

Abstract
A family of heteroleptic RuII coordination complexes containing substituted 1,10-phenanthroline (phen) ligands with extended conjugation was found to exhibit two simultaneously emissive excited states at room temperature in fluid solution. These systems demonstrate a breakdown of the standard non-radiative decay pathways that normally lead to a single, dominant, lowest energy emissive excited state in RuII complexes and most other chromophores. The structural requirements for dual emission were explored through the synthesis and characterization of isomeric systems. Two features were found to be primarily responsible for resolvable dual emission. Extended conjugation at the 4-position of the 1,10-phenanthroline ligand was identified as an essential feature, and asymmetry in the phenanthroline ligand substitutions appears to greatly facilitate the production of these two non-equilibrated emissive states. Additional complexes were studied which displayed “tunable” emissive characteristics for the two excited states as a function of covalent and non-covalent modification.
Introduction
Luminescence-based sensors are important analytical tools. In addition to their historical significance, they have also gradually displaced radiologic methods, making them useful in both basic research laboratories and routine assays.1 Yet, the chromophores used in these applications have been, for the most part, minor modifications of dyes dated to the early years of the synthetic dye industry.2 Recent advances in molecular design promise significantly more sophisticated probes for powerful future applications. One theme will likely be the introduction of species that exhibit dual emission, where distinct optical signals reflect different states of the sensing moiety. In basic research, linking two chromophores within a protein or a supramolecular assembly permits exploration of geometric organization and energy or charge migration, and in essence represents an extreme case of a potential dual emitter. For diagnostic assays, the use of dual-emitters will permit ratiometric techniques that simplify standardization. A fundamental question that occurs to us is, how small can a molecule be and still display easily observed, long-lived dual emission at ambient conditions? Among candidate systems, charge transfer excited states in transition metal complexes, such as those of RuII, are attractive in being fairly bright and having microsecond lifetimes that are sufficiently long-lived to be perturbed by their microenvironment.3
To observe long-lived dual photoluminescence from an individual molecule, one must have two excited electronic states that (i) can both be populated by the same absorption event (although somewhat distinct excitation spectra will be useful in practical applications), (ii) are separated by an energy barrier that prevents interconversion of populations, and (iii) exhibit sufficiently different luminescence properties in energy and/or excited state lifetime to allow distinction in practical experiments. For proteins and large supramolecular assemblies, these requirements can be met by attaching distinct chromophores separated either by long distances or by rigidly defined angular relationships. In typical small molecules, however, excited states usually involve occupation of orbitals extending over the entire molecule and spaced at very modest energy separations, making it impossible to achieve requirement (ii) and sometimes difficult to accomplish (iii).
In retrospect, metal-to-ligand charge-transfer (MLCT) states in transition metal complexes might have been expected to constitute an exception. Picosecond studies established that many, perhaps most, MLCT excited states become essentially localized onto one ligand extremely rapidly, even when the ligands are identical, as in Ru(bpy)32+.4 Such studies did not, however, probe the issue important to us: do such states remain localized indefinitely or do they exhibit “exciton hopping” on a time scale faster than the luminescence lifetime? It is entirely possible that dual emission from non-equivalent MLCT states has been observed all along, but gone unrecognized because of a failure to meet condition (iii) in the most studied RuII complexes; but it is also possible that condition (ii) is rarely met. Certain low temperature experiments on RuII complexes have reported dual emission, where barrier crossing would be slowed.5–7
Recently, we reported the observation of dual emission from a family of dimetallic, alkyne-linked RuII complexes.8 These complexes exhibited two emission features that were well resolved both in energy and lifetime, and were the first systems described that possess two such luminescent states in fluid solution at room temperature. From this study, we concluded that appropriately substituted phen complexes can defy Kasha’s rule,9 and avoid standard non-radiative decay pathways. In an effort to better define the structural features that produce such anomalous, intriguing behavior, we have undertaken the synthesis and photophysical characterization of monometallic RuII complexes, containing alkyne-substituted phen ligands. These compounds offer a simpler model for multiple excited states, as there is a single metal center. In the study reported here, we have systematically varied the substitution position on the 1,10-phenanthroline (phen) ligand, and explored the effects of electronic manipulation of the appended conjugated moiety. We find that dual emission is exclusively limited to RuII complexes containing 4-substituted phen ligands, and is, in fact, a consistent feature for these compounds. The two emission features can be modulated by synthetic modification of the extended phen ligand. Non-covalent interactions can also be detected by the appropriately substituted system. The symmetry of the complex appears to play a role in the relaxation pathways that lead to the final emissive state(s). In short, we report dual emission in an extensive array of compounds and show it to be robust, tunable, and clearly correlated to specific structural features of one of the coordinating ligands.
Experimental Section
General
Reagents and solvents were purchased from Aldrich and Acros, and used as received. NMR spectra were obtained on a Varian Mercury 400 MHz spectrometer. The 1H chemical shifts are expressed relative to the residual solvent signal of acetonitrile at δ 1.93. The 13C chemical shifts are reported relative to CD3CN at δ 1.30. Absorption spectra were collected using a Hewlett-Packard 8452A diode array spectrophotometer. Emission spectra were recorded using a Perkin-Elmer LS 50B luminescence spectrophotometer, equipped with an optional, red-sensitive photomultiplier. Lifetime measurements used excitation by a pulsed dye laser at 455 nm pumped by an XeCl laser, at about one pulse per second. The emission wavelength was defined by a small monochromator (JY H20) along with low-emission glass filters. Luminescence was detected with a high-current photomultiplier tube (Amperex TUVP56). The signal was digitized using a LeCroy 9361 oscilloscope. No deconvolution was warranted, so decays were simply fit to a sum of two exponentials, using nonlinear least squares fitting based on the Marquardt algorithm.10 Crude time-resolved emission spectra were constructed point-by-point from the decomposition of the decay curves.
Synthesis
Compound 3 was synthesized as reported previously.11 For all other metal complexes, Sonogashira coupling reactions were carried out in the same manner. The RuII complex, containing a halo-substituted phen (50 mg for Br-phen complexes, 48 mg for Cl-phen complexes, 0.05 mmol), Pd(dppf)Cl2·CH2Cl2 (4 mg, 0.005 mmol) and CuI (1 mg, 0.005 mmol) were dissolved in a degassed solution of anhydrous DMF (5 mL) and triethylamine (2 mL) under argon. A solution of the substituted alkyne (0.5 mmol), dissolved in the same solvent mixture, was added via cannula, and the solution stirred at room temperature. When the reaction was complete the solvents were removed under reduced pressure, and the product purified by flash chromatography (silica gel, 0.1% saturated KNO3, 2% water in acetonitrile ramped to 5% water). The product fractions were combined and solvents removed under reduced pressure. A saturated aqueous solution of KPF6 was added, and the metal complex extracted into CH2Cl2. The solvents were removed under reduced pressure to give the product.
