

Abstract
“Smart tattoo” sensors – fluorescent microspheres which can be implanted intradermally and interrogated noninvasively using light – are being developed as potential tools for in vivo biochemical monitoring. In this work, a platform for enzymatic tattoo-type sensors is described, and prototype devices evaluated using glucose as a model analyte. Sensor particles were prepared by immobilizing Pt(II) octaethylporphine (PtOEP), a phosphorescent dye readily quenched by molecular oxygen, into hybrid silicate microspheres, followed by loading and subsequent covalent immobilization of glucose oxidase (GOx). Rhodamine B (RITC)-doped multilayer nanofilms were subsequently assembled on the surfaces of the particles to provide a reference signal and provide critical control of glucose transport into the particle. The enzymatic oxidation of glucose within the sensor results in the glucose concentration-dependent depletion of local oxygen levels, enabling indirect monitoring of glucose by measuring relative changes in PtOEP emission. A custom testing apparatus was used to monitor the dynamic sensor response to varying bulk oxygen and glucose levels, respectively. For the prototypes tested, dynamic test results indicate that the sensors respond rapidly (t95 = 84 sec) and reversibly to changes in bulk glucose levels, while demonstrating high baseline stability. The sensitivity (change in intensity ratio) of these devices was determined to be 4.16 ± 0.57 %/mg dL−1. The analytical range for the prototypes was determined to be 2 to 120 mg/dl, though this can be extended to cover the physiologically relevant range by tailoring the nanofilm coatings. These findings confirm the potential for enzymatic microscale optical, and pave the way for extension of this initial demonstration with glucose to target other biochemical species relevant to metabolic monitoring.
Introduction
Developing methods for monitoring biochemical concentrations in real-time has become a medical priority due to the potential for improved diagnostics and management of conditions where chemical imbalances are manifest. Optical monitoring techniques are attractive for frequent or continuous monitoring and as a result, techniques including fluorescence,1 absorbance,2 and Raman3 spectroscopy4 have been heavily researched as approaches to monitor biochemical levels. The high sensitivity and superior selectivity associated with fluorescence sensing techniques, due to fluorescence assays involving specific interactions between target analytes and receptor molecules, have led to increased interest by many groups.1,5 A particularly interesting concept which has the potential to provide both spot and continuous biochemical measurements are “smart tattoo” type sensors, which consist of populations of micron-scale luminescent particles which could be implanted and noninvasively interrogated with light.6–9
To realize such an ideal, the aforementioned assay chemistry must be packaged into an appropriate carrier. Due to the huge population of diabetics that could benefit from improved analysis methods, the work in this field has primarily focused on developing and entrapping glucose-sensitive assays within microparticles for use in transdermal glucose monitoring; however, the approaches is general and may be used with assays sensitive to other analytes. To date, the majority of assays developed for use in smart tattoo sensors are competitive-binding based, which rely on changes in fluorescence emission properties modulated by resonance energy transfer (RET) to optically transduce glucose levels.10–14 The RET approach hinges upon the competitive binding of a fluorescent ligand and target analyte to receptor sites on lectins, such that the ligand is displaced from the receptor when in the presence of the analyte, causing measurable changes in emission spectra.15–17 To achieve high sensitivities with RET-based schemes, the free movement of analyte, ligand and receptor is desired, requiring considerable effort to be focused on not only assay development but also designing the appropriate sensor architecture. This concept was demonstrated by using hollow capsule-based containers to facilitate free movement of assay components, resulting in increased device sensitivity when compared to previous systems based on assay entrapment within hydrogel spheres.6,10,18
An alternative approach to competitive binding, which requires mobility of at least one component of the two-molecule system, is to use substrate-specific enzymatic reactions. This is commonly done for electrochemical sensors, where enzymes are immobilized on electrodes and an outer coating is applied to control relative diffusion rates.19 Enzymatic assays using optical transduction, which typically comprise an oxygen-quenched luminescent dye and an oxidase with affinity to the analyte of choice (e.g. glucose oxidase may be used in glucose sensing applications), have also been developed for use in smart tattoo technologies.9,20,21 When successfully entrapped within a microscale container, local oxygen levels are proportionally reduced and relayed through the fluorescence emission of the oxygen reporter as the analyte diffuses into the sensor, providing an indirect means to monitor analyte levels.22,23 Although the integration of an enzyme into the sensing scheme endows the sensor with innate selectivity, as well reversibility, controlling the reaction-diffusion kinetics of the reactants are key to the realization of optimized sensors.24 Previous reports have described fiber optic biosensors with sensor components immobilized in transport limiting matrices;25–27 however, applying a transport-limiting film is peculiarly difficult when working with microparticles, as compared to the simple dip- or spin-coating methods used to modify electrochemical sensors or optical fibers. Thus, designing appropriate sensor architecture to control substrate transport properties, and processing methods to achieve this architecture, is of utmost importance when downscaling biosensors.
A particularly promising approach to fabricating precise nanoscale thin films capable of controlling the transport of low molecular weight species—biochemical target analytes—is the deposition of polyelectrolyte nanofilms using layer-by-layer (LbL) self-assembly.28–30 Utilizing surface adsorbed polyelectrolyte nanofilms to control substrate diffusion, a more complex microparticle biosensor system based on calcium alginate hydrogel microspheres that contained and an oxygen-quenched ruthenium compound was demonstrated.9,24,31 In this system, which used glucose as a model analyte, local oxygen levels (internal to the particle) are proportionally reduced through glucose oxidase (GOx)-initiated catalysis (Scheme 1) and tracked through relative changes in ruthenium emission properties. A key finding was that altering the physical properties of the nanofilm coatings results in modification of the analyte transport properties into to the microsphere, such that control of overall response characteristics, including sensitivity and linear range, may be achieved.24
Scheme 1.

Consumption of glucose and oxygen resulting from GOx catalyzed glucose oxidation.
Although the results reported to date are promising, previous work employed phosphorescent ruthenium complexes as the reporter molecules, resulting in low sensitivities (0.02 % change/mg dL−1) over the analytical range of 0 – 140 mg/dL.21 Given that ruthenium complexes have a much lower sensitivity to oxygen than metalloporphyrins and the overall response properties of smart tattoo sensors using oxidoreductase enzymes are dependent on the oxygen probe, it is hypothesized that the integration of a more sensitive Pt(II) porphyrin dye should result in an overall increase in sensitivity.5,27,32,33 To our knowledge there has only been one report utilizing Pt(II) porphyrins in enzymatic sensors, in which case the protein and indicator were immobilized together at the end of an optical fiber.27 The lack of work with porphyrin indicators could be due to their extreme hydrophobicity, causing difficulties in aqueous applications.5 To overcome this, immobilization matrices comprising silicon, organic glassy polymers, cellulose derivatives, or fluoro-polymers have been developed. Pt(II) porphyrins can be adsorbed to these materials, allowing dissolved oxygen measurements in aqueous environments.33,34 It is well documented that the immobilization matrix can drastically affect the oxygen sensitivity and that silica-containing matrices produce high sensitivities, although the exact mechanisms are still debated.32,35
We recently introduced a facile approach to self-assemble mesoporous silicate particles comprising homogeneously distributed hydrophilic and hydrophobic domains as a platform technology for biosensor development.24 All required steps are performed at room temperature and independent of surfactants, so it is inherently bio-friendly. As a first step in preparing enzymatic smart tattoos, Pt(II) octaethylporphine (PtOEP) was adsorbed into alginate-modified mesoporous silica particles, followed by characterization of the oxygen-induced quenching effect.36 As an extension of that work, this report focuses developing glucose-sensitive smart tattoos by focusing on the adsorption of the critical glucose sensor components – GOx and PtOEP – into alginate-modified mesoporous silica particles, which are subsequently coated with fluorescent nanofilms using the LbL technique. These or similar systems may potentially be used with minimally-invasive monitoring devices for diabetics, cancer patients, soldiers, livestock, allowing continuous and spot measurements of biochemical levels without the pain and bother associated with blood withdrawal. This work lays the foundation for the development of additional highly-sensitive biochemical monitors in a “mix and match” approach, allowing new sensors to be rapidly prototyped by incorporating specific enzymes and appropriate transport-controlling coatings.
