

Abstract
The importance of global influenza surveillance using simple and rapid diagnostics has been frequently highlighted. For influenza type B, the need exists for discrimination between the two currently circulating major lineages, represented by virus strains B/Victoria/2/87 and B/Yamagata/16/88, as only one of these lineages is represented in seasonal influenza vaccines. Here, the development and characterization of a low-density DNA microarray (designated BChip) designed to detect and identify the two influenza B lineages is presented. The assay involved multiplex nucleic acid amplification and microarray hybridization of viral RNA. Detection and lineage identification was achieved in less than 8 hours. In a study of 62 influenza B virus samples from 19 countries, dating from 1945 to 2005, as well as 5 negative control samples, the assay exhibited 97% sensitivity and 100% specificity. Furthermore, application of a trained artificial neural network to the pattern of relative fluorescence signals resulted in correct lineage assignment for 94% of 50 applicable influenza B viruses, with no false assignments.
Keywords: influenza B, DNA microarray, influenza diagnostic, virus lineage, Yamagata lineage, Victoria lineage
Introduction
Influenza viruses are segmented, negative-sense, RNA viruses that exhibit considerable antigenic diversity. Of the three existing types, influenza A, B, and C, which are distinguished by serological responses to their internal proteins, only types A and B have significant potential to cause severe disease and recurrent annual epidemics in humans.1, 2 Although the influenza B virus is often associated with limited outbreaks of relatively mild disease, it may occasionally cause severe epidemics of considerable morbidity and mortality. In the last decade, B viruses have tended to be prominent and sometimes even dominant every 2–3 years.3, 4
While the natural hosts for influenza A viruses are aquatic birds, with various mammals including humans also being infected, influenza B viruses are almost entirely restricted to humans. Relative to influenza A viruses, B viruses do not show the same degree of antigenically distinct subtypes. However, like influenza A, influenza B viruses are subject to antigenic drift through the accumulation of point mutations, with a slightly lower evolutionary rate than type A.5 Since the early 1980’s, two distinct antigenic and evolutionary lineages of influenza B have co-circulated in humans.5 These lineages are antigenically related to the prototype strains B/Victoria/2/87 (Vic87) and B/Yamagata/16/88 (Yam88). With the continued evolution of co-circulating strains and multiple genotypes of influenza B viruses, the issues associated with viral reassortment have become a greater concern.6, 7
During the 1990s, Vic87-like viruses were isolated infrequently and were limited almost entirely to eastern Asia8 until they reappeared in North America and Europe in 2001.9 Although not considered subtypes, Yam88 and Vic87-like viruses are antigenically different, producing little or no post-infection cross-neutralizing antibody response in ferrets.10 In immunologically unprimed children, vaccination with a Yam88-like strain did not induce detectable hemagglutination inhibiting or neutralizing antibody to Vic87-like viruses.11 This lack of antigenic cross-reactivity has made the designation of a type B vaccine strain problematic, since current influenza vaccines are formulated to include only a single strain of influenza B. After the lineage split occurred in the early 1980’s, influenza B vaccine strains have changed with increasing frequency between Yam88-like and Vic87-like viruses, as shown graphically in Figure 1. Continued surveillance of influenza type B viruses is critical in order to ensure that future vaccines contain the most appropriate strain of virus.10
Figure 1.
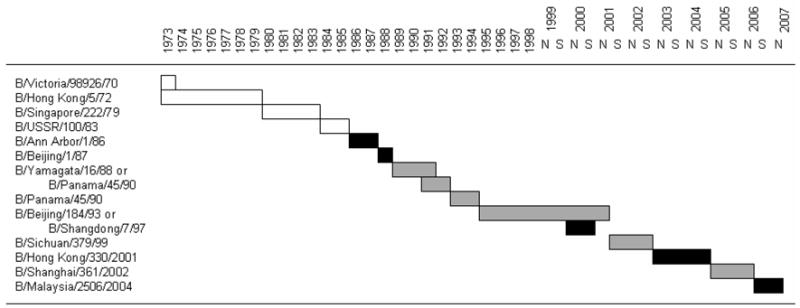
Changes in the influenza type B vaccine strain recommended by the WHO during 1973–2007. Since 1999 there have been two recommendations per year, one for the northern hemisphere (N) and the other for the southern hemisphere (S). White bars indicate virus strains before the lineage split occurred; grey bars represent Yam88-like and black bars Vic87-like strains.
A number of diagnostic methods are available for the detection of influenza viruses. Virus culture is still considered to be the “gold standard”, but is highly time-consuming (7–14 days) and has a low sample throughput even in light of recent rapid culture methods.12 Although a number of point-of-care rapid diagnostic tests are also available, many do not even detect influenza B.13, 14 Other available methods are based on either amplification of viral nucleic acid (RNA) utilizing real-time reverse-transcription polymerase chain reaction.15–18 serological diagnosis, such as hemagglutination inhibition (HI), enzyme immunoassays, complement fixation, and neutralization tests.14, 19 However, fewer tests are available for lineage determination of influenza viruses, the two most common methods being HI assay (antigenic characterization) and sequencing (phylogenetic characterization) after culture, both of which are time-consuming.
A number of microarray-based methods for influenza detection have been reported,20–22 most of which can detect influenza B virus but do not provide lineage information. There is only one study by Ivshina et al.23 that reported a microarray method for mapping genomic segments of influenza B virus strains. They demonstrated the microarray’s ability to distinguish between the two influenza B reference strains B/Beijing/184/93 (Yam88-like) and B/Shangdong/7/97 (Vic87-like) and reassortants of these two viruses. However, the gene-mapping approach uses capture probes of high specificity to each virus, and, therefore, is of limited utility as a diagnostic tool to be applied to a wide range of viruses.
