

Abstract
OBJECTIVE—To determine whether interindividual heterogeneity in the erythrocyte (red blood cell [RBC]) transmembrane glucose gradient might explain discordances between A1C and glycemic control based on measured fructosamine.
RESEARCH DESIGN AND METHODS—We modeled the relationship between plasma glucose and RBC glucose as the concentration distribution (Ci-to-Co ratio) of a nonmetabolizable glucose analog 14C-3-O-methyl glucose (14C-3OMG) inside (Ci) and outside (Co) RBCs in vitro. We examined the relationship between that distribution and the degree of glycation of hemoglobin in comparison with glycation of serum proteins (fructosamine), the glycation gap. A1C, fructosamine, and in vitro determination of the 14C-3OMG distribution in glucose-depleted RBCs were measured in 26 fasted subjects.
RESULTS—The Ci-to-Co ratio 0.89 ± 0.07 for 3-O-methyl-d-glucopyranose (3OMG) ranged widely (0.72–1.04, n = 26). In contrast, urea Ci-to-Co (1.015 ± 0.022 [range 0.98–1.07], P < 0.0001) did not. Concerning mechanism, in a representative subset of subjects, the Ci-to-Co ratio was retained in RBC ghosts, was not dependent on ATP or external cations, and was reestablished after reversal of the glucose gradient. The 3OMG Ci-to-Co ratio was not correlated with serum fructosamine, suggesting that it was independent of mean plasma glucose. However, Ci-to-Co did correlate with A1C (R2 = 0.19) and with the glycation gap (R2 = 0.20), consistent with a model in which differences in internal glucose concentration at a given mean plasma glucose contribute to differences in A1C for given level of glycemic control.
CONCLUSIONS—The data demonstrate interindividual heterogeneity in glucose gradients across RBC membranes that may affect hemoglobin glycation and have implications for diabetes complications risk and risk assessment.
A1C is the current gold standard for determination of chronic glycemic control in people with diabetes. Yet it is common to find hematologically normal people with diabetes in whom A1C appears discordant from other measures of glycemic control. Some have suggested the notion of a “hemoglobin glycation index” to assess A1C discordance from mean blood glucose (1–3). We have quantitated this discordance with the use of the glycation gap (GG), a measure of the disparity between two integrated measures of glycemic control, one intracellular in red blood cells (RBCs), A1C, and the other extracellular, glycated serum protein (GSP) measured as fructosamine (4). A non-zero GG could result from differences between the ambient glucose concentrations or rates of glycation in the intracellular and extracellular compartments, and/or interindividual differences in the turnover/metabolism of underlying proteins. We have demonstrated that the GG is reproducible within subjects over time and is associated with important clinical end points. In longstanding type 1 diabetes, there is a over a twofold rise in prevalence of nephropathy in patients with high GG versus low GG (4). GG is also a predictor of retinopathy (5). Furthermore, approximately one-third of the heritable component of A1C (6) is shared in common with a heritable component of the GG (7), suggesting a genetically determined mechanism for discordance between fructosamine and A1C.
As indicated by Higgins et al. (8), two factors other than plasma glucose influence the level of glycated hemoglobin: lifespan and “glucose permeability” of the RBC. Yet there has been little systematic investigation of how interindividual variability in these parameters may affect the relationship between measured A1C levels and plasma glucose. In another study, we examined differences in RBC survival and reported variability that can produce significant deviations of A1C from expected values (9). We focus here on glucose permeability, i.e., the concentration of glucose within versus outside the RBC. Glucose transport across the RBC membrane mediated by the GLUT1 transporter is complete within seconds at body temperature (10). Thus, in addition to plasma glucose and RBC lifespan, “equilibrium” intracellular glucose, rather than the rate of glucose transport, is likely the determining factor in hemoglobin glycation.
Over 80 years ago, Somogyi (11) found that the “corpuscular sugar–to–serum sugar” ratio averaged 0.77. Although it varied among healthy subjects between 0.66 and 0.95, the ratio did not change with glucose challenge and was similar between a diabetic and nondiabetic subject. In the Higgins et al. (8) description of the variation in A1C between animal species in relation to glucose permeability of the RBC, the erythrocyte glucose–to–plasma glucose concentration ratio was 0.67 ± 0.14, demonstrating greater variation than expected for experimental technique or RBC water space, which is very tightly regulated (mean corpuscular hemoglobin concentration [MCHC] range 32–35%) (12). This suggests the possibility of substantial interindividual variation in erythrocyte glucose. Similarly, Gould and Yudkin and coworkers (2,3) reported systematic variation in the relationship between A1C and fasting plasma glucose in a nondiabetic population that correlated with erythrocyte glucose. A significant difference in erythrocyte glucose–to–plasma glucose concentration ratio was found under non–steady state conditions: 0.74 ± 0.12 in “low glycators” and 0.98 ± 0.06 in “high glycators” (P < 0.01) (3) after accounting for cell water (B. Gould, personal communication). The similarity among the results of Somogyi, Higgins et al., and Gould et al. are sufficient to propose that there are individual differences in how sugar distributes into the RBC. Yet, one of the key assumptions in the assessment of glycemic control by A1C is that glucose in the intra-erythrocyte space (which determines hemoglobin glycation) bears a common relationship to glucose in the plasma space (which defines glycemic control). A systematic difference in glucose permeability would constitute a source of intersubject differences in A1C at a given average plasma glucose concentration.