Experimental Details
Materials
Sodium Alginate (low viscosity, 250 cps, MW 12–80 kDa), (3-glycidyloxypropyl) trimethoxysilane (GPTS), and ammonium hydroxide were obtained from Sigma and used for the synthesis of mesoporous alginate-silica particles. Platinum(II) octaethylporphine (PtOEP, Frontier Scientific), tetrahydrofuran (THF, Fluka), glucose oxidase (GOx, type VII from Aspergillus niger, 198 k units/g solid, Sigma), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC, Fluka), N-hydroxysulfosuccinimide sodium salt (NHSS, Toronto Research Chemicals Inc.), and sodium acetate (Sigma) were used to prepare PtOEP/GOx-doped alginate-silica particles. Fluorescein iosothiocyanate (FITC, Sigma) was conjugated to GOx for use in fluorescence imaging. Poly(allylamine hydrochloride) (PAH, MW = 70 kDa, Aldrich), poly(sodium 4-styrene sulfonate) (PSS, MW = 70 kDa, Aldrich), and sodium chloride (Sigma) were used during the deposition of multilayer thin films. Additionally, rhodamine B isothiocyanate (RITC, Aldrich) was conjugated to PAH and used in thin film deposition. β-D-glucose (MP Biomedicals, Inc.), O2 and N2 gas (Air Liquide), and phosphate buffered saline (PBS, Sigma) were used during dynamic testing. All necessary titrations were performed using 1.0 M HCl and 1.0 M NaOH, which were obtained from Fluka. All chemicals listed above were reagent grade and used as received. Spectroscopic grade potassium bromide (KBr, Thermo Electron) was used to prepare infrared spectroscopy samples. Throughout all experimental procedures, ultra pure water with a resistivity of greater than 18 MΩ was used. Unless otherwise stated, all experimental processes were conducted at 25 °C.
Preparation of mesoporous alginate-silica particles
Alginate-silica (“algilica”) particles were prepared using a procedure similar to that which was previously reported.24,36 Briefly, the precursor was prepared by stirring a solution comprised of 1.5 wt% aqueous alginate solution and glycidyl silane (GPTS) in a 1:1 volumetric ratio for at least 4 hours, resulting in an alginate-modified silanol. While stirring, 2 mL of the precursor silanol was added to 3 mL of water. To initiate the sol gel process, 1.25 mL of 10 M NH4OH was added and stirred for 20 min, followed by the addition of 10 mL of water and an additional stirring time of 40 min. An extra 40 mL of water was added and the suspension stirred for at least an additional 4 hours. The resulting particle suspension was rinsed with DI water using four sequential centrifugation cycles and diluted to a total aqueous suspension volume of 1.5 mL. A Beckman Coulter counter (Z2) equipped with a 100 μm aperture was used to obtain the mean diameter and concentration of particles comprising the stock suspension. Fourier transform infrared spectroscopy (FTIR, Nexus 470) was used to characterize the chemical makeup of the particles. Samples for FTIR analysis were prepared by using a hand press with accompanying die and anvil (Thermo Electron) where approximately 1 mg of dried particles were mixed with 15 mg of KBr and pressed into pellet form. After the spectrometer was purged with N2, IR spectra were acquired using the prepared pellets.
pH effect on GOx immobilization
For use in fluorescence imaging, FITC-labeled GOx (FITC-GOx) was prepared using 1:1 molar ratio during the labeling reaction, which was performed using a standard amine labeling protocol.37 The final solution was diluted to a final concentration of 2 mg/mL in 0.05 M sodium acetate. Separate FITC-GOx solutions were titrated to pH of 3.0, 4.0, 5.0, and 6.0, confirmed with an electrochemical pH meter (270 A+, Thermo Orion). Approximately 5 mg of dried alginate-silica particles were added to 250 μL of the respective FITG-GOx solutions. Solutions were allowed to equilibrate overnight prior to analysis. Samples were prepared for image analysis by placing 10 μL of each particle suspension onto a coverslip (24×60 mm No. 1.5, VWR). Image analysis was performed on a Leica TCS 2 (Leica Microsystems) confocal imaging system equipped with a 63× HCX PL APO oil immersion lens (1.4 – 0.60 NA) and 488 nm Ar/Kr excitation.
Preparation of PtOEP and GOx-doped particles
Approximately 250 μL of stock particle suspension was placed in a microcentrifuge tube and dried under streaming N2, followed by adding 250 μL of a 500 μM PtOEP solution prepared in THF. The container was sealed to prevent volatilization of THF and stirred for 30 min, after which 30 μL of water was added and the suspension stirred for an additional 30 min. The water added to the suspension initiated a solvent-mediated controlled precipitation of PtOEP into the mesoporous particles, a technique used in previous reports to aid in the immobilization of desired molecules.38 The suspension was subsequently rinsed with DI water four times. Following the last rinse cycle, the supernatant was removed and 300 μL of 35 mg/mL GOx prepared in 0.05 M pH 4 sodium acetate buffer was added. The pH value was selected based on experimental results from the previous section. The suspension was stirred for 1.5 hours, followed by two rinsing cycles with DI water. The supernatant was subsequently removed and replaced with 250 μL of solution comprised of 30 mg/mL and 10 mg/mL of EDC and NHSS in 0.05 M sodium acetate (pH = 5.0) to catalyze amide bond formation between amine and carboxylic acid functionalities on GOx and alginate, respectively.24 Finally, the suspension of PtOEP/GOx-doped particles was stirred for 4 hours then rinsed two times with DI water. The Lowry method was used to determine the amount of GOx immobilized per particle.39 The Lowry results suggest that an average of 165 pg of GOx was immobilized in each sensor, equivalent to an internal concentration of 720 μM. For imaging purposes, a separate batch of PtOEP/GOx-doped particles were prepared using FITC-GOx and imaged with confocal microscopy. Sequential scanning mode was used, with 488 nm Ar/Kr and 543 nm He/Ne excitation, respectively.
Adsorption of nanofilm surface coatings
RITC was conjugated to PAH (PAH-RITC) according to a standard amine conjugation protocol.37 Prior to multilayer film deposition, solutions of PSS, PAH, and PAH-RITC were prepared in 0.2 M NaCl at a concentration of 2 mg/mL. The supernatant of the PtOEP/GOx-doped particles was removed and the particles resuspended in PAH-RITC. Following a 15 min adsorption period, during which the particles were continuously vortexed and protected from light with aluminum foil, the particles were rinsed three times with DI water and subsequently resuspended in PSS solution. To determine the overall number of PAH-RITC/PSS bilayers required for a strong reference signal, fluorescence emission spectra were acquired after each bilayer deposition using the sample chamber detailed in the next section. When the PtOEP emission maxima (645 nm) was approximately 75% of the RITC emission maxima (580 nm), which in this case was three bilayers of PAH-RITC and PSS (denoted {PAH-RITC/PSS}3), PAH-RITC deposition was halted. To adjust the transport properties of the coatings, an additional seven PAH/PSS bilayers were deposited using the same protocol, making the total film architecture deposited to the PtOEP/GOx-doped particles [{PAH-RITC/PSS}3/{PAH/PSS}7] and completing sensor fabrication. The complete sensors were imaged using confocal microscopy with 543 nm He/Ne excitation.