Recently, we reported the development of DNA microarrays for detection and identification of influenza A viruses.24–26 Here we report the development a microarray (BChip) that was designed to target the influenza B gene segments HA, NA, and M and provide lineage information regarding the two currently circulating lineages, Yam88 and Vic87. The study presented herein included 62 different influenza B virus samples, as well as negative control samples of influenza A and parainfluenza 1.
Materials and Methods
Assay design
To detect unlabeled viral RNA from influenza virus samples a 2-step sandwich process was used (Figure 2a). This 2-step sandwich process involves the hybridization of non-labeled viral RNA to a DNA capture sequence on a microarray and the visualization of the bound RNA by a second hybridization step using a specific 5′-Quasar Q570 dye containing DNA label sequence. For reasons described in reference27 the capture and the label sequence were designed to hybridize in close proximity (separated by a single nucleotide gap) on the viral RNA to achieve a positive hybridization signal, as poor or no hybridization signals are obtained when capture and label sequence hybridize to positions far from one another on the viral RNA.
Figure 2.
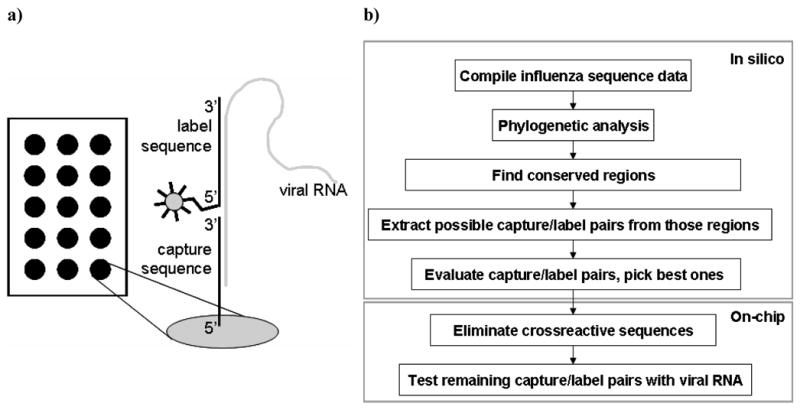
(a) Scheme of the 2-step hybridization assay. The non-labeled viral RNA hybridizes to a DNA capture sequence on a microarray and the binding event is visualized by a second hybridization step using a specific 5′-Quasar Q570 dye containing DNA label sequence. (b) Scheme of the sequence selection process.
Capture/label sequence selection
The method of capture/label sequence selection for the influenza microarray was described in detail in Mehlmann et al.24 A schematic of the sequence selection process is shown in figure 2b. Briefly, gene-specific databases of each of the three gene segments HA, NA, and M of influenza B were compiled using the Los Alamos National Laboratories (LANL) influenza database (http://www.flu.lanl.gov) and the Centers for Disease Control and Prevention influenza databases. The HA database was limited to influenza B viruses from the years 2000–2005. Following phylogenetic analysis of each of the three databases, conserved regions were identified for sub-portions of the database, allowing discrimination between the different lineages. Capture/label sequences were obtained from conserved regions of at least 45 nt in length; both capture and label sequences were between 16 and 25 nt in length, and the capture/label pair was designed to be separated by a single nucleotide gap when hybridized to the viral RNA.27 Capture/label pairs were designed to be employed in a two-step hybridization method. The surface-bound “capture” sequences (Operon Biotechnologies, Inc., Huntsville, AL) were used to capture amplified viral RNA and the 5′-Quasar 570-modified “label” sequences (Biosearch Technologies, Inc., Novato, CA) served as the fluorescence probe. Possible cross-reactive capture/label pairs (i.e., capture sequences that hybridized to label sequences resulted in a false positive signal) were identified experimentally and were excluded from the final microarray layout. A positive control (PC) capture sequence and a fluorescence-labeled complementary label sequence were added to the set of capture/label pairs for use as an internal control for hybridization efficiency. All sequences are available for research purposes upon request from the Technology Transfer Office at the University of Colorado.
Microarray layout
As shown in Figure 3A, the influenza B microarray contained 36 capture probes spotted in triplicate, as well as a positive control (PC) sequence that also served as a position marker. Two identical microarrays were printed on each slide. The 5′-amino-C6-modified capture probes were spotted onto aldehyde-modified glass microscope slides VALS-25 (CEL Associates, Inc., Pearland, TX) under optimized conditions described in Dawson et al.28 using a Genetix OmniGrid microarray spotter (Genetix, Boston, MA).
Figure 3.
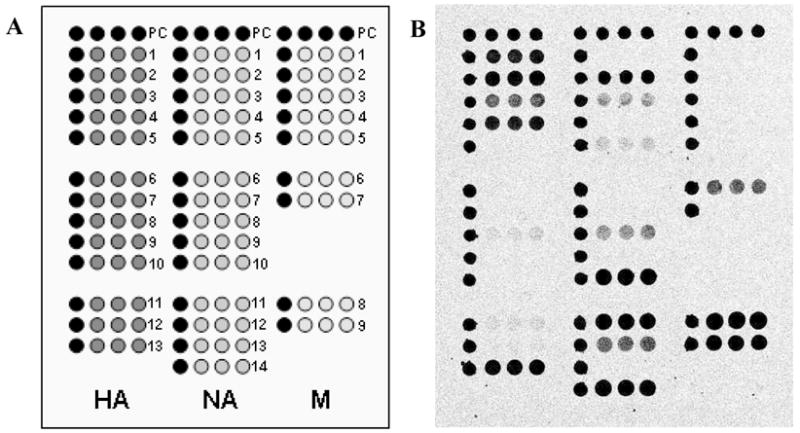
Microarray layout of the BChip (a) and example image (b) of virus sample B/Fujian/437/2004. Black symbols represent the positive control sequences. Each capture sequence was spotted in triplicate. For the fluorescence images, darker shades represent higher fluorescence.