In this study, we sought to test the hypothesis that interindividual heterogeneity of the intracellular-to-extracellular glucose ratio contributes to variability in the relationship of A1C to fructosamine. We confirmed the existence of a glucose gradient across the human RBC membrane by measuring the distribution of the nonmetabolizable glucose analog, 14C-3-O-methyl glucose (14C-3OMG) into the RBC, which eliminated the potential contribution of metabolism of glucose to such measurements. We validated our methodologies by demonstrating the expected equilibration of another small molecule, urea, and eliminated intracellular hemoglobin, ATP, and transmembrane cation gradients as contributors to the glucose gradient. Finally, we demonstrate that the variation of the transmembrane glucose gradient among individuals correlates with the glycation gap and A1C but not with fructosamine.
RESEARCH DESIGN AND METHODS
3-O-methyl-d-glucopyranose (3OMG), 3-O-(14C-methyl)-d-glucose, mercuric chloride, phloretin, cytochalasin B, Drabkin's reagent, and hemoglobin standards were obtained from Sigma Chemical (St. Louis, MO). PBS, perchloric acid, Scintisafe Plus cocktail, and STANBIO cyanmethemoglobin standards were from Fisher Scientific (Pittsburgh, PA).
Subjects recruited were ≥14 years of age (Table 1). Exclusion criteria included baseline serum creatinine >1.5 mg/dl, urine albumin >200 μg/min (timed collection) or >179 μg/mg creatinine (spot collection), transaminases more than three times the upper limit of normal, New York Heart Association heart failure stage ≥3, hematocrit <34%, reticulocyte count >2%, evidence of hemoglobinopathy on high-performance liquid chromatography (HPLC) analysis of hemoglobin, active infection, or an underlying illness known to be associated with body wasting (e.g., malignancies or tuberculosis). All research procedures were approved by the University of Cincinnati Institutional Review Board.
TABLE 1.
Study subject characteristics
| Age (years) | 44 ± 11 |
| N | 26 |
| Sex | |
| Female | 13 |
| Male | 13 |
| Glucose tolerance status and diabetes treatment | |
| Nondiabetic | 5 |
| Type 1 diabetic | 10 (all insulin only) |
| Type 2 diabetic | 11 (6 insulin, 7 metformin, 2 sulfonylurea, 4 thiazolidinediones) |
| Race (n) | |
| Caucasian | 20 |
| African American | 5 |
| South Asian | 1 |
| Glycemic control | |
| Fasting plasma glucose (mg/dl) | 168 ± 76 (56–309) |
| A1C (%) | 7.6 ± 2.1 (4.6–12.4) |
| Fructosamine (μmol/l) | 322 ± 82 (191–505) |
Data are means ± SD and means ± SD (range) unless otherwise indicated.
Preparation of erythrocytes and ghosts.
Blood was drawn in heparin-coated tubes and processed as previously described (13) with modifications from Klepper et al. (14). Blood specimens were centrifuged at 2,500 rpm for 5 min to sediment RBCs. Serum and buffy coat were aspirated. To minimize differences in cellular sugar transport experiments resulting from diabetic subjects’ plasma glucose in vivo, erythrocytes were incubated at 37°C twice for 30 min in a 10-fold excess of PBS, then washed twice in PBS at ice temperature. Assays were standardized by measurement of hemoglobin spectrophotometrically with Drabkin's or Stanbio reagents and adjusting all erythrocyte suspensions to hemoglobin 15 g/dl for 3OMG transport. To prepare pink ghosts, packed RBCs were lysed in 10 volumes of 5 mmol/l phosphate buffer, pH 7.5. For white ghosts, erythrocytes were lysed in 50 V of the hypotonic buffer. Ghosts were centrifuged; pink ghosts were washed twice with cold PBS, and white ghosts were washed with lysis buffer until the supernatant was clear. For the depletion of intracellular ATP, 8 units/ml of the enzyme apyrase was included in the lysis buffer. Ghosts were resealed with 150 mmol/l KCl in lysis buffer at 37°C for 1 h. Resealed ghosts were sedimented, the supernatant was aspirated, and the ghosts were decanted (to remove the nonlysed cell button adhering to the bottom of the centrifuge tube) and subjected to an additional wash/centrifugation cycle. The resulting cells are impermeable to 14C-maltose (the 3OMG-to-maltose space ratio of RBC ghosts is 100:1) (15).
Erythrocyte membrane glucose gradient in vitro methods.
Erythrocyte membrane glucose gradient in vitro methods were developed from those used for zero-trans glucose influx kinetics (10,14,16,17). 3OMG, which undergoes phosphorylation several orders of magnitude more slowly than glucose and is virtually nonmetabolizable (18–20), has been used extensively in glucose transport studies (10). Aliquots of erythrocytes (100 μl), prepared as above, were suspended in 200 μl 14C-labeled (0.5 μCi/ml final) and unlabeled 3OMG, yielding a final concentration of 10 mmol/l for single Ci-to-Co ratio determinations, or 0.3–20 mmol/l to measure Ci-to-Co ratio versus 3OMG. Incubations were stopped by the addition of ice-cold stop solution consisting of 50 μmol/l phloretin and 100 μmol/l mercuric chloride in PBS at various time points. Zero time values were obtained by the addition of stop solution to the RBC suspension before addition of 14C-3OMG. Cells were centrifuged at 14,000 rpm for 30 s, and 100-μl aliquots of the supernatant were removed to sample extracellular counts. The RBC pellet was washed twice with stop solution on ice and then lysed with 0.5 ml 3% perchloric acid. After centrifugation at 14,000 rpm for 10 min at room temperature, 200-μl aliquots of the perchloric acid extract were collected to sample intracellular counts. After scintillation counting, the concentration of 14C-3OMG (in counts per minute per milliliter) outside the cell (Co) and inside the cell (Ci) was then calculated. Ci was determined as counts per minute per milliliter cell water. Assuming identical internal- and external-specific activities, the ratio counts of Ci to Co would then be equivalent to the ratio of inside to outside concentrations of unlabeled compound.