Dynamic testing apparatus
A custom dynamic testing apparatus and associate virtual instrument software control program (LabVIEW, National Instruments) was developed to monitor real-time changes in sensor response (Figure 1). The system allows control of bulk oxygen concentration by means of regulated mixing of oxygen and nitrogen through a pair of individually addressable pressure controllers (Model 00122QA, Cole-Parmer Instrument Company) for the buffer and glucose reservoir, respectively. Equilibration of oxygen levels within each reservoir was hastened with gas diffusers (ChemGlass, fine frit) and magnetic assisted stirring (Dylastir, VWR). Desired glucose concentrations in the reaction chamber were obtained through mixing of glucose and buffer stock solutions extracted from the reservoirs by peristaltic pumps (MasterFlex L/S 7550 pump drive with MasterFlex Easy Load 3 pump heads). Prior to entering the reaction chamber, the bulk oxygen level of the glucose solution was recorded using an in-line oxygen microelectrode and picoammeter (OX 500 and PA2000, Unisense). The reaction chamber consists of a custom designed garolite (Cole Parmer) flow cell, which accepts a standard microscope slide (25×75×1 mm, VWR) with the sensors immobilized to the surface. Additionally, the reaction chamber contains a port to interface the sample slide with a custom optical fiber. The optical fiber probe was comprised of one delivery fiber and six collection fibers (400 μm multi-mode, BFH37-400, Thor Labs), which was used to deliver excitation light from a Hg-Xe arc lamp (Model 68811, Oriel) containing a 530 ± 5 nm interference filter (Thor labs). Sensor emission was subsequently delivered to a diode array spectrometer (USB 2000, OceanOptics) through the collection bundle. Following passage through the reaction chamber, outlet solution oxygen levels were recorded using an additional inline microelectrode chamber prior to being collected as waste and discarded. It was determined that tubing in direct contact with solution (Norprene®, MasterFlex) exhibited negligible oxygen permeability, thereby minimizing changes in oxygen levels as the solution traveled through the apparatus.
Figure 1.

Schematic of dynamic testing apparatus used to quantify sensor response properties.
Oxygen sensitivity testing
The testing apparatus was set up to pump only buffer (0.01 M PBS, pH = 7.4) into the reaction chamber (Figure 1). To prepare a sample for testing, double-sided pressure-sensitive polyacrylate adhesive (3M) was placed on the surface of a standard microscope slide followed by adding 10 μL (~ 106) of sensors onto the adhesive surface. The sample was dried under streaming N2 and fixed into the reaction chamber. Randomized oxygen levels ranging from oxygen depleted to oxygen saturated conditions were obtained by bubbling O2 and N2 at varied partial pressures into the buffer reservoir. With the solution flow rate set at 4 mL/min, the oxygen-dependent emission spectra of the sensors were collected when the oxygen level at the reaction chamber inlet reached steady-state, as indicated by the in-line oxygen microelectrode.
Dynamic glucose sensitivity testing
Large volume reservoirs (4 L) supplied the testing apparatus with buffer and glucose, respectively (Figure 1). The buffer reservoir contained 0.01 M PBS (pH = 7.4), while the glucose reservoir contained 600 mg/dL of β-D-glucose dissolved in and 0.01 M PBS (pH = 7.4). The oxygen levels in both reservoirs were equilibrated to air saturated conditions. Sample preparation was performed as described above. Random triplicate glucose levels, ranging from 25 mg/dL to 300 mg/dL in 25 mg/dL increments, were flowed through the reaction chamber at a rate of 4 ml/min for approximately 8 min at each concentration, parameters which were experimentally predetermined to minimize convective glucose transport while maintaining bulk oxygen levels. To check reversibility of the sensors and assess baseline stability, buffer was flowed at the same rate between the glucose step changes for the same time allotment. Spectral data were collected in real-time throughout the course of the experiments.
Results and Discussion
Preparation of algilica particles
Following particle fabrication, the infrared spectrum of the particles were acquired using FTIR spectroscopy, and compared to previously reported spectra.24 As expected, the spectrum confirmed alginate-GPTS conjugation indicated by absorbance bands present at 1630cm−1 – contributed by the carboxylic acid groups on alginate – and 1730 cm−1 – contributed by the conversion of carboxylic acid groups into ester bonds during the conjugation. Furthermore, the spectrum possessed a very strong Si-O bond absorbance band between 1100 and 1000 cm−1, confirming successful condensation of the silanol and formation of the silicate structure. After confirming successful formation of the silicate structure, particle sizing analysis was performed, yielding an average diameter of 14 ± 3 μm.
Sensor component immobilization
With enzymatic smart tattoos, it is important to have a uniform distribution of the enzyme and indicator dye so that a homogeneous sensing environment is achieved. For the reported case, that involved achieving a uniform distribution of GOx and PtOEP throughout the microparticle matrix. During preliminary experimentation, the pH of the GOx loading solution was observed to have a profound effect on the enzyme distribution within the particles. To characterize this, GOx immobilization within algilica particles was performed at various pH levels and imaged using confocal microscopy (Figure 2). The distribution profile of GOx within the particles was obtained by line scan analysis of the confocal micrographs (Figure 2 – insets). By inspecting the relative fluorescence intensities of the line scan data corresponding to the particle center and comparing those to the intensities at the surface, it is evident that solution pH determines whether GOx distribution is uniform (Figure 2, pH 3 and 4) or non-uniform (Figure 2, pH 5 and 6). To quantitatively compare the profiles, the following equation was introduced to calculate an associated distribution index, Dr
Figure 2.
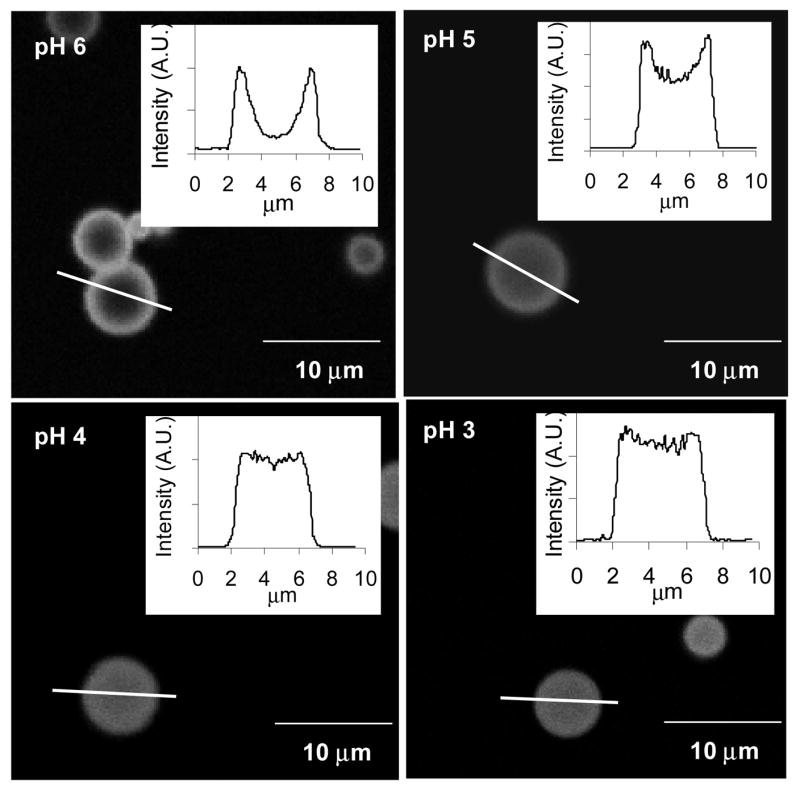
Confocal micrographs of GOx loading at different pH levels. Insets for the respective micrographs depict the intensity profile along the given line.