Virus samples
Influenza virus samples were provided by the Centers for Disease Control and Prevention (CDC), Atlanta. A human parainfluenza virus sample was provided by the Colorado Department of Health and Environment. All samples were viral isolates, propagated either in embryonated eggs or in MDCK cell cultures.29 Virus type and lineage was determined by hemagglutination inhibition (HI) assay at the CDC.
RNA isolation and amplification
RNA was selected as the target on the microarray in anticipation that improvements in sample handling and sensitivity may allow the direct capture of viral RNA in the future. Viral RNA was extracted from influenza virus samples using either the MagNA Pure LC system (Roche, Indianapolis, IN) or the RNeasy kit (Qiagen, Valencia, CA). Extracted RNA was stored at −80°C until further use. Reverse-transcription polymerase chain reaction (RT-PCR), followed by run-off transcription, was employed to amplify extracted viral RNA. The RT step was performed using SuperScript II Reverse Transcriptase (Invitrogen Corp., Carlsbad, CA) and SZA+/SZB+ universal influenza primers as described by Zou et al.30 Subsequently, the HA, NA, and M gene segment were amplified in a multiplex PCR step using Taq Polymerase (Invitrogen Corp., Carlsbad, CA) and gene-specific primers (HA forward: ATC CAC AAA ATG AAG GCA; NA forward AGC AGA AGC AGA GCA TCT TCT CAA; HA/NA reverse: AGT AGT AAC AAG AGC ATT TTT C; M forward: AGC AGA AGC ACG CAC TTT C; M reverse: AAA CAA CGC ACT TTT TCC). PCR amplification was confirmed by identifying DNA of appropriate length on a 1 % agarose gel (35 min at 100 V) stained with ethidium bromide. PCR reverse primers contained a T7 promoter site that allowed subsequent runoff transcription using T7 RNA polymerase (Invitrogen Corp., Carlsbad, CA). Transcribed RNA was kept at −20°C for immediate use and at −80°C for long-term storage.
RNA fragmentation and hybridization
Transcribed RNA was fragmented prior to microarray hybridization as described by Mehlmann et al..31 Fragmented RNA was mixed with label sequences and hybridized to the microarray as described by Townsend et al..25 Fluorescence read-out was conducted with a VersArray ChipReader (Bio-Rad, Hercules, CA) using the 532 nm excitation channel, a laser power of 60 %, a PMT sensitivity of 700 V, and 5 μm resolution. The resulting images were processed with VersArray Analyzer software (Bio-Rad, Hercules, CA). Images shown in this study were contrast-enhanced for improved visualization.
Data analysis
For each capture probe, background-corrected mean intensity values and signal-to-noise (S/N) ratios (mean net intensity/standard deviation of background) were obtained. The test for influenza B was considered positive when the S/N values were >10 for at least one capture probe. For artificial neural network-based lineage analysis, in order to minimize the influence of slide-to-slide variations, relative intensity values were calculated for the 13 HA capture probes, assigning 100 % to the highest mean HA spot intensity on each image.
Influenza B lineage discrimination using an artificial neural network (ANN)
The commercially available software package EasyNN-Plus 7.0c (Neural Planner Software, Cheshire, England) was used to develop the ANN model, using a feed-forward method with weighted back-propagation. The ANN utilized 14 input nodes (13 relative intensities and the highest mean intensity of HA capture probes), a hidden layer with 8 nodes, and 2 output nodes (“Yam88” and “Vic87” with values of 1 or 0, designating true or false, respectively).
Initial type and lineage assignments were conducted on blinded samples by visual inspection of the microarray images, with excellent results. However, the purpose of the ANN was to automate the process and remove any user subjectivity. Of the 62 influenza B viruses initially processed, it was necessary to exclude 12 from the neural network analysis. Specifically, 7 older influenza B viruses that originated in years before the lineage split occurred, and 5 influenza B viruses that had S/N<10 for HA sequences were excluded from the ANN analysis. Thus, 50 influenza B viruses, some of which were processed in duplicate, as well as some negative controls, were used in combination with the ANN. Two separate experiments were conducted in order to test all of the virus samples. In one experiment half of the over 60 images were randomly selected and used to train and validate the ANN, the other half were then tested as “unknowns”. In the second experiment the two sets of data were reversed (i.e., the previous training/validation set was treated as unknowns). Learning rate and momentum were both optimized by the software. The learning process was stopped after 101 cycles when the average target error was below 0.005. The average training error was found to be ~1.5 × 10−4. A minimum output value of 0.9 was set as a threshold for a positive assignment as either Yam88 or Vic87.
Results and Discussion
Figure 3 is a graphical representation of BChip microarray layout. The microarray contained three sections: in the left section are 13 capture sequences, each in triplicate, that target different regions of the HA gene segment of influenza B viruses; the middle section contains 14 NA capture sequences; and sequences in the right section target the M gene segment. Positive control (PC) sequences serve both as position markers and to ensure that the hybridization step worked properly. Briefly, the assay involves extraction of viral RNA, nucleic acid amplification through RT-PCR followed by run-off transcription, and finally fragmentation and hybridization of amplified viral RNA to the microarray.