An alternative indirect method was developed for comparison of Ci to Co for 14C-3OMG and 14C-urea (final unlabeled urea, 2 mmol/l). Tracer was added to RBC suspensions adjusted to 15 g/dl hemoglobin (45% hematocrit) and incubated to steady state. After centrifugation, aliquots of supernatant and 14C-tracer stock solution were counted for radioactivity. From the supernatant and stock solution counts, the hematocrit, and the cell water fraction estimated from MCHC, the concentration of counts in cell water Ci and the corresponding Ci-to-Co ratio were calculated.
Results are reported as the mean of triplicate measurements. In representative experiments, there was no change in the hematocrit from the beginning to the end of the incubation, despite mild visible evidence of hemolysis (<0.6% by absorbance; no difference in hemolysis between 3OMG and urea incubations).
Calculation of cell water.
The cell water was calculated for each sample from the MCHC determined on a complete blood count sent simultaneously to the clinical laboratory according to the following equation (21):
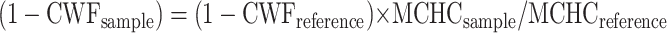 |
where cell water fraction (CWF)reference was measured as 0.669 (volume of solvent water per volume of cells) on a sample of normal cells with measured MCHCreference = 33.5 g/dl. In preliminary experiments, CWF was determined experimentally on several blood samples and was comparable with that calculated by MCHC.
Analytic methods.
A1C was measured using HPLC ion exchange (Tosoh, Tokyo, Japan) (interassay coefficient of variation [CV] 2.9% at A1C 5.5% and 1.9% at A1C 10.0%). Fructosamine was measured at Quest Diagnostics by autoanalyzer (Roche Diagnostics) with a nitroblue tetrazolium reaction, interassay CV 2% (4).
Statistical analysis.
The GG was computed based on a reference regression line relating A1C and fructosamine as previously reported (4). All data are reported as means ± SD, unless otherwise indicated. We used Pearson's correlation coefficients and linear regression to evaluate relationships between variables. Analyses were conducted using SPSS version 15.0 (SPSS, Chicago, IL) and JMP (SAS Institute, Cary, NC).
RESULTS
Evidence for a glucose concentration gradient in human erythrocytes under conditions of steady state.
Human erythrocytes incubated with a series of concentrations of 3OMG rapidly reached a steady state within 5 min at 37°C (Fig. 1A). This was stable well beyond the 60-min time point chosen for ongoing comparative analysis (data not shown). However, 14C-3OMG inside (Ci) and outside (Co) the cell did not equalize, and the Ci-to-Co ratio at steady state at 37°C departed significantly from unity. Ci-to-Co increased with increasing 3OMG (Fig. 1B), but the glucose gradient expressed as the difference between inside and outside was actually higher at higher concentrations. Ci-to-Co measured at physiological concentrations of 10 mmol/l 3OMG was <1 in 25 of 26 subjects, with a broad distribution of values (0.89 ± 0.07) (Fig. 1C). For determinations of Ci-to-Co on fresh samples assayed over a 12-day period, within subject CV ranged from 5 to 7%. There was no correlation of Ci-to-Co with mean corpuscular volume (MCV) or MCHC.
FIG. 1.

A: Time course of 14C-3OMG uptake into glucose-depleted human erythrocytes at 37°C at a series of initial external sugar concentrations. Values shown are mean of triplicate incubations. B: Dependence of the 3OMG gradient on external 3OMG. The concentration gradient decreases (i.e., Ci-to-Co approaches 1) with increasing 3OMG across a concentration range broader than the physiologic (n = 7; different symbols and lines denote individual subjects). C: Distribution of Ci-to-Co determined at steady state in human subjects (Table 1).
Comparison of 3OMG and urea gradients.
To test whether the transmembrane asymmetry would also be observed with a distinct small molecule thought to fill the RBC water space, parallel incubations were conducted simultaneously with 14C-3OMG and 14C-urea, each in quadruplicate (Fig. 2). Since rapid efflux of urea from RBC precluded washing of cells, Ci for both compounds in these studies was calculated from measured counts in the 14C-labeled stock solution added to the suspension and in the supernatant representing the extracellular space (see research design and methods). A wide distribution and departure from unity of 3OMG Ci-to-Co ratios is apparent, confirming the previous observation (Fig. 1C). In contrast, urea Ci-to-Co was tightly clustered at 1.015 ± 0.022. The calculations of Ci for both 3OMG and urea used the same estimates of cell volume, MCHC, and cell water; therefore, the Ci-to-Co ratio of unity for urea validates these estimates. In eight subjects, Ci-to-Co ratios were assessed using both direct and indirect measures for 14C-3OMG, along with simultaneous measurement of the 14C-urea concentration ratio (indirect) on the same blood samples. The results were significantly lower (P < 0.0001 each; paired t test) for 14C-3OMG with both the direct (0.93 ± 0.02) and indirect methods (0.86 ± 0.05) compared with 14C-urea (1.04 ± 0.04). Ci-to-Co ratios for 14C-glucose, 14C-galactose, and 14C-2-deoxy-glucose were similar to those with 14C-3OMG (data not shown), providing confirmation among a series of sugars transported by GLUT1. These results are consistent with previous findings that the “nonsolvent” water compartment in red cells is very small, i.e., all the water of RBCs is available as solute for small molecules such as urea. Bookchin et al. found similar results for glucose (22). Thus, Ci-to-Co ratios <1 are not due to variations or error in estimation of water content of the cells.