| (1) |
where ν̄ is the average fluorescence intensity of the center 10% of the particle and ᾱ and β̄ are the average fluorescence intensities of the outer 10% of the distribution profile, located at the right and left boundaries of the particle surface, respectively. As the value of Dr approaches unity, GOx is uniformly distributed throughout the particle, and as Dr approaches 0, the distribution of GOx becomes inhomogeneous, with bias toward the surface boundaries. The calculated mean Dr values were ~0.92, 0.92, 0.73, and 0.22 for solution pH of 3, 4, 5, and 6, respectively. Dr values close to unity were observed for pH 3.0 and 4.0 with no statistical difference between the two (student’s t test, α= 0.1), indicating that uniform distributions were obtained when GOx was loaded under these conditions. Conversely, the Dr values significantly decreased when the bulk pH was increased to 5.0 and 6.0, respectively, signifying an increased preference for GOx adsorption to the outer surface of the particle. This observed trend could be explained through pH-induced modulation of attractive and repulsive electrostatic forces. As the solution pH is reduced below the pI of GOx (4.2)40 and above the algilica pI (estimated to be 2 from reports on similar organically-modified silicates) 41–43, GOx exhibits a cationic surface potential, while the net surface potential of algilica remains anionic. Opposite surface potentials elicit attractive electrostatic forces between GOx and algilica, allowing GOx to readily penetrate and adsorb throughout the microparticle. Such behavior was observed at bulk pH levels of 3.0 or 4.0. However, as bulk pH values are elevated above the GOx pI, the surface potential of both GOx and algilica become increasingly anionic, correspondingly increasing repulsion. These repulsive forces incrementally reduce GOx penetration throughout the particle, while progressively confining adsorption to the particle surface, as seen in loading solutions of pH 5.0 and 6.0, respectively. Similarly, other reports have observed the same phenomenon and have used pH induced electrostatic loading as a means to immobilize enzymes.44 Therefore, to obtain a uniform distribution of GOx within the sensors, a pH 4.0 loading solution was used. Additionally, it is noteworthy that post-loading exposure to elevated pH levels resulted in no significant changes of the GOx distribution profile.
Following the controlled precipitation of PtOEP, algilica particles were doped with GOx, and subsequently imaged using confocal microscopy (Figure 3A and B). Dr values calculated for GOx (Figure 3A) and PtOEP (Figure 3B) were 0.87 ± 0.11 and 0.86 ± 0.08, respectively, suggesting that the precipitation technique used to adsorb PtOEP has no significant effect on GOx immobilization. We also observed that the distribution profiles of GOx or PtOEP remained unchanged if the order of immobilization was reversed (e.g. GOx loaded prior to PtOEP), suggesting interaction with different domains of the hybrid material.. The ability to obtain uniform immobilization of both GOx and PtOEP is in part due to the unique chemical and structural properties of the mesoporous algilica matrix, which is comprised of a homogeneous arrangement of hydrophilic and hydrophilic functionalities that provide thermodynamically stable environments for both GOx and PtOEP.24
Figure 3.

Confocal images depicting the distribution of FITC-GOx (A) and PtOEP (B) within algilica microparticles, obtained with sequential excitation at 488 and 543 nm. Images C and D are of completed sensors, depicting the location of fluorescent components – RITC-doped nanofilms (C) and PtOEP (D) – as obtained with 543 nm excitation.
In fluorescence sensing applications, it is common to incorporate a reference dye which is non-sensitive to the measured analyte into the sensor, allowing ratiometric read-out. Ratiometric monitoring of sensor response provides a signal independent to instrumental drift, namely light source fluctuations, and the number of sensors interrogated. To provide a reference signal, RITC-doped multilayer nanofilms were adsorbed to the outer surface of the GOx/PtOEP-doped particles using the well established layer-by-layer technique. The completed ratiometric sensors were imaged using confocal microscopy (Figure 3C and D), and the resulting line scans used to calculate Dr. The Dr value calculated for the RITC-nanofilms was 0.22 ± 0.03, confirming surface limited adsorption of the nanofilms.
Sensor response to bulk oxygen changes
The reported sensors indirectly measure local glucose levels by monitoring changes in local oxygen levels elicited through GOx catalyzed glucose oxidation. Therefore, it is critical to quantify the oxygen response characteristics of the sensors prior to performing glucose sensitivity experiments. To accomplish this, the dynamic testing apparatus (Figure 1) was used to vary bulk oxygen levels and collect emission spectra after the establishment of concentration equilibrium. To depict the oxygen-induced quenching effect observed at extreme oxygen levels - oxygen and nitrogen saturated conditions, respectively – with air saturated conditions as a reference, selected spectra normalized to the RITC emission maxima at 585 nm are presented in Figure 4A. A more concise view of the oxygen-quenching effect is demonstrated by plotting the PtOEP/RITC peak ratio, which was obtained by normalizing the intensity observed at the PtOEP maxima (645 nm) to that of the RITC maxima (585 nm), versus bulk oxygen concentration (Figure 4B). These data suggest the sensors are highly sensitive to oxygen levels between depleted and atmospheric (277 μM) levels, such that approximately 95% of the total change in ratiometric intensity occurs within this region. For enzymatic smart tattoo, applications high oxygen sensitivity within this region is critical to optimal sensor performance. Reported reaction-kinetic simulations of similar systems under physiological conditions show that local oxygen levels within the sensor can be reduced to 10 μM (assuming intradermal oxygen levels of 90 μM) when elevated blood-glucose levels (350 mg/dL) are present.24,45
Figure 4.