As can be observed in Figure 3, all three sections of the microarray exhibited fluorescence signals for B/Fujian/437/2004, indicating successful multiplex RT-PCR amplification and surface-capture for all 3 gene segments. Note that the relative fluorescence intensities of the different capture sequences vary, and that, as designed, not all capture sequences show hits with this particular sample. The presence or absence of signal for specific capture sequences was used for lineage discrimination.
Detection of influenza type B viruses
A total of 65 samples were analyzed on the BChip, including 62 different influenza B positive samples that originated from locations worldwide and covered the years 1945 to 2005. Additionally, two samples of influenza A, representing the two subtypes currently circulating in humans (H3N2 and H1N1), and one sample of human parainfluenza virus type 1, a common virus causing influenza-like illness, served as negative control samples.
A summary of microarray and the artificial neural network results is given in Table 1. The decision whether a sample tested positive or negative was based on the highest S/N value of all capture probe signals on the microarray. A threshold of S/N > 10 for a positive test was used. As can be seen in Table 1, in some cases the signals were below the threshold. The source of assay failure was identified by agarose gel analysis as failed amplification of one or more gene segments during multiplex RT-PCR. In the event that at least one gene segment was detected, the sample was considered positive for influenza type B. Overall, for detection of influenza B virus BChip assay resulted in a clinically defined sensitivity of 97 % and a specificity of 100 % for the detection of influenza B (although it should be noted that the samples were viral isolates and not clinical samples).
Table 1.
BChip and Neural Network Results.
| Sample information
|
S/N values
|
ANN output
|
||||||
|---|---|---|---|---|---|---|---|---|
| ID | HA lineage | HA | NA | M | pos/neg | Yam88 | Vic87 | |
| B/Baker/45 | NA | 1.6 | 2.2 | 2.1 | − | |||
| B/Muelder/45 | NA | 0.8 | 25.6 | 6.9 | + | |||
| B/Peacock/45 | NA | 1.3 | 2.2 | 1.7 | − | |||
| B/Colorado/1/65 | NA | 1.0 | 296.0 | 80.6 | + | |||
| B/Michigan/1/66 | NA | 3.9 | 452.5 | 159.1 | + | |||
| B/Ann Arbor/2/74 | NA | 73.2 | 465.0 | 123.6 | + | |||
| B/Ann Arbor/1/76 | NA | 348.8 | 705.1 | 154.5 | + | |||
| B/Panama/45/90 | Yam88 | 51.0 | 70.5 | 27.5 | + | 1.00 | 0.00 | + |
| B/Argentina/218/97 | Yam88 | 425.4 | 436.2 | 335.5 | + | 1.00 | 0.00 | + |
| B/Paris/459/99 | Yam88 | 889.9 | 744.7 | 557.4 | + | 1.00 | 0.00 | + |
| B/Johannesburg/5/99 | Yam88 | 328.4 | 231.0 | 79.8 | + | 0.99 | 0.01 | + |
| B/Hawaii/2/2000 | Yam88 | 13.6 | 45.4 | 5.7 | + | 0.99 | 0.01 | + |
| B/Moscow/4/2000 | Yam88 | 148.0 | 427.4 | 82.6 | + | 0.99 | 0.01 | + |
| B/Guangdong/120/2000 | Yam88 | 60.4 | 377.5 | 65.5 | + | 0.98 | 0.02 | + |
| B/Guangdong/299/2001 | Yam88 | 157.2 | 152.5 | 55.0 | + | 1.00 | 0.00 | + |
| B/Bucharest/676/2001 | Yam88 | 404.9 | 532.4 | 26.2 | + | 1.00 | 0.00 | + |
| B/Sichuan/34/2001 | Vic87 | 65.2 | 385.2 | 45.9 | + | 0.05 | 0.95 | + |
| B/Minnesota/14/2001 | Yam88 | 2.6 | 54.8 | 13.8 | + | LS | ||
| B/Taiwan/1484/2001 | Vic87 | 4.0 | 7.8 | 32.0 | + | LS | ||
| B/Wuhan/359/2001 | Yam88 | 73.8 | 90.0 | 8.7 | + | 1.00 | 0.00 | + |
| B/Chile/5068/2001 | Yam88 | 161.7 | 72.1 | 18.0 | + | 1.00 | 0.00 | + |
| B/Hawaii/35/2001 | Vic87 | 37.5 | 95.0 | 21.1 | + | 0.06 | 0.93 | + |
| B/Singapore/67204/2001 | Yam88 | 154.7 | 78.0 | 18.8 | + | 1.00 | 0.00 | + |
| B/Philippines/70299/2001 | Yam88 | 13.4 | 11.9 | 1.7 | + | 1.00 | 0.00 | + |
| B/Mississippi/3/2001 | Yam88 | 61.2 | 93.0 | 25.3 | + | 1.00 | 0.00 | + |
| B/Texas/11/2001 | Yam88 | 495.4 | 264.3 | 75.7 | + | 1.00 | 0.00 | + |
| B/Thailand/80835/2001 | Yam88 | 105.1 | 177.4 | 52.5 | + | 0.97 | 0.03 | + |