FIG. 2.

Paired values of the Ci-to-Co ratio determined for 3OMG vs. urea within subject are different (P < 0.0001). The Ci-to-Co ratio approximates unity and has a narrow distribution for urea in all subjects but is below unity and more heterogeneous in the same population for 3OMG.
Transmembrane gradient is maintained in RBC ghosts.
Some studies in the literature suggest that the high concentration of hemoglobin may “compartmentalize” glucose, generating an apparent 3OMG gradient (23). To test this possibility, Ci-to-Co determinations were conducted in “pink” and “white” ghosts of red cells. Pink ghosts represent the resealed product of lysing red cells in a 10-fold excess of 5 mmol/l sodium phosphate hypotonic buffer, reducing the MCHC from 33.3 to 1.6–2.6 g/dl; white ghosts result from conducting analogous lysis in a 50-fold excess of hypotonic solution, further reducing MCHC to 0.011–0.037 g/dl. These procedures raise the CWF from ∼0.675 to 0.973 and 0.99, respectively. Preparation of each ghost form did not result in disappearance of the Ci-to-Co gradient (both Ci and Co measured directly using stop solution). In fact, the Ci-to-Co gradient became more pronounced (Fig. 3A and B). This excludes hemoglobin as a source of the Ci-to-Co gradient.
FIG. 3.

3OMG (3-O-methyl-glucose) Ci-to-Co dependence on external 3OMG in pink (PG) (A) and white (WG) (B) erythrocyte ghosts compared with intact RBCs (S1–S5 denote different human subjects). The 3OMG concentration gradient was not reduced, and in fact was more pronounced as hemoglobin decreased. A is representative of 17 experiments in eight subjects, and B is representative of one experiment in each of three subjects.
Effect of perturbing energy status on observed gradient.
The Ci-to-Co gradient was not dependent on the normal cellular Na/K gradient. Cells incubated in either isotonic NaCl or isotonic KCl had identical Ci-to-Co gradients (Fig. 4A). The Ci-to-Co gradient was also not dependent on ATP. Figure 4B demonstrates that Ci-to-Co was the same in ghosts made with and without the ATP-destroying enzyme apyrase, which is present during resealing.
FIG. 4.

A: Effect of transmembrane cation gradient on the 3OMG Ci-to-Co dependence on external 3OMG. The dependence of 3OMG Ci-to-Co on 3OMG was compared in extracellular medium containing 137 mmol/l KCl vs. the standard PBS containing 139 mmol/l NaCl. The use of KCl buffer failed to overcome the gradient. The data are representative of one experiment from each of three subjects. B: ATP effects. Preparations of pink ghosts (PG) sealed with the addition of the enzyme apyrase to deplete intracellular ATP failed to shift Ci-to-Co toward unity. However, the preparation of ghosts reduced the 3OMG dependence of the Ci-to-Co ratio and was further reduced by apyrase. The data are representative of one experiment in each of four subjects.
Testing for an “impurity” in the labeled 3OMG.
Quality control analysis on the 14C-3OMG provided by the manufacturer using HPLC indicated a homogeneous species. It could be hypothesized that another molecular species that fails to cross the red cell membrane, e.g., an enantiomer, might not be detected by that technique, giving rise to an artifactual 3OMG Ci/Co gradient (24,25). We chose a functional approach to test for such an impurity in the tracer. If a nontransportable impurity were responsible for the Ci-to-Co gradient, incubation with RBCs would result in uptake solely of transportable “pure” tracer. Incubation of those cells with fresh tracer-free medium would then elute the tracer from the cells and result in an “impurity-depleted” eluate. Subsequent incubation of fresh RBCs with this impurity-depleted eluate should therefore eliminate the Ci-to-Co gradient, if the impurity were the source of the apparent gradient. Therefore, a standard zero-trans experiment was performed with and without the following modification: the supernatant from the initial incubation for 1 h was removed by centrifugation and replaced with 200 μl fresh tracer-free medium for 60 min at 37°C. This medium (termed the “eluate”) was then separated from the cells by centrifugation, and 200 μl was added to a fresh suspension of 100 μl RBCs in 10 mmol/l 3OMG. Similar Ci-to-Co gradients were observed with medium containing fresh unmodified tracer (0.92 ± 0.01) and with the eluate (0.91 ± 0.04, n = 11, P = NS) whose tracer had to have been able to enter and then exit from cells to which it had previously been exposed. This indicates that a nontransportable impurity is unlikely to account for the deviation of the Ci-to-Co ratio from 1.0.
Effect of reversal of initial glucose gradient.