Compound 4
4 was obtained from (4-chlorophen)Ru(bpy)2·2PF6− in 85% yield after 1 hr. 1H NMR (CD3CN) δ 8.65 (dd, J=8.8, 2.9 Hz, 2H), 8.54–8.50 (m, 4H), 8.33 (d, J=9.2 Hz, 1H), 8.13–8.11 (m, 4H), 8.10–7.99 (m, 2H), 7.84–7.75 (m, 6H), 7.60 (d, J=4.76 Hz, 1H), 7.54–7.44 (m, 6H), 7.23–7.22 (m, 2H). 13C NMR (CD3CN) δ 157.79, 157.71, 157.50, 153.46, 152.57, 152.50, 152.46, 148.43, 148.19, 138.55, 138.43, 137.55, 132.91, 131,85, 131.16, 130.63, 129.62, 129.43, 128.24, 128.22, 128.08, 127.03, 127.01, 126.76, 124.92, 124.85, 121.75, 103.56, 84.35. UV (CH3CN) λmax nm (ε× 10−4) 286 (8.4), 324 (2.7), 460 (2.1). ESI-MS calcd for C40H28F6N6PRu [M]+ 839.1, found 838.9 [M]+.
Compound 5
5 was obtained from (5-chlorophen)Ru(bpy)2·2PF6− in 65% yield after 8 hr. 1H NMR (CD3CN) δ 8.99 (dd, J=8.4 1.1 Hz, 1H), 8.57–8.45 (m, 7H), 8.12 (dd, J=5.1, 1.1 Hz, 1H), 8.10–8.06 (m, 3H), 7.98 (t, J=6.6 Hz, 2H), 7.83–7.79 (m, 3H), 7.76–7.70 (m, 3H), 7.56 (d, J=5.1 Hz, 1H), 7.53 (d, J=5.1 Hz, 1H), 7.49–7.42 (m, 4H), 7.23–7.21 (m, 2H). 13C NMR (CD3CN) δ 157.87, 157.58, 153.64,153.57, 152.62, 148.33, 148.00, 138.56, 138.44, 137.17, 136.07, 132.62, 132.15, 131.43, 131.22, 130.55, 129.59, 128.24, 128.13, 127.20, 124.96, 124.88, 122.38, 122.33, 98.19, 84.75. UV (CH3CN) λmax nm (ε × 10−4) 284 (7.35), 332 (2.10), 450 (1.66). ESI-MS calcd for C40H28F6N6PRu [M]+ 839.1, found 838.9 [M]+.
Compound 6
6 was obtained from (4-chlorophen)Ru(bpy)2·2PF6− in 72% yield after 2 hr. 1H NMR (CD3CN) δ 8.64 (d, J=7.4 Hz, 2H), 8.55–8.48 (m, 4H), 8.32 (d, J=9.2 Hz, 1H), 8.12–8.08 (m, 3H), 8.03–7.97 (m, 3H), 7.84 (d, J=5.5 Hz, 2H), 7.72–7.71 (m, 4H), 7.59 (d, J=5.5 Hz, 1H), 7.54 (d, J=5.5 Hz, 1H), 7.47–7.43 (m, 2H), 7.25–7.20 (m, 2H), 7.07–7.04 (m, 2H), 3.86 (s, 3H). 13C NMR (CD3CN) δ 162.54, 158.22, 158.16, 157.95, 153.78, 152.97, 152.94, 152.91, 152.73, 148.76, 148.62, 138.89, 138.77, 137.89, 135.15, 132.19, 131.58, 131.45, 129.57, 128.55, 128.44, 128.41, 128.16, 127.29, 127.19, 125.25, 125.18, 115.64, 113.87, 104.86, 83.93, 56.32. UV (CH3CN) λmax nm (ε × 10−4) 286 ESI-MS calcd for C41H30F6N6PRu [M]+ 868.8, found 869.4 [M]+.
Compound 7
7 was obtained from (4-chlorophen)Ru(bpy)2·2PF6− in 68% yield after 4 hr. 1H NMR (CD3CN) δ 8.65 (d, J=7.8 Hz, 2H), 8.54 (d, J=8.1Hz, 2H), 8.51–8.48 (m, 2H), 8.35 (d, J=8.8 Hz, 1H), 8.13–8.08 (m, 4H), 8.03–7.94 (m, 4H), 7.87–7.81 (m, 5H), 7.79–7.58 (dd, J=8.1, 5.1 Hz, 1H), 7.59 (d, J=5.1 Hz, 1H), 7.53 (d, J=5.9 Hz, 1H), 7.48–7.44 (m, 2H), 7.25–7.20 (m, 2H). 13C NMR (CD3CN) δ 158.21, 158.10, 157.94, 153.94, 152.98, 152.92, 152.89, 148.92, 148.58, 138.97, 138.86, 137.97, 133.84, 132.25, 131.52, 130.19, 129.96, 128.94, 128.60, 128.48, 128.42, 127.43, 127.02, 126.81, 126.77, 126.73, 125.28, 125.20, 101.58, 86.44, 47.98. UV (CH3CN) λmax nm (ε × 10−4) 286 (8.7), 324 (3.1), 348 (1.7), 464 (2.1). ESI-MS calcd for C41H27F6N6PRu [M]+ 906.7, found 907.1 [M]+.
Compound 8
8 was obtained from (4-chlorophen)Ru(bpy)2·2PF6− in 65% yield after 4 hr. 1H NMR (CD3CN) δ9.23 (s, 1H), 9.04 (d, J=2.5 Hz, 1H), 8.76 (s, 1H), 8.64 (d, J=8.8 Hz, 1H), 8.57–8.49 (m, 5H), 8.37 (d, J=7.3 Hz, 1H), 8.27 (d, J=8.8 Hz, 1H), 8.13–8.09 (m, 4H), 8.04–7.99 (m, 4H), 7.98–7.85 (m, 3H), 7.74–7.63 (m, 4H), 7.49–7.45 (m, 2H), 7.30–7.22 (m, 2H). 13C NMR (CD3CN) δ 158.34, 158.21, 158.00, 157.99, 153.99, 153.34, 153.13, 153.08, 152.98, 152.47, 151.56, 148.99, 148.62, 146.63, 146.51, 140.82, 139.06, 138.95, 138.92, 137.92, 137.46, 132.25, 131.48, 130.79, 130.19, 129.92, 128.91, 128.69, 128.65, 128.57, 127.45, 127.28, 127.00, 125.37, 125.29, 124.96, 100.68, 88.28. UV (CH3CN) λmax nm (ε × 10−4) 284 (11.3), 334 (4.7), 356 (4.1), 476 (2.7). ESI-MS calcd for C46H30F6N6PRu [M]+ 940.8, found 941.3 [M]+.