| B/Mexico/418/2001 | Yam88 | 340.4 | 133.4 | 28.1 | + | 1.00 | 0.00 | + |
| B/Oman/16304/2001 | Yam88 | 25.4 | 146.9 | 12.4 | + | 0.95 | 0.04 | + |
| B/India/7600/2001 | Vic87 | 80.2 | 224.8 | 71.1 | + | 0.00 | 1.00 | + |
| B/Brisbane/32/2002 | Vic87 | 111.9 | 334.4 | 345.0 | + | 0.00 | 1.00 | + |
| B/China/118180/2002 | Yam88 | 3.1 | 36.2 | 21.9 | + | LS | ||
| B/China/109892/2002 | Yam88 | 111.6 | 492.1 | 481.7 | + | 1.00 | 0.00 | + |
| B/Egypt/2267/2002 | Vic87 | 118.0 | 660.1 | 368.7 | + | 0.18 | 0.82 | NA |
| B/Taiwan/143999/2002 | Yam88 | 60.3 | 344.6 | 116.2 | + | 0.92 | 0.07 | + |
| B/South Carolina/3/2003 | Vic87 | 18.7 | 395.3 | 37.2 | + | 0.86 | 0.12 | NA |
| B/Hong Kong/553/2003 | Vic87 | 7.0 | 527.9 | 84.0 | + | LS | ||
| B/South Carolina/4/2003 | Vic87 | 313.8 | 1375.9 | 532.7 | + | 0.00 | 1.00 | + |
| B/Washington/3/2003 | Yam88 | 173.0 | 356.7 | 138.7 | + | 1.00 | 0.00 | + |
| B/Fujian/437/2004 | Yam88 | 61.2 | 220.3 | 66.4 | + | 1.00 | 0.00 | + |
| B/Hong Kong/310/2004 | Vic87 | 46.7 | 185.4 | 69.9 | + | 0.40 | 0.61 | NA |
| B/HongKong/64/2004 | Yam88 | 4.3 | 23.0 | 20.7 | + | LS | ||
| B/HongKong/64/2004* | Yam88 | 14.8 | 23.5 | 26.6 | + | 1.00 | 0.00 | + |
| B/Shizuoka/02/2004 | Yam88 | 41.4 | 223.0 | 112.6 | + | 0.98 | 0.02 | + |
| B/Shizuoka/02/2004* | Yam88 | 178.4 | 522.0 | 647.7 | + | 0.99 | 0.01 | + |
| B/Egypt/2040/2004 | Yam88 | 536.0 | 1074.2 | 39.6 | + | 1.00 | 0.00 | + |
| B/Hawaii/10/2004 | Vic87 | 52.9 | 143.0 | 15.8 | + | 0.01 | 0.99 | + |
| B/Florida/7/2004 | Yam88 | 414.9 | 275.9 | 119.2 | + | 1.00 | 0.00 | + |
| B/Hawaii/33/2004 | Vic87 | 126.9 | 265.3 | 215.6 | + | 0.03 | 0.97 | + |
| B/Colorado/13/2004 | Yam88 | 91.0 | 75.8 | 41.4 | + | 1.00 | 0.00 | + |
| B/Malaysia/2506/2004 | Vic87 | 100.7 | 168.3 | 148.9 | + | 0.00 | 1.00 | + |
| B/Kansas/01/2005 | Yam88 | 163.1 | 227.0 | 56.8 | + | 1.00 | 0.00 | + |
| B/Kansas/01/2005* | Yam88 | 105.3 | 130.1 | 83.3 | + | 1.00 | 0.00 | + |
| B/Kansas/01/2005* | Yam88 | 134.1 | 76.6 | 27.6 | + | 1.00 | 0.00 | + |
| B/Kentucky/04/2005 | Yam88 | 46.1 | 180.5 | 27.5 | + | 1.00 | 0.00 | + |
| B/Kentucky/04/2005* | Yam88 | 101.0 | 82.6 | 10.0 | + | 1.00 | 0.00 | + |
| B/Mexico/19/2005 | Yam88 | 90.9 | 285.7 | 31.4 | + | 1.00 | 0.00 | + |
| B/Mexico/19/2005* | Yam88 | 110.7 | 70.2 | 9.8 | + | 1.00 | 0.00 | + |
| B/Texas/10/2005 | Yam88 | 61.0 | 76.1 | 34.1 | + | 1.00 | 0.00 | + |
| B/Texas/10/2005* | Yam88 | 166.1 | 308.2 | 86.4 | + | 1.00 | 0.00 | + |
| B/Alaska/06/2005 | Yam88 | 65.8 | 91.1 | 34.4 | + | 1.00 | 0.00 | + |
| B/Alaska/06/2005* | Yam88 | 392.5 | 381.3 | 100.0 | + | 1.00 | 0.00 | + |
| B/Brazil/136/2005 | Vic87 | 6.7 | 32.2 | 18.2 | + | LS | ||
| B/Brazil/136/2005* | Vic87 | 21.1 | 62.0 | 51.1 | + | 0.00 | 1.00 | + |
| B/Brazil/136/2005* | Vic87 | 50.5 | 144.3 | 62.0 | + | 0.00 | 1.00 | + |
| B/Illinois/36/2005 | Vic87 | 66.6 | 410.2 | 98.6 | + | 0.00 | 1.00 | + |
| B/Illinois/36/2005* | Vic87 | 184.7 | 463.0 | 89.8 | + | 0.00 | 1.00 | + |
| B/Georgia/02/2005 | Vic87 | 26.1 | 177.2 | 71.2 | + | 0.00 | 1.00 | + |
| B/Georgia/02/2005* | Vic87 | 53.4 | 138.6 | 41.8 | + | 0.00 | 1.00 | + |
| B/North Carolina/01/2005 | Yam88 | 94.1 | 226.4 | 81.5 | + | 1.00 | 0.00 | + |
| B/North Carolina/01/2005* | Yam88 | 461.6 | 303.9 | 138.6 | + | 1.00 | 0.00 | + |
| B/Mississippi/4/2005 | Yam88 | 230.0 | 238.7 | 63.0 | + | 1.00 | 0.00 | + |
| B/Mississippi/4/2005* | Yam88 | 131.1 | 127.2 | 71.8 | + | 1.00 | 0.00 | + |
| B/Ohio/1/2005 | Vic87 | 70.8 | 110.7 | 70.6 | + | 0.01 | 0.99 | + |
| B/Illinois/47/2005 | Vic87 | 186.8 | 233.1 | 189.5 | + | 0.08 | 0.92 | + |
| B/Utah/1/2005 | Yam88 | 1.7 | 185.7 | 68.5 | + | LS | ||
| Parainfluenza | 2.5 | 3.0 | 1.6 | − | ||||
| A/H3N2 | 0.7 | 1.2 | 1.5 | − | ||||
| A/H3N2 ** | 1.2 | 2.4 | 1.7 | − | ||||
| A/H1N1 | 0.3 | 1.0 | 0.5 | − | ||||
| A/H1N1 ** | 1.8 | 4.3 | 1.5 | − | ||||
NA = not assigned; LS = low signal in HA region, therefore not used for ANN analysis;