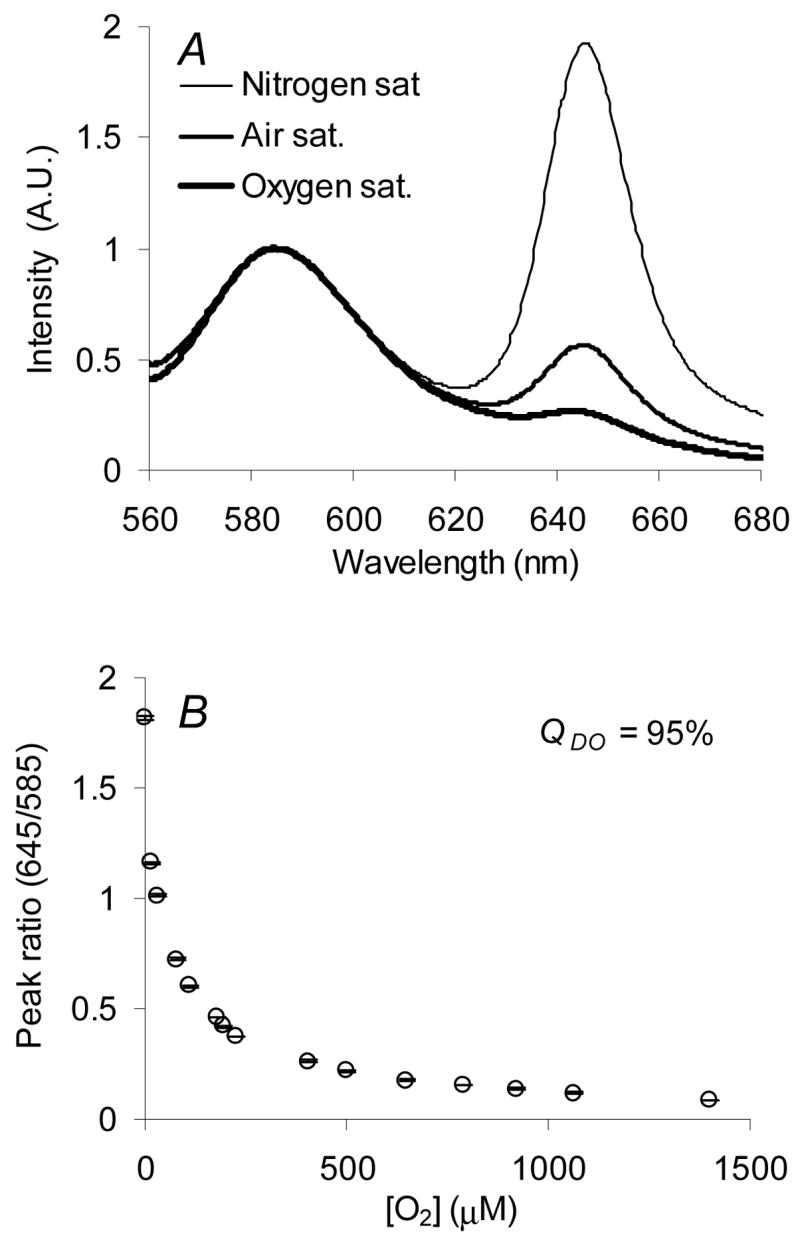
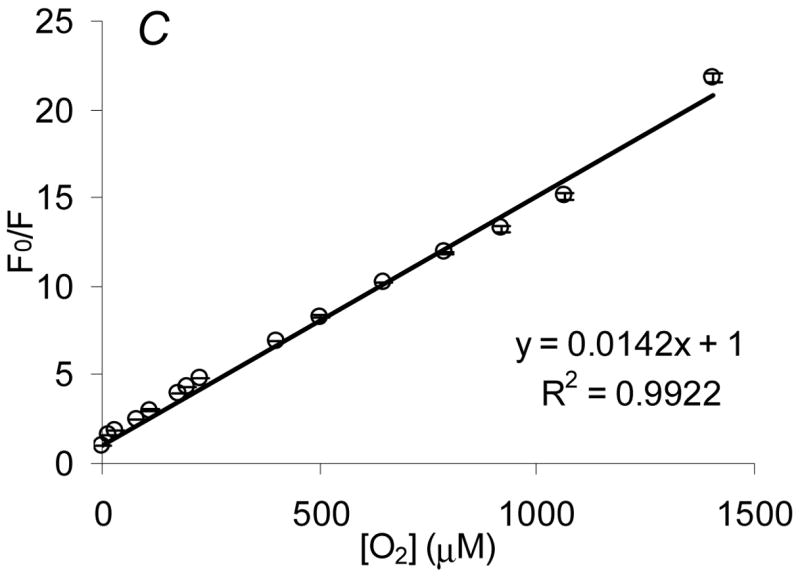
Sensor response to variations in bulk oxygen levels: spectral response to air, nitrogen, and oxygen saturated environments (A), peak ratio response to randomized oxygen levels ranging from oxygen depleted to oxygen saturated conditions (B), and the Stern-Volmer quenching characteristics (C). Error bars denote on standard deviation from triplicate measurements.
To quantify the sensitivity of the overall oxygen-induced quenching response (QDO), the following relationship is commonly used:
| (2) |
where RN2 and RO2 are the PtOEP/RITC peak ratios observed under nitrogen and oxygen-saturated conditions, respectively. As expected, QDO was calculated to be 95%, a sensitivity equivalent to that observed in our previous work on algilica-based dissolved oxygen sensors which did not contain GOx.36 This finding indicates that GOx adsorption does not result in an impenetrable barrier that prevents fractal diffusion of oxygen and quenching of PtOEP, but rather a situation that allows oxygen to readily diffuse throughout the matrix. Although the steady-state response is not altered by GOx adsorption, current studies are underway to examine whether the absolute transport rate of oxygen is affected. Additionally, a linear Stern-Volmer relationship (R2 = 0.99) was observed (Figure 4C), confirming PtOEP adsorption within a homogeneous environment, a characteristic also observed in previous work.32,36 These data show high sensitivity to oxygen levels within predicted sensor operation limits, while exhibiting linear quenching characteristics.
Sensor response to bulk glucose changes
Although the reported system demonstrated high sensitivity to oxygen, a response to glucose is not necessarily guaranteed. Since the sensors are designed to indirectly measure glucose levels through monitoring glucose-limited consumption of local oxygen, the system must adequately control glucose diffusion relative to that of oxygen. The glucose response characteristics were monitored using the dynamic testing apparatus (Figure 1), which allowed real-time monitoring of sensor performance as step changes in bulk glucose levels were introduced. In this report, dynamic testing was used to efficiently evaluate critical sensor characteristics such as stability, response time, reversibility, sensitivity, detection limit, and analytical range.
Dynamic response data collected during sensor exposure to random bulk glucose levels ranging from 0 to 300 mg/dL are shown in Figure 5. It is important to note that the data displayed in Figure 5 are only that of the initial 150 min of the experiment, which totaled 500 min in duration. The data displayed in Figure 5A are the respective maximum peak intensities of RITC and PtOEP, which were presented individually to highlight the advantages of ratiometric monitoring. Figure 5A depicts three key properties about the sensor fluorimetrics: (1) step changes in glucose concentration elicit a significant increase in PtOEP emission (Figure 5A, denoted “Glucose step”), (2) flushing the system with buffer (Figure 5A, marked “Buffer flush”) caused the PtOEP intensity to return to the baseline value, and (3) RITC emission intensity remains independent of modulations in glucose concentration. Using ratiometric analysis, it is possible to account for source fluctuations and changes in sensor concentration. One such case can be observed in Figure 5A, where after 10 min had elapsed, the peak intensities of PtOEP and RITC began to decline as a result of sensor detachment (Figure 5A, marked as “Sensor detachment”), causing a decrease in sensor concentration; however, after normalizing the peak intensities, the concentration dependent artifact is eliminated and the baseline prior to glucose introduction (t = 0–45 min) becomes stable (Figure 5B). Additionally, it is noteworthy that after approximately 50 min, the RITC peak also becomes stable (Figure 5A), signifying the end of detachment of weakly adhered sensors.
Figure 5.

Dynamic sensor response data shown in raw intensity units (A) and ratiometric intensity (B). Computing the ratio of the intensity at 645nm to that at 585nm accounts for changes in particle concentration due to sensor detachment.
While Figure 5 is instrumental in depicting the advantages of ratiometric monitoring, it also illustrates the fundamental sensor operating principles. Following a step change in environment from pure buffer to buffer with glucose, an increase in ratiometric intensity is observed, resulting from depletion of internal oxygen levels as glucose diffuses from the bulk into the sensor and is oxidized by GOx. As the balance between diffusive glucose delivery, consumption of glucose and oxygen via glucose oxidation, and diffusive oxygen replenishment begin to equilibrate, the rate of change in the ratiometric signal is significantly reduced; however, it is important to note that, in this experiment, the intensity does not reach steady-state before the chamber is flushed with buffer. Initially, it was hypothesized that the drift observed my be due to depletion of local oxygen levels in the sensor microenvironment; however, additional experiments performed with extended exposure times of 10 minutes yielded stable intensity values. Thus, it appears that this drift is simply indicative of the time required for the diffusion of glucose and oxygen and reaction to reach equilibrium over the population of sensors. Additional discussion on how this drift may affect results will be given in the following sections. Furthermore, the ratiometric intensity returns to baseline after glucose is removed from the bulk and local oxygen levels within the sensor approach those of bulk levels (277 μM).