The previous experiments represented influx of glucose into the cell down an outside-to-inside concentration gradient. We attempted to reverse this gradient by first equilibrating cells in 14C-3OMG with 0.2 mmol/l 3OMG and then expanding the extracellular space with an equal volume of sugar-free buffer (which thereby dilutes the extracellular sugar concentration from 0.2 to 0.1 mmol/l) to examine the steady-state gradient achieved when flux is in the opposite direction. The lower concentration of 3OMG was used to allow for studying the effects of cytochalasin B, a well-documented inhibitor of GLUT1-mediated glucose transport (26,27). Despite reversing the direction of the initial gradient, the final steady-state Ci-to-Co ratio was not significantly altered (Fig. 5) and there was no effect of incubation with cytochalasin B (data not shown). This finding is striking, in that cellular glucose concentration falls below extracellular glucose concentration. We do not have a mechanistic explanation for this phenomenon, given the independence of Ci-to-Co ratios from ATP and cation gradients. However, this finding suggests that the Ci-to-Co gradient would persist in vivo, tracking changes in plasma glucose concentrations whether rising or falling (see discussion).
FIG. 5.

Membrane gradient reversal. In this experiment, RBCs were first incubated with 3OMG until an apparent steady-state outside-to-inside gradient was reached. Next, the samples (arrow) were diluted twofold to expand the extracellular volume. If the observed gradient was due to a failure to reach true equilibrium, then the gradient would be expected to reverse after dilution. However, after dilution, there was a prompt reestablishment of essentially the same steady-state outside-to-inside gradient, indicating that the sugar moved up a concentration gradient. The data are representative of one experiment in each of 15 subjects.
Relationship of A1C and GG with Ci-to-Co ratio.
Ci-to-Co ratio, which varied among individuals, correlated positively and significantly with both A1C and GG (Fig. 6). Higher A1C levels were associated with higher Ci-to-Co ratios. Likewise, individuals with higher Ci-to-Co ratios had higher GG values, i.e., when the intra-erythrocyte concentration of sugar was higher relative to extracellular sugar (represented by higher Ci-to-Co ratios), so was the glycation of the intracellular protein hemoglobin in comparison with the glycation of extracellular proteins (reflected in higher GG). This is consistent with variation in the Ci-to-Co ratio among individuals contributing to A1C levels and to the discordance between A1C and glycemic control. In contrast, there was no correlation of either Ci-to-Co or GG with serum fructosamine (Table 2), indicating that Ci-to-Co and GG were not influenced by glycemic control per se. In statistical terms, the GG is the residual of the regression of A1C on fructosamine. It should be randomly distributed with respect to fructosamine, hence, as observed, not correlated with fructosamine. On the other hand, GG would be expected to correlate with A1C because a high GG suggests that the A1C is higher than would be predicted and a low GG suggests that the measured A1C is lower than predicted from fructosamine. The correlation of Ci-to-Co with GG is consistent with the GG being determined in part by the Ci-to-Co ratio.
FIG. 6.

A1C (A) and GG (B) rise as the 14C-3OMG Ci-to-Co ratio increases. The GG is a measure of variance in A1C relative to glycated serum proteins. Across the population, intracellular sugar rises relative to extracellular, as does hemoglobin glycation relative to the glycation of extracellular proteins. Values of r2 are shown to demonstrate the fraction of the variance in A1C and GG accounted for by Ci-to-Co ratio. In contrast (Table 2), fructosamine has no significant slope relative to Ci-to-Co ratio.
TABLE 2.
Correlation coefficients (r) and P values between A1C, fructosamine, GG, and Ci-to-Co ratio*
| r | P | |
|---|---|---|
| A1C vs. fructosamine | 0.77 | <0.001 |
| A1C vs. GG | 0.76 | <0.001 |
| GG vs. fructosamine | 0.17 | 0.40 |
| Ci-to-Co vs. A1C* | 0.44 | 0.025 |
| Ci-to-Co vs. GG | 0.45 | 0.023 |
| Ci-to-Co vs. fructosamine | 0.23 | 0.26 |
Ratio of sugar concentration inside to outside measured as 14C-3OMG CPM inside vs. outside (see Fig. 6 legend).
DISCUSSION
The key findings in this study are that 1) at steady state, there is a sugar concentration gradient across the human erythrocyte membrane that varies between individuals; 2) the gradient is not dependent on glucose metabolism, hemoglobin concentration, ATP, or the transmembrane cation gradient; 3) the glucose gradient correlates with A1C but is not related to a non-RBC glycated protein measure of glycemic control, suggesting that it reflects an influence on A1C unrelated to plasma glucose. Taken together, these data are consistent with the hypothesis that variability in the intracellular (erythrocyte glucose) relative to extracellular glucose significantly contributes to interindividual variation in A1C. This has direct implications for the mechanisms determining A1C and its clinical interpretation as a measure of glycemic control and risk prediction.
Although use of a nonphysiologic sugar to exclude the effects of glucose metabolism and defined media are potential limitations to generalizability in vivo, results were similar with physiologic sugars. We eliminated a smaller distribution space for glucose in the RBC than the cell water volume estimated from the MCHC as a potential explanation of a steady-state transmembrane glucose gradient. Cell water volume estimated from MCHC and validated by urea Ci-to-Co = 1.0, is consistent with the long-standing observation that virtually all water in RBCs was available for solvation of small molecules (28,29). More recently, Bookchin et al. (22) showed that even the water contained in the highly structured dense protein domain of intracellular polymerized hemoglobin S was accessible to small molecules, including glucose. Furthermore, the Ci-to-Co gradient did not depend on high intracellular hemoglobin concentrations. The presence of intracellular membrane vesicles could conceivably represent a compartment inaccessible to glucose. Such endocytic vesicles have been demonstrated in sickle RBCs but not in normal RBCs (30). Thus, there is no evidence in RBCs for a substantial water compartment inaccessible to glucose that could explain Ci-to-Co ratios <1.