Compound 9
9 was obtained from (4,7-dichlorophen)Ru(bpy)2·2PF6− in 75% yield after 1 hr. 1H NMR (CD3CN) δ 8.72 (s, 1H), 8.54–8.48 (m, 3H), 8.12–8.08 (m, 3H), 8.00 (td, J=8.0, 1.5 Hz, 2H), 7.84–7.77 (m, 6H), 7.59 (d, J=5.1, 1H), 7.55–7.44 (m, 6H), 7.24 (td, J=5.5, 1.1 Hz, 2H). 13C NMR (CD3CN) δ 157.78, 157.58, 152.74, 152.60, 148.65, 138.30, 138.69, 133.01, 131.50, 131.37, 130.89, 129.79, 128.63, 128.41, 128.27, 127.52, 125.01, 121.83, 103.98, 84.41. UV (CH3CN) λmax nm (ε × 10−4) 288 (9.7), 325 (5.2), 545 (3.2). ESI-MS calcd for C48H32F6N6PRu [M]+ 938.8, found 938.9 [M]+.
Compound 10
10 was obtained from (4-chlorophen)Ru(bpy)2·2PF6− in 95% yield after 1 hr. 1H NMR (CD3CN) δ 8.66 (d, J=8.05 Hz, 1H), 8.58–8.52 (m, 5H), 8.35 (d, J=9.15 Hz, 1H), 8.16–8.07 (m, 4H), 8.03–7.99 (q, J=4.4 Hz, 2H), 7.86 (d, J=5.1 Hz, 2H), 7.80–7.77 (d, J=5.5 Hz, 1H), 7.73 (d, J=5.5 Hz, 1H), 7.59 (d, J=5.5 Hz, 1H), 7.55 (d, J=5.5 Hz, 1H), 7.40–7.47 (m, 2H), 7.24 (q, J=7.3 Hz, 2H), 0.36 (s, 9H). 13C NMR (CD3CN) δ 157.77, 157.70, 157.47, 153.52, 152.59, 152.55, 152.49, 152.48, 148.40, 148.15, 138.58, 138.45, 137.56, 131.82, 131.40, 130.03, 129.66, 128.60, 128.26, 128.14, 128.09, 127.09, 126.50, 124.93, 124.85, 110.97, 98.87. UV (CH3CN) λmax nm (ε × 10−4) 280 (9.5), 452 (2.0). ESI-MS calcd for C37H32F6N6PRuSi [M]+ 834.8, found 834.8 [M]+.
Results and Discussion
Synthesis
Previous work had shown that functionalized polypyridyl metal complexes can be used as modular “building blocks” for the construction of substituted complexes and multimetallic arrays, utilizing palladium-mediated cross-coupling reactions.12–15 We employed this same approach for the construction of the acetylene-containing systems, where the halo-substituted ligands were coordinated to the desired RuII center before further chemistry was performed on the complex. The resulting building blocks were thoroughly purified by flash chromatography to remove any contamination by the free ligand. Subsequently, subjecting each of the halo-phenanthroline-containing RuII complexes to Sonogashira cross coupling conditions with an acetylene-containing moiety afforded the desired systems in less than an hour at room temperature in good yield. Only in the case of the 5-chloro-1,10 phenanthroline complex were extended reaction times and elevated temperatures required to drive the reaction; this is to be expected, due to the lower reactivity of the aryl chloride.
Effect of Substitution Position: Appended phenyl acetylene systems
Our earlier study focused on dimetallic systems linked with acetylene spacers,8 with the objective of constructing multimetallic arrays. Acetylene linkages were chosen because they have been shown to behave as “molecular wires,” providing effective pathways for electron and energy transfer between metal centers.16 Appending acetylene functionalities also profoundly increases electronic delocalization in the excited state of conjugated systems, inducing significant red-shifts in emission wavelength, and increasing emission intensity.17 An additional advantage was that the rigid carbon-carbon triple bond allowed construction of stereochemically well defined systems. The combination of these features made acetylenes the optimal initial functionality to explore the effect of ligand modification on emission properties. Fortuitously, the extended conjugation of the phenanthroline ligand at a particular position produced dual emission that was readily resolved in both energy and lifetime.8 The emissive features appear to originate from spatially separated MLCT excited states, one localized on the extended bridging phenanthroline (resulting in the lower energy emission) and the other on the bipyridine coligands (correlated to the higher energy emission).8 This is in marked contrast to nearly all other reports of heteroleptic RuII complexes, where only the lowest energy emissive state is observed. The occurrence of dual emission in those initial studies seemed to be specific to chemical substitution at the 4-position of the phenanthroline ligand.
To follow up on the discovery of dual emission in acetylene-linked dimetallic complexes, we chose to use similar phenyl acetylene substituents with monometallic systems. The phenylacetylene functionality was chosen for these systems as it is capable of profound electronic delocalization, but is also rigid, eliminating complication in analysis due to conformational (rather than just electronic) dynamics in the excited state. We expand our studies now to compare the substitution effects at the other available positions of the phenanthroline ring. Modification at the 2- position was excluded from the study as it produces distorted octahedral geometries,18 but the 3-, 4-, and 5- positions were all investigated (compounds 3, 4, and 5, respectively; see Figure 1). In addition, we tested complexes with substituents that allow for tunable emissive characteristics, and explored the possibility that symmetric and asymmetric substitution patterns might be an important factor governing dual emission. A combination of ground and excited state spectroscopy was used to investigate these systems.
Figure 1.