= duplicate experiment to examine slide-to-slide variations;
HA, NA, and M gene segments were amplified by RT-PCR using primers specific for influenza A. Numbers in
Out of the 62 influenza B viruses tested, only two samples, both dating from 1945, were not detected on the microarray. These two samples also showed no signal when tested by gel electrophoresis, indicating that multiplex RT-PCR amplification had failed for all three gene segments. The experiment was repeated using RT-PCR amplification of only the HA gene segment, and both samples showed positive signal on the microarray (data not shown). The fact that the BChip assay detected viruses over 60 years old is noteworthy, considering that selection of capture/label sequences for the HA segment as well as primer design was based on an influenza HA gene database containing viruses from the years 2000–2005. This suggested that the sequences, which were chosen because of their high degree of conservation, might have extended usefulness even as the influenza B virus continues to evolve.
All negative control experiments using influenza A, parainfluenza, and multiple negative controls without RNA template (data not shown) were correctly identified as negative for influenza B, i.e. the method produced no false positive results. In an additional negative control experiment, RT-PCR was performed on two influenza A samples utilizing HA, NA, and M gene primers specific for influenza A. The amplified influenza A viral RNA was analyzed on the microarray and resulted in a negative for influenza B. The lack of cross-reactivity between influenza A viral RNA and influenza B capture sequences is encouraging for future influenza diagnostics as it would be necessary for any combined test for influenza of both types, A and B.
Lineage determination of influenza B
In addition to the detection of influenza B with high accuracy, the BChip was designed to discriminate between the two currently circulating lineages of influenza B (Yam88-like and Vic87-like viruses). In order to do so, the relative fluorescence signal intensity pattern of the HA section of the microarray was utilized. The HA capture/label pair sequences were derived from conserved regions of the B/HA gene segment that were either specific for Yam88-like viruses, specific for Vic87-like viruses, or broadly reactive for all influenza B viruses. Therefore, differences in the relative signal pattern were expected to occur, allowing for lineage identification of the sample tested.
Figure 4 shows the HA region of representative microarray images for Yam88-like and Vic87-like viruses. Visual inspection revealed a distinct difference in relative signal patterns between the lineages. Comparing these HA patterns, it can be seen that capture sequences HA-1, 2, and 3 exhibited strong signals for Yam88-like viruses (keep in mind that the top row is the positive control), while capture sequences HA-6 and 9, while weaker, were indicative of Vic87-like viruses. The HA-4, 8, and 13 sequences were broadly reactive and consistently exhibited medium to strong signals with nearly all influenza B samples. Other capture sequences, e.g. HA-11 and 12, were occasionally detected with varying intensities. Although lineage discrimination by pattern recognition could easily be accomplished by a trained eye, a more quantitative approach was developed in order to avoid user subjectivity. An artificial neural network (ANN) was trained and evaluated for lineage discrimination using on the HA portion of the microarray.
Figure 4.
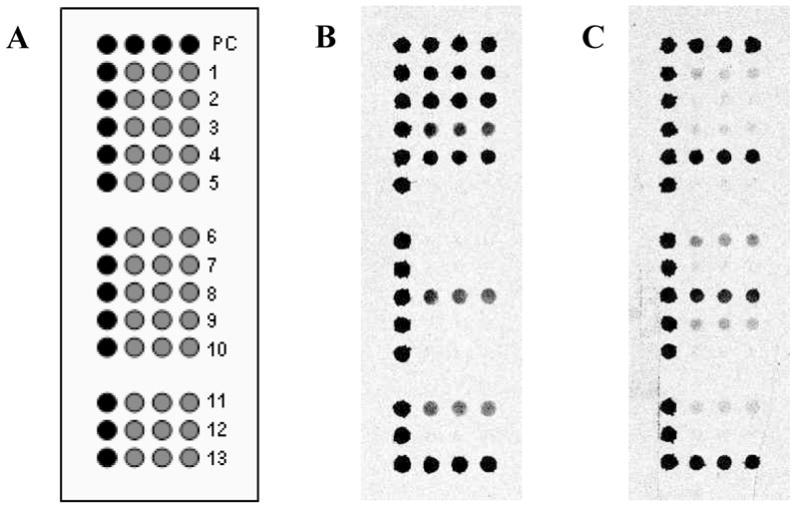
Discrimination between the two major influenza B virus lineages, Yam88 and Vic87, using the HA section of the BChip. (a) Microarray layout of the HA section; (b) sample B/Johannesburg/5/99 (Yam88-like); (c) sample B/South Carolina/4/2003 (Vic87-like). For the fluorescence images, darker shades represent higher fluorescence.