Another potential explanation of the failure of glucose concentrations to equilibrate across the membrane is that kinetic characteristics of the glucose transport system limits transport before equilibration occurs, i.e., glucose influx is shut down before equal concentrations are reached. However, the reestablishment of Ci-to-Co when external glucose concentrations were diluted (Fig. 5) is inconsistent with this mechanism. This experiment in fact suggests a third possibility: a mechanism for net export of glucose from the cell. There has been no evidence to date for active transport of glucose in or out of the RBC, and the Ci-to-Co gradient was not dependent on ATP or cation gradients that might serve as energy sources; there is not an obvious alternative candidate in the artificial media used for these 3OMG uptake experiments. Other molecules share the GLUT1 transporter (31,32), but competition is unlikely in the artificial media used, especially given the wide concentration range over which the sugar Ci-to-Co gradient is preserved (Fig. 1B). Nevertheless, there are a number of aspects of the glucose transport system that suggest more complex characteristics and regulation than that of a simple “facilitated diffusion” mechanism. Red cell sugar transport displays kinetic asymmetry in which Vmax and Km for sugar exit are greater than Vmax and Km for sugar entry (16,17,33–35). This does not result in net glucose export when intracellular sugar = extracellular sugar because the ratio of Vmax to Km for net exit and entry are identical (36). Nevertheless, the transient fall in 3OMG concentration below external concentrations has been seen in rapid counter-flow studies, and the rapid 3OMG exit observed in human RBCs and ATP-containing RBC ghosts (A.C. and J.M. Leitch, unpublished observations) supports this alternative possibility that RBCs contain a second glucose transport system that actively exports sugars (37). The identity of the putative glucose exporter is not known (37), and the energy source driving glucose exit is unclear.
As has recently been indicated, the GG (or the analogous hemoglobin glycosylation index) (1,38–40) is not independent of A1C (41,42). It does however allow quantitation of sources of variation in A1C (43). In this construct, in which the GG is defined as the difference between measured and predicted A1C, the Ci-to-Co ratio is linked not only to A1C but also with the fraction of the variance in A1C that is not shared in common with the integrated extracellular measure of glycemic control, fructosamine.
Whatever the molecular mechanism involved in establishing the glucose gradient of RBCs, the observed variation among individuals in Ci-to-Co ratios from ∼0.7 to 1.0, could lead to substantial differences in hemoglobin glycation for a given plasma glucose, with significant clinical implications. Critical clinical decisions are made based on A1C in the 6–8% range (44–48). The difference between one individual with a Ci-to-Co of 0.75 and another with Ci-to-Co of 0.99 could be an ∼25% difference in the level of hemoglobin glycation for a given mean plasma glucose, which translates to a difference of 1.5–2.0 A1C percentage points. We speculate that the interindividual variation in Ci-to-Co may contribute to the observed genetic variation in the incidence of diabetes complications. It will be important to understand whether the phenomenon we are describing in RBCs extrapolates to other cell types, notably endothelial cells. If so, then the Ci-to-Co glucose gradient may have implications not only for A1C, but also for those intracellular glucose concentrations that affect the rate of progression of diabetes complications themselves.
In conclusion, we have identified a variable in the RBC—the trans-membrane glucose gradient—that is a strong candidate to introduce interindividual variability into the rate of A1C formation. Recognizing this variable has important implications for clinical interpretation of this widely used test. Understanding the underlying mechanism may have fundamental implications for interindividual variation in the frequency of diabetes complications in relation to apparent glycemic control.
Acknowledgments
These studies were supported by a grant from the Central Ohio Diabetes Association (not affiliated with the American Diabetes Association), National Institutes of Health Grants PHS RO1 DK-63088 and PHS RR-08084 (to the Cincinnati Children's Hospital, University of Cincinnati General Clinical Research Center), and the Cincinnati Veterans Affairs Medical Center.
Parts of this study were presented in abstract form at the 61st, 62nd, and 64th annual meetings of the American Diabetes Association, Philadelphia, Pennsylvania, 22–26 June 2001, San Francisco, California, 14–18 June 2002, and Orlando, Florida, 4–8 June 2004, respectively.
We acknowledge helpful discussions with Drs. John C. Winkelmann, Steven D. Chernausek, James A. Fagin, and David D'Alessio.