Sensor stability
In this report, the sensors were immobilized on a glass slide and continuously illuminated for approximately 500 min, during which time approximately 5000 spectra were acquired. The power density of the excitation source at the sample was measured to be 36 mW cm−2. Baseline values were defined as the average ratiometric intensity values occurring at 0 mg/dL glucose throughout the experiment. A statistical comparison of the baseline values indicate insignificant drift had occurred through the course of the experiment (p = 0.07). It is also noteworthy to state that the data were not corrected for photobleaching, indicating these systems exhibit a high degree of photostability.
Response time and reversibility
Response time is a critical figure of merit for diabetic monitoring, as sensors must respond within the physiological time scale of glucose fluctuations to ensure accuracy. Using the dynamic data acquired, sensor response time was defined as the time required for the PtOEP/RITC peak ratio to reach 95% of its maximum value (as determined by the average of the 25 data points collected before buffer flushing), in response to a step change in glucose concentration. As previously discussed, the response does not entirely reach steady-sate conditions before flushing the chamber with buffer. However, upon comparison between data sets acquired over extended glucose exposure times, the variation between response times averaged less than 5%. Using the data presented in Figure 5B, the average response time was determined to be 86 ± 12 seconds, making these sensors adequate to monitor fluctuations in blood glucose, which usually occur over a period of 30 min.46 Upon comparison, the measured response time is similar to other systems based on optical oxygen transduction of glucose levels.23 Additionally, ongoing experiments are in progress to quantify the effect of fluidic latency of the test chamber on response time.
Reversibility is a figure of merit that assesses baseline change before and after exposure to the analyte and was quantified by flushing the reaction chamber with buffer prior to subsequent glucose step changes and the calculating the baseline variation. Figure 6 shows sensor response to several step changes in glucose concentrations. It is important to note that after glucose is flushed from the bulk solution (Figure 6B), the PtOEP/RITC peak ratio returns to the baseline value observed prior to the glucose step (Figure 6A). The average percent change in the baseline value before and after a step change in glucose concentration was determined to be 0.75 ± 0.6 %, indicating a high degree of reversibility. This result is not surprising, as enzymatic based sensors usually display a high degree of reversibility due to consistent analyte consumption.9,21,23
Figure 6.
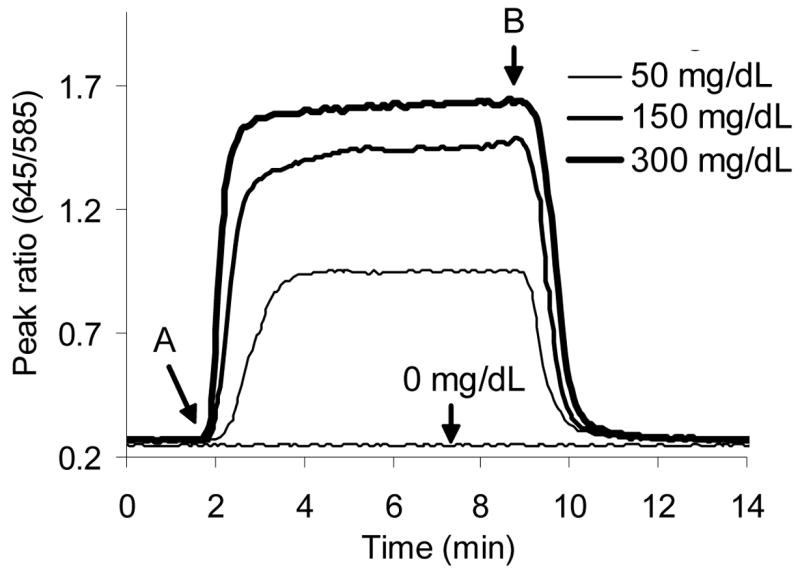
Sensor response to step changes in glucose concentration. Sensors were exposed to a step change in glucose at (A) and the chamber subsequently flushed with buffer to remove glucose (B). The peak ratio returns to baseline (0 mg/dL) value following glucose removal.
Sensitivity
Sensitivity is a critical sensor performance parameter that is used to quantitatively describe the magnitude of change in output signal due to an input change in analyte concentration. A sensitivity curve was generated by plotting the percent change in PtOEP/RITC peak ratio versus bulk glucose concentration (Figure 7). The response sensitivity was determined by taking the slope of the linear region, which was defined as the region within the sensitivity curve that exhibited a R2 value greater that 0.95. Using linear regression, with the y-intercept forced to 0, the response sensitivity was determined to be 4.16 ± 0.57 %/mg dL−1 (assuming 95% confidence bounds, Figure 7), which is at least one order of magnitude greater than those of similar systems.10,21,23,27 However, it should be noted that high sensitivity is often associated with a reduction of the analytical range; therefore, the sole objective is not high glucose sensitivity, but to obtain the highest sensitivity while staying within the confines of the desired analytical range. Additional discussion on the analytical range and how multilayer thin films can be used to tailor the response sensitivity will be given later.
Figure 7.
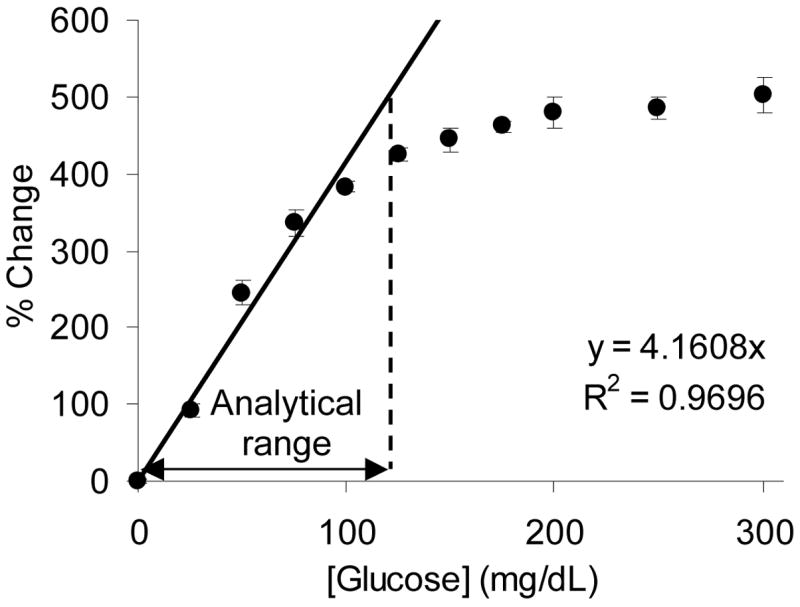
Percent change in PtOEP/RITC peak ratio values following step changes in bulk glucose levels. Regression line used to depict linear operation range, while analytical range demarks a 10% response deviation from linearity. Error bars denote one standard deviation of random triplicate measurements.
Detection limits
Detection limits are used to quantify the lowest concentration of analyte that can be readily detected. For this work, the detection limit used is the quantification limit, which is defined as the lowest analyte concentration in which there is at least 90% certainty that the analyte is present, and was used to determine if these sensor prototypes could detect the lower limit of the clinically accepted glucose monitoring rage, 40 mg/dL.47 The quantification limit, CQL, is defined as:
| (2) |
where σbaseline is the standard deviation of the baseline (e.g. glucose concentration equal to 0 mg/dL) and S is the response sensitivity defined above. CQL was determined to be 1.5 ± 0.2 mg/dL. The low CQL is a result of the high signal-to-noise ratio, which was determined to be 175, and the high response sensitivity of the sensors. Given that the hypoglycemic comas are commonly observed when blood glucose levels drop below 30 mg/dL,48 the low detection limit of these devices are not advantageous for diabetic monitoring devices; however, other applications requiring low detection limits could find this characteristic useful.