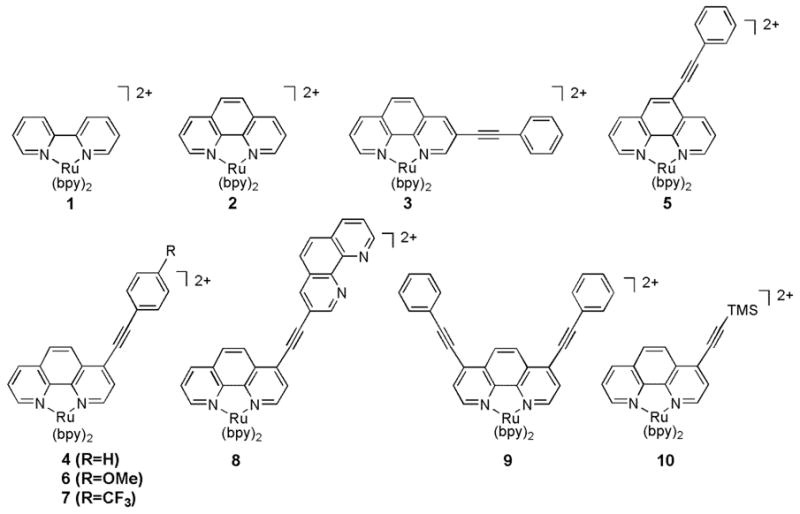
RuII complexes used in this study.
13C NMR was employed as one method to measure the effect of the conjugated phenylaetylene moiety on the electronic character of the complexes’ ground states. There are two resonances for the acetylene carbons; one assigned to the more electron deficient carbon attached to the ligand (identified as C1), and one for the more electron rich carbon, adjacent to the phenyl ring (identified as C2). The chemical shift of the acetylene carbons reflects the electron demand from the metal ion through the sigma framework (which should produce a down-field shift of those acetylenic resonances). This essentially reflects the degree of electronic delocalization in the ground state. These studies show that the chemical shift for the carbon adjacent to the phenyl group, C2, was essentially unchanged for the three isomers 3, 4 and 5, with a resonance at about 85 ppm (see Table 1). In contrast, C1 was significantly affected by the point of substitution on the ring: for the 3-substituted coordination complex 3, it appeared at 96.14 ppm, while for the 4-substituted system 4, it was downfield shifted by over 7 ppm to 103.56 ppm. Compound 5 exhibited an intermediate chemical shift, with the C1 acetylene carbon at 98.19 ppm. The most pronounced downfield shift for the 4-substituted system demonstrates more electronic delocalization through that position to the coordinated nitrogen, resulting in larger sigma electron donation to the metal center. Interestingly, only the most proximal carbon in the alkyne showed a significant change. This indicates that, in the ground state, the electron delocalization does not extend through the acetylene bond and into the phenyl ring. For all three systems, the ground state involves a delocalized π system that extends (to different degrees) to the acetylene bond, but not through that bond into the conjugated phenyl ring.
Table 1.
Chemical Shifts for Acetylene Carbons for Model Compounds.a
| Compound | 13C C1 (ppm) | 13C C2 (ppm) |
|---|---|---|
| 3 | 85.32 | 96.14 |
| 4 | 84.35 | 103.56 |
| 5 | 84.75 | 98.19 |
| 9 | 84.41 | 103.98 |
Measurements were performed in room temperature CD3CN
The ground state UV/Vis absorption spectra of compounds 3–5 provided additional insight into how the position of substitution modifies the electronic environment (Figure 2). For the parent compound Ru(phen)(bpy)2 (2) the spectrum exhibits three main features: two ligand-centered π–π* transitions of the aromatic bpy and phen ligands at 266 and 286 nm, and a broad transition having a maximum at 450 nm and a higher energy shoulder at 440 nm (Figure 2). The peak and the shoulder are indicative of strongly overlapped transitions to different MLCT states, with the state, or degenerate states, at higher energy localized on the bipyridines. Extending the conjugation of the phenanthroline ligand by the phenylacetylene substituent in compounds 3, 4 and 5, resulted in spectra with marked changes. Although the π–π* transition at 286 nm of the bipyridine ligand was unaltered, there were additional peaks in the 300–360 nm region, which we attribute to π–π* transitions on the extended phenanthroline.19 For 3 and 5, this transition was broad and essentially featureless, while for 4 there were some discernable fine features. The MLCT transitions for 3 and 5 were similar to those of the parent complex, with λmax at 450 nm, although there was a reduction of the extinction coefficient in both 3 and 5. Compound 4, in contrast, exhibited a much broader, red-shifted MLCT transition at 460 nm (indicating profound electronic delocalization) and an extinction coefficient similar to that of 2.
Figure 2.

UV/Vis (left) and emission (right) spectra of compounds 2 (green), 3 (red), 4 (purple), and 5 (blue)
The steady state emission spectra display marked differences in the excited state behavior of the three isomers (see Figure 2). Compound 3 exhibited a red shift in the emission maximum of about 20 nm from 2, and there was a significant increase in the luminescent quantum yield to Φem = 0.103 (Φem = 0.060 for the parent complex 2). Compound 4 displayed a much greater red shift of about 60 nm, a very broad peak profile, and a substantial decrease in the quantum yield, to 0.023. In addition, 4 was found to be profoundly sensitive to quenching by molecular oxygen; in air-equilibrated samples, the complex was practically nonemissive. In contrast to these two compounds, 5 exhibited luminescent properties almost indistinguishable from those of 2, with neither a significant change in the quantum yield nor modulation of the triplet energy; the additional conjugation does not seem to have any impact on the coordination complex when placed at the 5-position. For all three systems, there was good agreement between the absorption and excitation spectra, indicating that the extended-phenanthroline based MLCT may be populated either by direct dπ–π*(phenanthroline) excitation or by internal sensitization following higher energy excitation.
Results for time-resolved luminescence studies paralleled results for the RuII dimers reported previously. Compound 5 behaved most like the parent compound 2, with a single-component lifetime of 1.31 μs in deoxygenated acetonitrile (see Table 2). Compound 3 also displayed monoexponential decay, but with a markedly longer lifetime (2.46 μs). This increase in lifetime with a decrease in emission energy is in opposition to the energy gap law,20 but this is a common observation in RuII complexes containing ligands with extended conjugation and delocalized excited states.20–22 Under our experimental conditions at room temperature, these two systems exhibit a single emissive excited state (or, conceivably, two very similar states in thermal equilibrium). In contrast, the luminescence decay of 4 required fitting to a biexponential equation, resulting in excited state lifetimes of 6.55 and 1.21 μs. Although the two discrete excited states could not be resolved in energy in the steady state emission spectra (for which only a single, broad emission feature was observed), their distinct lifetimes readily allowed their resolution using time resolved methods. The fractional contributions of the slow- and fast-decaying states to the luminescence decay varied with emission wavelength, as illustrated in Figure 3. The slow-decaying component contributed a larger fraction at longer emission wavelengths, proving that two emissive states coexist with long, but different, lifetimes and different, but overlapping, emission spectra. Excitation at higher energies produced both emissive states, showing that these states are both coupled to a higher energy state, but are not coupled to each other.