Published ahead of print at http://diabetes.diabetesjournals.org on 30 June 2008.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Hempe JM, Gomez R, McCarterRJ, Jr, Chalew SA: High and low hemoglobin glycation phenotypes in type 1 diabetes: a challenge for interpretation of glycemic control. J Diabetes Complications 16 :313 –320,2002 [DOI] [PubMed] [Google Scholar]
- 2.Yudkin JS, Forrest RD, Jackson CA, Ryle AJ, Davie S, Gould BJ: Unexplained variability of glycated haemoglobin in non-diabetic subjects not related to glycaemia. Diabetologia 33 :208 –215,1990 [DOI] [PubMed] [Google Scholar]
- 3.Gould BJ, Davie SJ, Yudkin JS: Investigation of the mechanism underlying the variability of glycated haemoglobin in non-diabetic subjects not related to glycaemia. Clin Chim Acta 260 :49 –64,1997 [DOI] [PubMed] [Google Scholar]
- 4.Cohen RM, Holmes YR, Chenier TC, Joiner CH: Discordance between A1C and fructosamine: evidence for a glycosylation gap and its relation to diabetic nephropathy. Diabetes Care 26 :163 –167,2003 [DOI] [PubMed] [Google Scholar]
- 5.Cohen RM, LeCaire TJ, Lindsell CJ, Smith EP, D'Alessio DJ: The relationship of prospective glycated hemoglobin to glycated serum proteins in incident diabetic retinopathy: implications of the glycation gap for mechanism of risk prediction. Diabetes Care 31 :151 –153,2007 [DOI] [PubMed] [Google Scholar]
- 6.Snieder H, Sawtell PA, Ross L, Walker J, Spector TD, Leslie RD: HbA1c levels are genetically determined even in type 1 diabetes: evidence from healthy and diabetic twins. Diabetes 50 :2858 –2863,2001 [DOI] [PubMed] [Google Scholar]
- 7.Cohen RM, Snieder H, Lindsell CJ, Beyan H, Hawa MI, Blinko S, Edwards R, Spector TD, Leslie RD: Evidence for independent heritability of the glycation gap (glycosylation gap) fraction of A1C in nondiabetic twins. Diabetes Care 29 :1739 –1743,2006 [DOI] [PubMed] [Google Scholar]
- 8.Higgins PJ, Garlick RL, Bunn HF: Glycosylated hemoglobin in human and animal red cells: role of glucose permeability. Diabetes 31 :743 –748,1982 [DOI] [PubMed] [Google Scholar]
- 9.Cohen RM, Ciraolo P, Palascak MB, Lindsell CJ, Khera PK, Smith EP, Joiner CH, Franco RS: Red blood cell (RBC) survival differences among hematologically normal people with diabetes (DM) make a clinically important difference in A1C (Abstract). Diabetes 56 (Suppl. 1):A116 ,2007 [Google Scholar]
- 10.Carruthers A: Facilitated diffusion of glucose. Physiol Rev 70 :1135 –1176,1990 [DOI] [PubMed] [Google Scholar]
- 11.Somogyi M: The distribution of sugar in blood. J Biol Chem 117 –127,1928
- 12.Chang H, Ewert SM, Bookchin RM, Nagel RL: Comparative evaluation of fifteen anti-sickling agents. Blood 61 :693 –704,1983 [PubMed] [Google Scholar]
- 13.Blodgett DM, Carruthers A: Quench-flow analysis reveals multiple phases of GluT1-mediated sugar transport. Biochemistry 44 :2650 –2660,2005 [DOI] [PubMed] [Google Scholar]
- 14.Klepper J, Garcia-Alvarez M, O'Driscoll KR, Parides MK, Wang D, Ho YY, De Vivo DC: Erythrocyte 3-O-methyl-D-glucose uptake assay for diagnosis of glucose-transporter-protein syndrome. J Clin Lab Anal 13 :116 –121,1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamill S, Cloherty EK, Carruthers A: The human erythrocyte sugar transporter presents two sugar import sites. Biochemistry 38 :16974 –16983,1999 [DOI] [PubMed] [Google Scholar]
- 16.Heard KS, Fidyk N, Carruthers A: ATP-dependent substrate occlusion by the human erythrocyte sugar transporter. Biochemistry 39 :3005 –3014,2000 [DOI] [PubMed] [Google Scholar]
- 17.Carruthers A: ATP regulation of the human red cell sugar transporter. J Biol Chem 261 :11028 –11037,1986 [PubMed] [Google Scholar]
- 18.Cortes S, Gromova M, Evrard A, Roby C, Heyraud A, Rolin DB, Raymond P, Brouquisse RM: In plants, 3-o-methylglucose is phosphorylated by hexokinase but not perceived as a sugar. Plant Physiol 131 :824 –837,2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malaisse-Lagae F, Giroix MH, Sener A, Malaisse WJ: Phosphorylation of 3-O-methyl-D-glucose by yeast and beef hexokinase. FEBS Lett 198 :292 –294,1986 [DOI] [PubMed] [Google Scholar]
- 20.Gatley SJ, Holden JE, Halama JR, DeGrado TR, Bernstein DR, Ng CK: Phosphorylation of glucose analog 3-O-methyl-D-glucose by rat heart. Biochem Biophys Res Commun 119 :1008 –1014,1984 [DOI] [PubMed] [Google Scholar]
- 21.Joiner CH, Lauf PK: Modulation of ouabain binding and potassium pump fluxes by cellular sodium and potassium in human and sheep erythrocytes. J Physiol 283 :177 –196,1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bookchin RM, Balazs T, Lew VL: Measurement of the hemoglobin concentration in deoxyhemoglobin S polymers and characterization of the polymer water compartment. J Mol Biol 244 :100 –109,1994 [DOI] [PubMed] [Google Scholar]
- 23.Levine KB, Robichaud TK, Hamill S, Sultzman LA, Carruthers A: Properties of the human erythrocyte glucose transport protein are determined by cellular context. Biochemistry 44 :5606 –5616,2005 [DOI] [PubMed] [Google Scholar]