Analytical range
The analytical range is the most critical figure of merit when evaluating the feasibility of a sensor. The analytical range was defined as the concentration range over which glucose levels can be statistically differentiated. Typically the lower limit of the analytical range is defined as CQL, while the upper limit is the analyte concentration observed when the response profile deviates 10 % from linearity (Figure 6). Using this definition, the analytical range of the sensors was determined to be 2 – 120 mg/dL of glucose, which does not cover the entire accepted range of 40 – 350 mg/dL required for self-testing systems.47 However, the reported system could adequately monitor hypoglycemic events (blood glucose levels below 70 mg/dL), which according to one report, is the most critical aspect of diabetic care.49 A detailed discussion on how the analytical range can be extended to cover a more clinically viable range using polyelectrolyte multilayers will be given later.
The fundamental operating principles of the sensors can be described through further analysis of Figure 7. As previously mentioned, these sensors operate on the principle of glucose-limited oxygen consumption within the smart tattoo. As glucose diffuses into the sensor, oxygen is consumed and rapidly replaced, allowing indirect glucose monitoring through an oxygen reporter. To achieve this essential behavior, the relative rate of oxygen into the sensor must be relatively large compared to that of glucose, such that local oxygen levels within the sensor are in excess, resulting in glucose-limited oxygen consumption. This condition can be observed in the linear response region ([β-D glucose] < 90 mg/dL, Figure 7), where steady-state oxygen levels are proportionally reduced in response to increasing bulk glucose levels. As bulk glucose levels are increased, the rate of glucose delivery into the sensor begins to eclipse the rate of oxygen replenishment, preventing the relative recovery of oxygen levels within the sensor. In this state of operation, which occurs for these sensors at bulk glucose levels between 90 and 150 mg/dL (Figure 7), the sensor response begins to deviate from linearity, indicating the onset of oxygen-limited glucose consumption. With additional increases in glucose levels above 150 mg/dL, the sensor does not respond to changes in bulk glucose concentrations (Figure 7). This phenomenon is due to internal depletion of oxygen levels, such that oxygen is readily consumed upon diffusion into the sensor, a condition elicited by the surplus of local glucose within the sensor and signifies absolute oxygen-limited catalysis and response saturation. Therefore, to extend the analytical range to cover both hypoglycemic and hyperglycemic events the mass transfer properties of oxygen and glucose must be altered to limit the glucose delivery rate into the tattoo particles. Limiting glucose diffusion allows oxygen levels within the sensor to remain relatively elevated, allowing the sensor to operate under glucose-limited conditions over a greater range of bulk glucose levels.24 In our approach, we utilize surface-immobilized multilayer nanofilm technology to control the analyte mass transfer and induce glucose-limiting kinetics. This technology allows exquisite control over substrate diffusivities through nanoscale changes in film thickness.9,24,50,51 We hypothesize that the analytical range of these sensors can be tuned by simply altering the thickness of the nanofilms, a hypothesis which is currently under examination and will be reported in a second installment.
It is also important to reiterate that the glucose sensitivity experiments in this work were performed at air-equilibrated conditions, in part because of the ease associated with establishing and maintaining bulk oxygen levels. However, the local oxygen supply within the dermis has been shown to be highly dependent on atmospheric conditions as well as depth, with concentration fluctuation from 215 μM (near the epidermis-dermis junction) to 90 μM (near vasculature).52 At elevated bulk oxygen levels, such as those at the epidermis-dermis junction, the average steady-state oxygen concentration within the sensors is elevated;53 therefore, higher incremental glucose levels are required to significantly reduce the elevated average oxygen level within the sensor. This occurrence ultimately results in an extension of the glucose-limited range, which in turn results in decreased device sensitivity and increased analytical range. At lower bulk concentration, such as those near hematogenic supplies, internal oxygen levels are depleted at lower bulk glucose concentrations, resulting in a reduction of the glucose-limited range. While current experimental work is quantifying the effect of bulk oxygen levels on response properties, direct oxygen monitoring functionality should be incorporated to allow compensation for fluctuations in bulk oxygen levels within the dermis for this approach to ultimately be successful in in vivo applications.
Conclusion
A highly sensitive microparticle based enzymatic glucose sensor technology has been demonstrated. The sensors were rely upon PtOEP adsorbed into silicate particles using a controlled precipitation technique, followed by the adsorption of GOx using electrostatic loading and subsequent chemical conjugation. Fluorescent nanofilms were deposited onto the particle to provide a reference signal and obtain glucose-limited oxygen consumption within the sensor by controlling glucose transport. The oxygen-dependent response of the sensors was quantified, indicating linear quenching characteristics and high sensitivity to oxygen within the expected range of physiological operation. Upon the addition of glucose, these-self referencing systems responded to step changes in glucose concentration in less than 90 seconds. The ratiometric monitoring approach resulted in excellent baseline stability and reversibility between alterations of bulk glucose levels, and the static sensitivity of the response was sufficient to accurately predict glucose concentrations over the hypoglycemic range (4 %/dL over 2 – 120 mg/dL). Although the prototypes demonstrated here are not sensitive over the entire range required for complete diabetic monitoring of glucose, which extends up to 600 mg/dL, they are indeed adequate to monitor hypoglycemia, which could be potentially useful for various research applications. Based on previous work in modeling the response properties of such sensing systems, it is believed that the multilayer nanofilms can be modified to control glucose diffusivity, allowing modulation of sensitivity and analytical range to cover both hypo- and hyperglycemic events. These positive results suggest that similar approaches may be used to develop enzymatic smart tattoos for a number of different applications, by using mesoporous microsphere platforms with enzymes, fluorescent indicators, and transport-limiting nanofilms.
Acknowledgments
The authors acknowledge the NIH (R01 EB000739) for financial support.