Table 2.
Selected Spectroscopic Information for RuII Complexes.a
| Compound | λmax | Φ b | τ1 (ns)c | τ2 (ns)c | Flongd | Fshortd |
|---|---|---|---|---|---|---|
| 1 | 610 | 0.062 | 894 (±4) | 1 | ||
| 2 | 610 | 0.060 | 798 (±3) | 1 | ||
| 3 | 630 | 0.103 | 2460 (±10) | 1 | ||
| 4 | 660 | 0.023 | 6550 (±300) | 1210 (±80) | 0.98 | 0.02 |
| 5 | 610 | 0.081 | 1310 (±20) | 1 | ||
| 6 | 662 | 0.016 | 11500 (±500) | 1060 (±60) | 0.93 | 0.07 |
| 7 | 675 | 0.021 | 6560 (±300) | 1300 (±80) | 0.97 | 0.03 |
| 8 | 679 | 0.016 | 6800 (±200) | 1200 (±50) | 0.92 | 0.08 |
| w/Zn2+ | 679 | 3650 (±200) | 900 (±100) | 0.30 | 0.70 | |
| w/H+ | 679 | 3700 (±200) | 950 (±100) | 0.30 | 0.70 | |
| 9 | 672 | 0.027 | 6400 (±300) | 1 | ||
| 10 | 660 | 0.023 | 6680 (±300) | 2200 (±100) | 0.97 | 0.03 |
Measurements were performed in 2–8 × 10−6 M solutions in room temperature CH3CN.
Emission maxima and yields following excitation at 450 nm
Lifetimes determined following pulsed excitation at 455 nm
Represents the fraction of the formation of short or long-lived state for each photon absorbed, corrected for PMT sensitivity at different wavelengths
Figure 3.
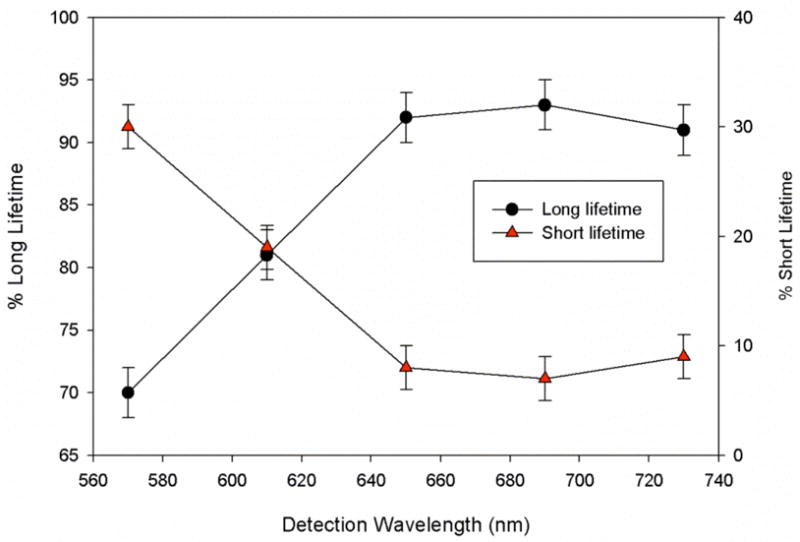
Detection wavelength dependence of fractional contributions of short- and long-lived components of fits to the excited state decays at each wavelength for compound 4
These results make it apparent that resolvable dual emission under these conditions only occurs in the case of complex 4. In these simple coordination complexes, it is the substitution position, rather than the actual substituent, that is the cause of the electronic perturbations that result in de-coupling the two excited states. It is interesting to note that this is not the first report of surprising substitution effects at this position of the phenanthroline ligand. Wallace, et al., discovered that substitution at the 4,7-positions had a far greater impact than any other substitution position on the MLCT energy at room temperature for rhenium complexes, and reported low temperature dual emission of the “distinct orbital type” for these systems.23–25 In contrast, in our system, both states display the characteristics of 3MLCT states, with regards to energy and excited state lifetime. We propose that this dual emission is the “spatially isolated” type, with one state (with the longer lifetime, more prominent at longer wavelengths) residing on the highly conjugated phenanthroline ligand, and the other (with the shorter lifetime) being localized on the bipyridine ligands. To confirm that the excited states we observe are MLCT in nature, we considered the possibility that one emissive feature might originate from a ligand-centered state on the extended phenanthroline ligand. Accordingly, we synthesized the 4-arylethynyl-1,10 phenanthroline free ligand and investigated its photophysical properties (see SI). Both the free ligand alone and in the presence of ZnII (used as a divalent ion surrogate to mimic the effect of coordinated RuII) exhibited absorption and emission maxima far to the blue of what we see in the RuII complex (see Figure S1). The time resolved luminescence decays were fit to sub-nanosecond lifetimes, in contrast to the observed μs lifetimes for the RuII complex 4. At room temperature, of course, triplet phosphorescence is not observed.26
Tunability of the excited state
We explored additional complexes containing appended phenylacetylene moieties to determine if the observed dual emission was both general and might be predicted and controlled. Complexes 6 and 7 represent two derivatives of 4 for which synthetic modification in the form of added electron donating and withdrawing groups, respectively, have been incorporated onto the peripheral phenyl ring. Both complexes exhibited two emissive states. Although the emission energy was not significantly perturbed by the nature of the para substituent, excited state lifetimes were affected, for both the shorter and longer-lived features (see Table 2). Remarkably, the electron-rich p-methoxyphenyl system displayed a lifetime of ca. 12 μs, nearly twice that observed for the complexes containing phenyl and p-trifluoromethylphenyl groups (in addition to a second, shorter lifetime of ca. 1 μs). The fractional populations of the two emitting states were also dependant upon the functionality on the phenyl ring. It would appear that subtle changes in the phen ligand are not sufficient to radically shift emission energy, but do strongly perturb excited state lifetimes and populations.
A complex containing an alkyne conjugated, uncoordinated phenanthroline ligand was also studied (compound 8). This extended ligand can, in principle, facilitate tuning of the RuII excited state behavior via non-covalent modification; addition of cations would be expected to bind to the free ligand, affecting the behavior of the metal complex and producing a simple prototypical sensor. Accordingly, we investigated the emissive properties of the complex both alone and in the presence of exogenous H+ and Zn2+ ions. This system validated our expectations, showing profound changes in lifetime and fraction population upon addition of both protons and the divalent metal ion (see Table 2). Clearly, any electronic perturbation of the attached phenanthroline ligand results in modulation of the excited state of the ligand to which it is appended. Interestingly, this effect is propagated through the central RuII ion to also affect the excited state residing on the coligands, as evidenced by a decrease in the lifetime associated with the bpy-based emissive state.