Of the 62 influenza B viruses used in this study, several were excluded from the neural network analysis. Specifically, 7 influenza B viruses that were collected in years before the lineage split occurred were not included. In addition, 5 influenza B viruses that resulted in a S/N value less than 10 for HA sequences were not considered for lineage assignment. Thus, 50 influenza B viruses were used in combination with the ANN. As there was no preexisting dataset to be used to train the ANN, half of the current dataset was used for training and the other half for querying and vice versa.
Normalized relative signal intensities were used as input values in order to eliminate slide-to-slide variations in absolute fluorescence intensity. Output values ranged between 0 and 1, corresponding to false and true, respectively, for the two distinct categories (i.e., Yam88 and Vic87). An output neuron was considered to be fully activated at values at or above 0.9 when using a logistic function, 32 and therefore an output value of 0.9 was used as the cutoff for positive assignments.
The ANN output values are summarized in Table 1. Of all examples (50 influenza B viruses) entered into the ANN, 94 % were identified correctly as Yam88-like or Vic87-like viruses, and only 3 cases resulted in no assignment. Interestingly, the only samples in which the ANN failed to yield a correct assignment were Vic87-like viruses. In the present case, the entire dataset consisted of 43 Yam88-like and only 19 Vic87-like examples; thus, the ANN may not have been sufficiently trained for Vic87-like viruses. Although the virus samples originating from years before the lineage split were not considered for lineage determination through ANN analysis, it is interesting to note that they did exhibit a distinct Yam88-like pattern in the HA region. Further exploration of the precursor viruses may be useful in understanding how influenza B evolved overtime and how lineage differentiation occurred.
While, this study served as proof-of-principle for rapid lineage discrimination, a logical next step is to explore the use of the NA gene for discriminating between different NA lineages. Although almost all of the currently circulating influenza B viruses contain Yam88-like NA gene segments (most Vic87-like viruses isolated since 2001 have been found to be reassortants having a Vic87 lineage HA and a Yam88 lineage NA gene segment9, 33), there remains the possibility that reassortment would allow a Vic87-like NA lineage to re-appear. On the other hand, with the HA lineage being the most important determinant of the virus’ antigenic behavior, a minimization of the microarray to only HA sequences poses a convenient alternative, gaining the benefits of a singleplex RT-PCR while retaining the possibility of HA lineage determination. This concept might prove advantageous for combining influenza A and B on a single microarray.
Conclusions
The detection and lineage determination of influenza B viruses using a DNA microarray-based diagnostic method was demonstrated. The BChip technology delivered results in less than 8 hours with a sensitivity of 97 % and a specificity of 100 % for the detection of influenza B. Additionally, a correct lineage assignment to either Vic87 or Yam88 was made for 47 out of 50 influenza B positive samples (94 %) using an artificial neural network. Since the BChip assay can be completed in less than 8 hours, it represents an attractive alternative method to current technologies that can require up to several days for determining lineage.
Acknowledgments
We gratefully acknowledge funding from NIH/NIAID (U01AI056528). We also thank Patricia Young and the Colorado Department of Public Health and Environment for providing samples of human parainfluenza virus type 1. The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention.
Abbreviations
- ANN
artificial neural network
- HA
hemagglutinin (gene segment)
- HI
hemagglutination inhibition (assay)
- M
matrix protein (gene segment)
- NA
neuraminidase (gene segment)
- RT-PCR
reverse transcription polymerase chain reaction
- S/N
signal-to-noise ratio
- Vic87
B/Victoria/2/87
- Yam88
B/Yamagata/16/88
References
- 1.Zambon MC. Journal of Antimicrobial Chemotherapy. 1999;44:3–9. doi: 10.1093/jac/44.suppl_2.3. [DOI] [PubMed] [Google Scholar]
- 2.Palese P, Young JF. Science. 1982;215:1468–1474. doi: 10.1126/science.7038875. [DOI] [PubMed] [Google Scholar]
- 3.Hay AJ, Gregory V, Douglas AR, Lin YP. Philosophical Transactions of the Royal Society of London, Series B: Biological Sciences. 2001;356:1861–1870. doi: 10.1098/rstb.2001.0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin YP, Gregory V, Bennett M, Hay A. Virus Research. 2004;103:47–52. doi: 10.1016/j.virusres.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 5.Rota PA, Wallis TR, Harmon MW, Rota JS, Kendal AP, Nerome K. Virology. 1990;175:59–68. doi: 10.1016/0042-6822(90)90186-u. [DOI] [PubMed] [Google Scholar]
