD+ in rat liver cells. Eur J Biochem. 2001;268:3943–3949. doi: 10.1046/j.1432-1327.2001.02306.x. [DOI] [PubMed] [Google Scholar]
- 50.Zocchi E, Usai C, Guida L, Franco L, Bruzzone S, Passalacqua M, De Flora A. Ligand-induced internalization of CD38 results in intracellular Ca2+ mobilization: role of NAD+ transport across cell membranes. FASEB J. 1999;13:273–283. doi: 10.1096/fasebj.13.2.273. [DOI] [PubMed] [Google Scholar]
- 51.Murayama T, Tsai SC, Adamik R, Moss J, Vaughan M. Effects of temperature on ADP-ribosylation factor stimulation of cholera toxin activity. Biochemistry. 1993;32:561–566. doi: 10.1021/bi00053a022. [DOI] [PubMed] [Google Scholar]