These results suggest the possibility of rationally designing dual emission systems in which the properties of the two excited states may be manipulated by functionalization of the “primary” ligand. In addition, we previously showed that the coligands may be altered to change excited-state properties;8 thus, these two approaches may be used in unison to optimize functional properties. With such promising results indicating that we can achieve good temporal resolution between the two excited states (with up to an order of magnitude difference in excited state lifetime), we have taken a step closer to creating systems with some viability as a switch or other device.
Symmetry Effects
To determine whether or not the overall symmetry of the complex had an effect on relaxation pathways to the emissive state(s), a symmetrical, di-substituted complex 9, which also contains the phenylacetylene functionality, was studied. This compound, analogous to compound 4, exhibited a single emission peak in the steady-state spectrum (see Figure S2). The time-resolved studies did suggest the possibility of a trace of a shorter-lived component appearing at shorter wavelengths, although the amount of this short-lived component as a fraction of the overall emission was at least an order of magnitude less than seen in other compounds displaying dual emission, and thus below the threshold value we deem necessary to report. Clearly, the asymmetric, singly substituted systems show the most pronounced dual emission, if it is not actually exclusively limited to them.
We conclude that the symmetry of the substituted phen ligand plays an important role in defining the decay pathways of the excited states, essentially shutting down the majority of the higher energy emission observed in the asymmetrically substituted systems (such as 4). This might be explained by a simple model of excited state manifolds for symmetrically and asymmetrically substituted complexes (see Figure 4). Upon excitation to the MLCT state, an electron is transferred from an orbital on the metal to a delocalized orbital extended over one ligand (after an initial relaxation process). The greater the difference in the overall electronic character of the excited state with regards to the ground state, the larger the structural perturbation along the specified nuclear coordinate.27 In the case of symmetrically substituted ligands, a symmetrical ground state is coupled to a symmetrical excited state, with little change in electronic geometries. This is not the case with asymmetric, highly conjugated ligands; if the excited state extends over the full ligand surface, a larger deviation from the ground state results. We know that the ground state of the extended ligand complexes includes a delocalized orbital that extends to, but not through the acetylene bond (from the trends in chemical shifts observed in the 13C NMR). In contrast, the excited state appears to extend though the acetylene bond — possibly in the form of a cumulene-type structure28 — to the appended phenyl (or phenanthroline) ring systems, as evidenced by the lower energy excited state, and the ability to tune that state through remote electronic manipulation, both seen in the luminescence experiments. As there is more deviation from the ground state geometry with such delocalization in the case of asymmetrically substituted ligands, there is greater translation of the excited state surface along the nuclear axis. This, in turn, affects the coupling of that MLCT state to other excited states and to the ground state. It seems likely to us that it is this effect that causes a decoupling of the two MLCT excited states, preventing the standard relaxation to a single, lowest energy emissive state so commonly seen.3,29 This might also explain the absence of previous reports of dual emission in systems similar to ours, as most ligands developed for coordination complexes are symmetrically substituted, a result of both synthetic considerations and ease of characterization of the resulting complexes. As our initial studies fortuitously focused on asymmetrically substituted and highly conjugated ligands, we were able to readily discern the presence of two emission features under typical, ambient conditions, and we subsequently were able to find evidence for minor amounts of dual emission in a wider range of complexes than we initially might have expected. It is also of interest to note that other reports of dual emission in RuII complexes involve heteroleptic complexes with asymmetric ligands.5–7
Figure 4.
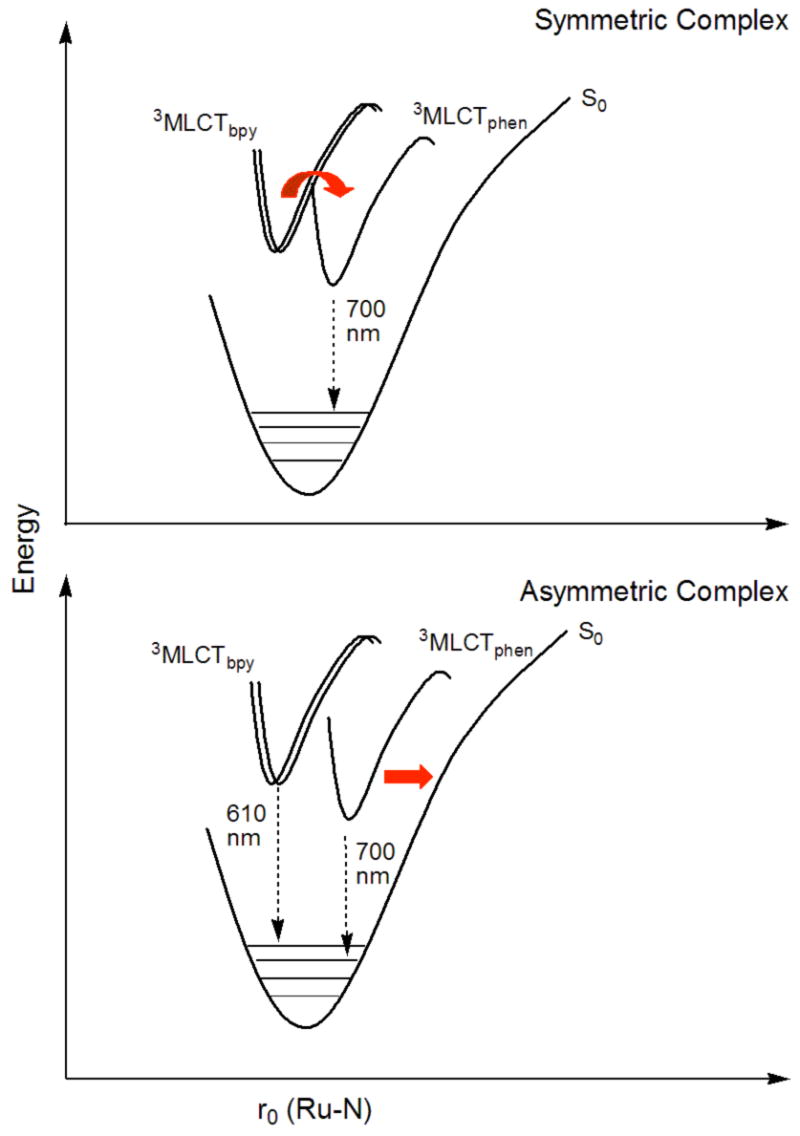
Proposed potential energy diagram for symmetric and asymmetric RuII complexes. Relaxation from a higher energy state is possible in the symmetrical complex (as indicated by the curved red arrow) but not in the asymmetrical case, as a result of subtle modulation of the excited state structure
Alternative Explanations for two emissive states
There are a few alternative scenarios that could be considered to explain our observation. We address two likely arguments below.