References
- 1.Pickup JC, Hussain F, Evans ND, Rolinski OJ, Birch DJS. Biosens Bioelectron. 2005;20:2555. doi: 10.1016/j.bios.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Amerov AK, Chen J, Small GW, Arnold MA. Anal Chem. 2005;77:4587–4594. doi: 10.1021/ac0504161. [DOI] [PubMed] [Google Scholar]
- 3.Stuart DA, Yonzon CR, Zhang X, Lyandres O, Shah NC, Glucksberg MR, Walsh JT, VanDuyne RP. Anal Chem. 2005;77:4013–4019. doi: 10.1021/ac0501238. [DOI] [PubMed] [Google Scholar]
- 4.Wan Q, Cote GL, Dixon JB. J Biomed Opt. 2005;10:024029. doi: 10.1117/1.1891175. [DOI] [PubMed] [Google Scholar]
- 5.Wolfbeis OS. J Mater Chem. 2005;15:2657–2669. [Google Scholar]
- 6.Russell RJ, Pishko MV, Gefrides CC, McShane MJ, Cote GL. Anal Chem. 1999;71:3126–32. doi: 10.1021/ac990060r. [DOI] [PubMed] [Google Scholar]
- 7.McShane MJ. Diabetes Technol Ther. 2002;4:533–8. doi: 10.1089/152091502760306625. [DOI] [PubMed] [Google Scholar]
- 8.McShane MJ, Rastegar S, Pishko M, Cote GL. IEEE T Biomed Eng. 2000;47:624–32. doi: 10.1109/10.841334. [DOI] [PubMed] [Google Scholar]
- 9.McShane MJ. In: Topics in Fluorescence Spectroscopy. Geddes CD, Lakowicz JR, editors. Vol. 11 Springer; New York: 2006. [Google Scholar]
- 10.Chinnayelka S, McShane MJ. Anal Chem. 2005;77:5501–5511. doi: 10.1021/ac050755u. [DOI] [PubMed] [Google Scholar]
- 11.D’Auria S, DiCesare N, Staiano M, Gryczynski Z, Rossi M, Lakowicz JR. Anal Biochem. 2002;303:138–44. doi: 10.1006/abio.2001.5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D’Auria S, Herman P, Rossi M, Lakowicz JR. Biochem Biophys Res Commun. 1999;263:550–3. doi: 10.1006/bbrc.1999.1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barone PW, Baik S, Heller DA, Strano MS. Nat Mater. 2005;4:86. doi: 10.1038/nmat1276. [DOI] [PubMed] [Google Scholar]
- 14.Ibey BL, Beier HT, Rounds RM, Cote GL, Yadavalli VK, Pishko MV. Anal Chem. 2005;77:7039–7046. doi: 10.1021/ac0507901. [DOI] [PubMed] [Google Scholar]
- 15.Chinnayelka S, McShane MJ. Diabetes Technol Ther. 2006;8:269–78. doi: 10.1089/dia.2006.8.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ballerstadt R, Polak A, Beuhler A, Frye J. Biosens Bioelectron. 2004;19:905. doi: 10.1016/j.bios.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 17.Ballerstadt R, Schultz JS. Anal Chem. 2000;72:4185. doi: 10.1021/ac000215r. [DOI] [PubMed] [Google Scholar]
- 18.Chinnayelka S, McShane MJ. J Fluoresc. 2004;14:585. doi: 10.1023/b:jofl.0000039345.57924.f3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gough DA, Lucisano JY, Tse PHS. Anal Chem. 1985;57:2351–2357. doi: 10.1021/ac00289a042. [DOI] [PubMed] [Google Scholar]
- 20.Brown JQ, Srivastava R, McShane MJ. Biosens Bioelectron. 2005;21:212–6. doi: 10.1016/j.bios.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 21.Brown JQ, Srivastava R, Zhu H, McShane MJ. Diabetes Technol Ther. 2006;8:288–295. doi: 10.1089/dia.2006.8.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moschou EA, Sharma BV, Deo SK, Daunert S. J Fluoresc. 2004;14:535–47. doi: 10.1023/b:jofl.0000039341.64999.83. [DOI] [PubMed] [Google Scholar]
- 23.Xu H, Aylott JW, Kopelman R. The Analyst. 2002;127:1471–1477. doi: 10.1039/b202782h. [DOI] [PubMed] [Google Scholar]
- 24.Zhu H, Srivastava R, Brown JQ, McShane MJ. Bioconjugate Chem. 2005;16:1451–1458. doi: 10.1021/bc050171z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosenzweig Z, Kopelman R. Anal Chem. 1996;68:1408. doi: 10.1021/ac950864g. [DOI] [PubMed] [Google Scholar]
- 26.Wolfbeis OS, Oehme I, Papkovskaya N, Klimant I. Biosens Bioelectron. 2000;15:69–76. doi: 10.1016/s0956-5663(99)00073-1. [DOI] [PubMed] [Google Scholar]
- 27.Papkovsky DB. Sensor Actuat B-Chem. 1993;B11:293. [Google Scholar]
- 28.Schonhoff M. Curr Opin Colloid In. 2003;8:86–95. [Google Scholar]
- 29.Liu X, Bruening ML. Chem Mater. 2004;16:351–357. [Google Scholar]
- 30.Miller MD, Bruening ML. Langmuir. 2004;20:11545–11551. doi: 10.1021/la0479859. [DOI] [PubMed] [Google Scholar]
- 31.Decher G. Science. 1997;277:1232–1237. [Google Scholar]
- 32.Koo YEL, Cao Y, Kopelman R, Koo SM, Brasuel M, Philbert MA. Anal Chem. 2004;76:2498–2505. doi: 10.1021/ac035493f. [DOI] [PubMed] [Google Scholar]
- 33.Papkovsky DB, O’Riordan TC. J Fluoresc. 2005;15:569–584. doi: 10.1007/s10895-005-2830-x. [DOI] [PubMed] [Google Scholar]
- 34.Lu X, Manners I, Winnik MA. Macromolecules. 2001;34:1917–1927. [Google Scholar]
- 35.Han BH, Manners I, Winnik MA. Chem Mater. 2005;17:3160–3171. [Google Scholar]
- 36.Stein EW, Zhu H, McShane MJ. JACS. 2006 Submitted. [Google Scholar]
- 37.Brinkley M. Bioconjugate Chem. 1992;3:2–13. doi: 10.1021/bc00013a001. [DOI] [PubMed] [Google Scholar]
- 38.Sukhorukov G, Dahne L, Hartman J, Donath E, Mohwald H. Adv Mater. 2000;12:112–115. [Google Scholar]
- 39.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 40.Pazur JH, Kleppe K. Biochemistry. 1964;3:578–83. doi: 10.1021/bi00892a018. [DOI] [PubMed] [Google Scholar]
- 41.McDonagh C, MacCraith BD, McEvoy AK. Anal Chem. 1998;70:45–50. doi: 10.1021/ac970461b. [DOI] [PubMed] [Google Scholar]
- 42.Tohver V, Smay JE, Braem A, Braun PV, Lewis JA. PNAS. 2001;98:8950–8954. doi: 10.1073/pnas.151063098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wirnsberger G, Yang P, Huang HC, Scott B, Deng T, Whitesides GM, Chmelka BF, Stucky GD. J Phys Chem B. 2001;105:6307–6313. [Google Scholar]
- 44.Coppi G, Iannuccelli V, Leo E, Bernabei MT, Cameroni R. J Microencapsul. 2002;19:37–44. doi: 10.1080/02652040110055621. [DOI] [PubMed] [Google Scholar]
- 45.Evans NT, Naylor PF. Respir Physiol. 1966;2:61–72. doi: 10.1016/0034-5687(66)90038-7. [DOI] [PubMed] [Google Scholar]
- 46.Kulcu E, Tamada JA, Reach G, Potts RO, Lesho MJ. Diabetes Care. 2003;26:2405–2409. doi: 10.2337/diacare.26.8.2405. [DOI] [PubMed] [Google Scholar]
- 47.Brunner GA, Ellmerer M, Sendlhofer G, Wutte A, Trajanoski Z, Schaupp L, Quehenberger F, Wach P, Krejs GJ, Pieber TR. Diabetes Care. 1998;21:585–590. doi: 10.2337/diacare.21.4.585. [DOI] [PubMed] [Google Scholar]
- 48.Ben-Ami H, Nagachandran P, Mendelson A, Edoute Y. Arch Intern Med. 1999;159:281–4. doi: 10.1001/archinte.159.3.281. [DOI] [PubMed] [Google Scholar]
- 49.Philip EC. Diabetes Metab Res. 1999;15:42–46. doi: 10.1002/(sici)1520-7560(199901/02)15:1<42::aid-dmrr1>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 50.Hu FB, van Dam RM, Liu S. Diabetologia. 2001;44:805–817. doi: 10.1007/s001250100547. [DOI] [PubMed] [Google Scholar]
- 51.Stanton BW, Harris JJ, Miller MD, Bruening ML. Langmuir. 2003;19:7038–7042. [Google Scholar]
- 52.Stucker M, Struk A, Altmeyer P, Herde M, Baumgartl H, Lubbers DW. J Physiol (Lond) 2002;538:985–994. doi: 10.1113/jphysiol.2001.013067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brown JQ, McShane MJ. Biosens Bioelectron. 2006;21:1760–1769. doi: 10.1016/j.bios.2005.08.013. [DOI] [PubMed] [Google Scholar]