- 6.McCullers JA, Saito T, Iverson AR. Journal of Virology. 2004;78:12817–12828. doi: 10.1128/JVI.78.23.12817-12828.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nerome K, Nerome R, Lindstrom SE, Hiromoto Y, Sugita S. Recent Advances in Influenza Virus Research. 2002:169–185. [Google Scholar]
- 8.Xu X, Shaw J, Smith CB, Cox NJ, Klimov AI. 2001:383–387. [Google Scholar]
- 9.Shaw MW, Xu X, Li Y, Normand S, Ueki RT, Kunimoto GY, Hall H, Klimov A, Cox NJ, Subbarao K. Virology. 2002;303:1–8. doi: 10.1006/viro.2002.1719. [DOI] [PubMed] [Google Scholar]
- 10.Rota PA, Hemphill ML, Whistler T, Regnery HL, Kendal AP. Journal of General Virology. 1992;73:2737–2742. doi: 10.1099/0022-1317-73-10-2737. [DOI] [PubMed] [Google Scholar]
- 11.Levandowski RA, Regnery HL, Staton E, Burgess BG, Williams MS, Groothuis JR. Pediatrics. 1991;88:1031–1036. [PubMed] [Google Scholar]
- 12.Ziegler T, Hall H, Sanchez-Fauquier A, Gamble WC, Cox NJ. Journal of clinical microbiology. 1995;33:318–321. doi: 10.1128/jcm.33.2.318-321.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nicholson KG, Wood JM, Zambon M. Lancet. 2003;362:1733–1745. doi: 10.1016/S0140-6736(03)14854-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cox NJ, Subbarao K. Lancet. 1999;354:1277–1282. doi: 10.1016/S0140-6736(99)01241-6. [DOI] [PubMed] [Google Scholar]
- 15.Daum LT, Canas LC, Schadler CA, Ujimori VAH, Huff WB, Barnes WJ, Lohman KL. Journal of Clinical Virology. 2002;25:345–350. doi: 10.1016/s1386-6532(02)00043-4. [DOI] [PubMed] [Google Scholar]
- 16.Lugovtsev VY, Vodeiko GM, Strupczewski CM, Levandowski RA. Journal of Virological Methods. 2005;124:203–210. doi: 10.1016/j.jviromet.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 17.Mackay IM, Arden KE, Nitsche A. Nucleic Acids Research. 2002;30:1292–1305. doi: 10.1093/nar/30.6.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ward CL, Dempsey MH, Ring CJA, Kempson RE, Zhang L, Gor D, Snowden BW, Tisdale M. Journal of Clinical Virology. 2004;29:179–188. doi: 10.1016/S1386-6532(03)00122-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellis JS, Zambon MC. Reviews in Medical Virology. 2002;12:375–389. doi: 10.1002/rmv.370. [DOI] [PubMed] [Google Scholar]
- 20.Li J, Chen S, Evans DH. Journal of Clinical Microbiology. 2001;39:696–704. doi: 10.1128/JCM.39.2.696-704.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sengupta S, Onodera K, Lai A, Melcher U. Journal of Clinical Microbiology. 2003;41:4542–4550. doi: 10.1128/JCM.41.10.4542-4550.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kessler N, Ferraris O, Palmer K, Marsh W, Steel A. Journal of Clinical Microbiology. 2004;42:2173–2185. doi: 10.1128/JCM.42.5.2173-2185.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ivshina AV, Vodeiko GM, Kuznetsov VA, Volokhov D, Taffs R, Chizhikov VI, Levandowski RA, Chumakov KM. Journal of Clinical Microbiology. 2004;42:5793–5801. doi: 10.1128/JCM.42.12.5793-5801.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mehlmann M, Dawson ED, Townsend MB, Smagala JA, Moore CL, Smith CB, Cox NJ, Kuchta RD, Rowlen KL. Journal of Clinical Microbiology. 2006;44:2857–2862. doi: 10.1128/JCM.00135-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Townsend MB, Dawson ED, Mehlmann M, Smagala JA, Dankbar DM, Moore CL, Smith CB, Cox NJ, Kuchta RD, Rowlen KL. Journal of Clinical Microbiology. 2006;44:2863–2871. doi: 10.1128/JCM.00134-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dawson ED, Moore CL, Smagala JA, Dankbar DM, Mehlmann M, Townsend MB, Smith CB, Cox NJ, Kuchta RD, Rowlen KL. Analytical Chemistry. 2006 doi: 10.1021/ac061739f. in press. [DOI] [PubMed] [Google Scholar]
- 27.Chandler DP, Newton GJ, Small JA, Daly DS. Applied and Environmental Microbiology. 2003;69:2950–2958. doi: 10.1128/AEM.69.5.2950-2958.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dawson ED, Reppert AE, Rowlen KL, Kuck LR. Analytical Biochemistry. 2005;341:352–360. doi: 10.1016/j.ab.2005.03.029. [DOI] [PubMed] [Google Scholar]
- 29.Kendal AP, Pereira MS, Skehel JJ. Department of Health and Human Services. Centers for Diseases Control; Washington, D. C.: 1982. [Google Scholar]
- 30.Zou S. Journal of Clinical Microbiology. 1997;35:2623–2627. doi: 10.1128/jcm.35.10.2623-2627.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mehlmann M, Townsend MB, Stears RL, Kuchta RD, Rowlen KL. Analytical Biochemistry. 2005;347:316–323. doi: 10.1016/j.ab.2005.09.036. [DOI] [PubMed] [Google Scholar]
- 32.Tan CNW. 1997 online: http://www.smartquant.com/references.php.
- 33.Puzelli S, Frezza F, Fabiani C, Campitelli L, Ansaldi F, Lin YP, Gregory V, Bennett M, D’Agaro P, Campello C, Crovari P, Hay A, Donatelli I. 2004:708–713. doi: 10.1002/jmv.20225. [DOI] [PubMed] [Google Scholar]