Presence of impurities
Positive identification of systems displaying authentic dual emission can be challenging, as possible contributions from luminescent impurities must be considered and eliminated. What we observed—distinct emission spectra, (sometimes) distinct excitation spectra, and distinct luminescence decay times—are exactly what one might see if a sample were contaminated with a luminescent impurity.30 Most such contaminations produce luminescence characterized by a multiexponential decay with components over the range 1–3 ns. We have never seen contamination give the microsecond lifetimes observed in this work. Any “impurity” would have to be another metal complex with CT luminescence; since we excited at maximums in the absorption spectra using quite weak excitation laser pulses, the “impurity” would have to be present in a concentration comparable to that of the sample molecule itself. We believe this is extremely unlikely for reasons also described in our first report.8 Briefly, we carefully tested blanks at all points of sample manipulation and data collection. A variety of different systems including both the bimetallic complexes reported earlier and the new complexes reported here were prepared by sometimes quite different synthetic routes, yet the dual emission correlates with a defined molecular structure, not with one or more particular synthetic procedures. A range of characterizations methods, including mass spectrometry, NMR, and analytical HPLC show no evidence of even trace contamination, within their detection limits.31 In the case of studies on compound 8, the impurity would have to be responsive to both protons and other metal ions. Finally, although the complexes are not particularly photosensitive, extended photolysis using the violet line of an argon laser was able to bleach both absorption and emission features—but bleached both the short wavelength and the long wavelength features identically. If there were two different molecules present, even two noninterconverting isomers, one would expect selective photobleaching of one complex over the other.
Isomerization
A possible way to have two different species responsible for the dual emission we observed is to have two isomers that interconvert on a time scale of seconds to minutes, that is, short compared to photobleaching, but long compared to luminescence studies. In such an interpretation of our observations, we would have two emissive MLCT states, but both MLCT excited states would be largely localized upon the phenanthroline ligand—each characteristic of a different conformation of the molecule. The two inequivalent states would be a result, presumably, of rotation along the arylalkyl axis (see Figure 5). One, the lower energy, longer lived state, could be attributed to the fully conjugated system, in which the phenyl group of the phenyl acetylene is coplanar and conjugated with the phenanthroline ligand. The other hypothetical state might result from the phenyl ring rotating out of the plane of the phenanthroline ligand, breaking conjugation and resulting in a more localized excited state that terminated with the acetylene bond. One might not normally expect rotation about the axis of the alkyne to be as slow as a millisecond at room temperature,32 but nevertheless we made a direct test of this possibility. We synthesized and investigated a system that was not capable of producing rotational isomers that are conjugatively distinct, namely, compound 10. In 10, the acetylene triple bond terminates in a silyl protecting group, instead of an additional conjugated moiety. Without any π orbitals on the terminal group, there should be no difference in the electronic states with rotation about the sp2-sp bonds. Steady-state emission studies showed a single (but broad) emission band for 10 (see Figure S2), as in the case of the other acetylene-containing complexes. As usual, it was necessary to carry out time-resolved emission studies to determine whether there might be more than one decay processes contributing to the emission spectra. Such studies showed unambiguously the presence of two excited states, well resolved in lifetime (see Table 2), proving that rotational isomers are not the source of the two emission features in this series of compounds.
Figure 5.

Hypothetical extreme excited-state rotamer structures for compound 4
Conclusion
The simplest explanation for our data is that there are two MLCT excited states localized on different ligands of certain RuII complexes, with the short-lived, short-wavelength component being essentially bipyridine-based, while the long-lived, long-wavelength component is localized predominately on the more conjugated phenanthroline ligand. One possible objection to this interpretation could be that the short-lived components are not exactly the same in all the molecules studied, and do not exactly match the lifetime of Ru(bpy)3. Given, however, that it is surprising to find co-existing states at all, because states on small molecules normally couple well, it should not be surprising that they are not entirely independent of each other. Modulation of each ligand’s electronic character and energy should have an impact on the energetics of the coligands, as well as on the exact spatial localization of electron density in molecular orbitals and, consequently, coupling between states. The photophysics of both excited states should be interdependent, and result in individual complexes with unique properties, but showing systematic trends, as we observe.
Initial attempts to tune the excited state behavior of the complexes by covalent modification or protonation/complexation have proven successful, indicating that we can create systems with variable lifetimes and degrees of population of the two excited states. Indeed, these complexes with “spatially isolated” excited states offer the tantalizing possibility of spatial, energetic and temporal control over high energy states that could be funneled in two different directions. If our hypothesis regarding the essential features of substitution position (for phenanthroline complexes) and asymmetry holds, rational design of these systems should be simple and accessible.
In summary, we have gradually come to the conclusion that even in species as small and as well studied as monometallic RuII complexes with rather simple ligands, we observe spatially localized excited states that persist for up to 10 μs or more with little or no population transfer between them. They are characterized by distinct emission spectra and distinct lifetimes, which would not be the case were the states in thermal equilibrium. The states both clearly have the properties of MLCT triplet states.33 The fact that luminescence quantum yields are considerably less than unity merely means that there are competitive nonradiative decay channels available for both the emitting triplet MLCTs. As to the formation of the emitting triplet states, we see no evidence for any rise time associated with either state (particularly, the lower energy state), as inferred from picosecond luminescence studies with a time resolution of about 10 ps. It seems likely that typical excitation photon energies are above the kinetic barrier separating the two states and that initial Franck-Condon states decay with some branching ratio into the two low-lying, triplet MLCT emissive states. It would be of interest in the future to use femtosecond transient absorption methods to explore further the earlier photophysical pathways between the photoexcited and the thermalized, coexisting luminescent states.