- 24.Rechthand E, Smith QR, Rapoport SI: Facilitated transport of glucose from blood into peripheral nerve. J Neurochem 45 :957 –964,1985 [DOI] [PubMed] [Google Scholar]
- 25.Talley CP, Clayborn H, Jewel E, McCarty R, Gold PE: Vagotomy attenuates effects of L-glucose but not of D-glucose on spontaneous alternation performance. Physiol Behav 77 :243 –249,2002 [DOI] [PubMed] [Google Scholar]
- 26.Cloherty EK, Heard KS, Carruthers A: Human erythrocyte sugar transport is incompatible with available carrier models. Biochemistry 35 :10411 –10421,1996 [DOI] [PubMed] [Google Scholar]
- 27.Cloherty EK, Sultzman LA, Zottola RJ, Carruthers A: Net sugar transport is a multistep process: evidence for cytosolic sugar binding sites in erythrocytes. Biochemistry 34 :15395 –15406,1995 [DOI] [PubMed] [Google Scholar]
- 28.Macleod J, Ponder E: Solvent water in the mammalian erythrocyte. J Physiol 86 :147 –152,1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cook JS: Nonsolvent water in human erythrocytes. J Gen Physiol 50 :1311 –1325,1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lew VL, Hockaday A, Sepulveda MI, Somlyo AP, Somlyo AV, Ortiz OE, Bookchin RM: Compartmentalization of sickle-cell calcium in endocytic inside-out vesicles. Nature 315 :586 –589,1985 [DOI] [PubMed] [Google Scholar]
- 31.May JM: Ascorbate function and metabolism in the human erythrocyte. Front Biosci 3 :d1 –d10,1998 [DOI] [PubMed] [Google Scholar]
- 32.Vera JC, Rivas CI, Fischbarg J, Golde DW: Mammalian facilitative hexose transporters mediate the transport of dehydroascorbic acid. Nature 364 :79 –82,1993 [DOI] [PubMed] [Google Scholar]
- 33.Carruthers A: Anomalous asymmetric kinetics of human red cell hexose transfer: role of cytosolic adenosine 5′-triphosphate. Biochemistry 25 :3592 –3602,1986 [DOI] [PubMed] [Google Scholar]
- 34.Leitch JM, Carruthers A: ATP-dependent sugar transport complexity in human erythrocytes Am J Physiol Cell Physiol 292 :C974 –C986,2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aubby DS, Widdas WF: Asymmetry of hexose transfer system in erythrocytes of fetal and new-born guinea-pigs. J Physiol 309 :317 –327,1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stein WD: In Transport and Diffusion Across Cell Membranes. New York, Academic Press,1986. , p.231 –305
- 37.Concha II, Velasquez FV, Martinez JM, Angulo C, Droppelmann A, Reyes AM, Slebe JC, Vera JC, Golde DW: Human erythrocytes express GLUT5 and transport fructose. Blood 89 :4190 –4195,1997 [PubMed] [Google Scholar]
- 38.McCarter RJ, Hempe JM, Gomez R, Chalew SA: Biological variation in A1C predicts risk of retinopathy and nephropathy in type 1 diabetes. Diabetes Care 27 :1259 –1264,2004 [DOI] [PubMed] [Google Scholar]
- 39.Chalew SA, McCarter RJ, Thomas J, Thomson JL, Hempe JM: A comparison of the glycosylation gap and hemoglobin glycation index in patients with diabetes. J Diabetes Complications 19 :218 –222,2005 [DOI] [PubMed] [Google Scholar]
- 40.McCarter RJ, Hempe JM, Chalew SA: Mean blood glucose and biological variation have greater influence on A1C levels than glucose instability: an analysis of data from the Diabetes Control and Complications Trial. Diabetes Care 29 :352 –355,2006 [DOI] [PubMed] [Google Scholar]
- 41.Lachin JM, Genuth S, Nathan DM, Rutledge BN: The hemoglobin glycation index is not an independent predictor of the risk of microvascular complications in the Diabetes Control and Complications Trial. Diabetes 56 :1913 –1921,2007 [DOI] [PubMed] [Google Scholar]
- 42.Genuth S, Lachin JM, Nathan DM: Biological variation in A1C predicts risk of retinopathy and nephropathy in type 1 diabetes: response to McCarter et al (Letter). Diabetes Care 28 :233 –235,2005 [DOI] [PubMed] [Google Scholar]
- 43.Cohen RM: A1C: does one size fit all? Diabetes Care 30 :2756 –2758,2007 [DOI] [PubMed] [Google Scholar]
- 44.Goodall I: A1C standardisation destination–global IFCC Standardisation. How, why, where and when–a tortuous pathway from kit manufacturers, via inter-laboratory lyophilized and whole blood comparisons to designated national comparison schemes. Clin Biochem Rev 26 :5 –19,2005 [PMC free article] [PubMed] [Google Scholar]
- 45.Goldstein DE, Little RR, Lorenz RA, Malone JI, Nathan D, Peterson CM, Sacks DB: Tests of glycemia in diabetes. Diabetes Care 27 :1761 –1773,2004 [DOI] [PubMed] [Google Scholar]
- 46.Nathan DM, Singer DE, Hurxthal K, Goodson JD: The clinical information value of the glycosylated hemoglobin assay. N Engl J Med 310 :341 –346,1984 [DOI] [PubMed] [Google Scholar]
- 47.Rohlfing CL, Wiedmeyer HM, Little RR, England JD, Tennill A, Goldstein DE: Defining the relationship between plasma glucose and HbA1c: analysis of glucose profiles and HbA1c in the Diabetes Control and Complications Trial. Diabetes Care 25 :275 –278,2002 [DOI] [PubMed] [Google Scholar]
- 48.The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus: the Diabetes Control and Complications Trial Research Group. N Engl J Med 329 :977 –986,1993 [DOI] [PubMed] [Google Scholar]