Supplementary Material
Additional synthetic details and UV/vis and fluorescence spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported in part by the National Institutes of Health (GM069773). D. M. thanks the late Professors K. Wilson and B. Zimm. E. C. G. thanks Dr. D. Jouvenot for insightful discussions and help with figures
References
- 1.Lakowicz JR. Principles of Fluorescece Spectrorcopy. 3. Springer; 2006. [Google Scholar]
- 2.For example, fluorescein, arguably the most widely used fluorescent dye, was first synthesized by Adolph Von Baeyer in 1871. See Huisgen R. Angew Chem Int Ed Engl. 1986;25:297–311.
- 3.Juris A, Balzani V, Barigelletti F, Campagna S, Belser P, von Zelewsky A. Coord Chem Rev. 1988;84:85–277. [Google Scholar]
- 4.Yeh AT, Shank CV, McCusker JK. Science. 2000;289:935–938. doi: 10.1126/science.289.5481.935. [DOI] [PubMed] [Google Scholar]
- 5.Keyes TE, O’Connor C, Vos JG. Chem Commun. 1998:889–890. [Google Scholar]
- 6.Keyes TE, O’Connor C, O’Dwyer U, Coates CG, Callaghan P, McGarvey JJ, Vos JG. J Phys Chem A. 1999;103:8915–8920. [Google Scholar]
- 7.Song L, Feng J, Wang X, Yu J, Hou Y, Xie P, Zhang B, Xiang J, Ai X, Zhang J. Inorg Chem. 2003;42:3393–3395. doi: 10.1021/ic026070j. [DOI] [PubMed] [Google Scholar]
- 8.Glazer EC, Magde D, Tor Y. J Am Chem Soc. 2005;127:4190–4192. doi: 10.1021/ja0440304. [DOI] [PubMed] [Google Scholar]
- 9.Kasha M. Faraday Soc Discuss. 1950;9:14–19. [Google Scholar]
- 10.Marquardt D. J Soc Indus Appl Math. 1963;11:431–441. [Google Scholar]
- 11.Hurley DJ, Tor Y. J Am Chem Soc. 2002;124:3749–3762. doi: 10.1021/ja0123103. [DOI] [PubMed] [Google Scholar]
- 12.Tzalis D, Tor Y. Chem Commun. 1996:1043–1044. [Google Scholar]
- 13.Connors PJ, Tzalis D, Dunnick AL, Tor Y. Inorg Chem. 1998;37:1121–1123. [Google Scholar]
- 14.Tzalis D, Tor Y. J Am Chem Soc. 1997;119:852–853. [Google Scholar]
- 15.Tor Y. Synlett. 2002:1043–1054. [Google Scholar]
- 16.Grosshenny V, Harriman A, Ziessel R. Angew Chem Int Ed Engl. 1995;34:1100–1102. [Google Scholar]
- 17.Maulding DR, Roberts BG. J Org Chem. 1969;34:1734–1736. [Google Scholar]
- 18.Baranoff E, Collin JP, Furusho J, Fususho Y, Laemmel AC, Sauvage JP. Inorg Chem. 2002;41:1215–1222. doi: 10.1021/ic011014o. [DOI] [PubMed] [Google Scholar]
- 19.Tzalis D, Tor Y. Tetrahedron Lett. 1995;36:6017–6020. [Google Scholar]
- 20.Treadway JA, Loeb B, Lopez R, Anderson PA, Keene FR, Meyer TJ. Inorg Chem. 1996;35:2242–2246. doi: 10.1021/ic950961s. [DOI] [PubMed] [Google Scholar]
- 21.Strouse GF, Schoonover JR, Duesing R, Boyde S, Jones WE, Meyer TJ. Inorg Chem. 1995;34:473–487. [Google Scholar]
- 22.Damrauer NH, Boussie TR, Devenney M, McCusker JK. J Am Chem Soc. 1997;119:8253–8268. [Google Scholar]
- 23.Wallace L, Rillema DP. Inorg Chem. 1993;32:3836–3843. [Google Scholar]
- 24.Wallace L, Woods C, Rillema DP. Inorg Chem. 1995;34:2875–2882. [Google Scholar]
- 25.Wallace L, Jackman DC, Rillema DP, Merkert JW. Inorg Chem. 1995;34:5210–5214. [Google Scholar]
- 26.While there was no evidence for phosphorescence at room temperature, it was observed at 77 K, with a lifetime of ca. 450 ms. This is in contrast to the two lifetimes we observed at 77 K for complex 4, which were fit to lifetimes of 4.5 and 23 μs.
- 27.While representational potential energy diagrams such as Figure 4 may appear to suggest that radiative and non-radiative processes are equally affected by the displacement along the specified nuclear coordinate, this is not necessarily the case. The two processes may occur along different coordinates, and thus, there may be significant displacement along a coordinate that impacts a radiative transition, but does not affect non-radiative decay; See Ref. 22. We hypothesize that this is the case with our system.
- 28.Polyansky DE, Danilov EO, Voskresensky SV, Rodgers MAJ, Neckers DC. J Am Chem Soc. 2005;127:13452–13453. doi: 10.1021/ja053120l.Note that there is some disagreement in the field about the contribution of cumulenic structures to the excited state of phenylenethynylenes; see Beeby A, Findlay KS, Low PL, Marder TB, Matousek P, Parker AW, Rutter SR, Towrie M. ChemComm. 2003:2406–2407. doi: 10.1039/b307005k.
- 29.Kalyanasundaram K. Photochemistry of Polypyridine and Porphyrin Complexes. 1. Academic Press Limited; San Diego: 1992. [Google Scholar]
- 30.Belser P, von Zelewsky A, Juris A, Barigelletti F, Balzani V. Chem Phys Lett. 1984;104:100–104. [Google Scholar]
- 31.We have also considered and eliminated the possibility of aggregation; there is no evidence of concentration effects in the luminescence experiments, and no aggregation observed in the NMR, which is measured at a concentration several orders of magnitude above that used in the spectroscopy experiments. Formation of transient aggregates in the form of eximers is eliminated due to the absence of a rise time in the time resolved experiments.
- 32.Of course, in the ground state, there is a very low barrier to rotation about the –C≡C- unit; see Miljaniæ Oš, Han S, Holmes D, Schaller GR, Volhardt KPC. ChemComm. 2005:2606–2608. doi: 10.1039/b503173g. and references therein.. This is not the case in the excited state
- 33.This is apparent from the excited state energies, lifetimes, and the fact that these states are populated with close to unit quantum efficiencies as judged from singlet dioxygen sensitization; B. Hernandez; M. Selke, unpublished results.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional synthetic details and UV/vis and fluorescence spectra. This material is available free of charge via the Internet at http://pubs.acs.org.