

Abstract
Tissue factor (TF) facilitates the recognition and rapid activation of factor X (fX) by factor VIIa (fVIIa) in the extrinsic Xase pathway. TF makes extensive interactions with both light and heavy chains of fVIIa, however, with the exception of a basic recognition site for the Gla-domain of fX, no other interactive-site on TF for the substrate has been identified. Structural and modeling data have predicted that a basic region of TF comprised of residues Asn-199, Arg-200 and Lys-201 located at a proper height on the membrane surface to interact with either the C-terminus of the Gla-domain or the EGF-1 domain of fX. To investigate this possibility, we prepared the Ala substitution mutants of these residues and evaluated their ability to function as cofactors for fVIIa in the activation of wild-type fX and its two mutants which lack either the Gla-domain (GD-fX) or both the Gla and EGF-1 domains (E2-fX). All three TF mutants exhibited normal cofactor activity in the amidolytic activity assays, but the cofactor activity of Arg-200 and Lys-201 mutants in fVIIa activation of both fX and GD-fX, but not E2-fX was impaired ~3-fold. Further kinetic analysis revealed that kcat with both TF mutants are impaired with no change in Km. These results suggest that both Arg-200 and Lys-201 of TF interact with EGF-1 of fX to facilitate the optimal docking of the substrate into the catalytic groove of the protease in the activation complex.
Tissue factor (TF)1 is an integral membrane cofactor that upon exposure to circulating blood and subsequent binding to factor VIIa (fVIIa) catalyzes the rapid activation of factors IX (fIX) and X (fX) to their catalytically active forms, thereby initiating the blood clotting cascade (2–4). The structure of TF is composed of two fibronectin type III-like extracellular domains, a single membrane spanning domain and a short cytoplasmic tail (5,6). All three domains of TF are required for the physiological function of the cofactor, however, the two extracellular domains expressed by recombinant DNA methods as a soluble protein (sTF) can bind to fVIIa with a high affinity to enhance both the amidolytic and proteolytic activities of the protease (7–10). TF improves the proteolytic activity of fVIIa toward both of its natural substrates fIX and fX more than 104-fold (11). Recent x-ray crystal structure of the fVIIa-sTF complex has provided some insight into the mechanism by which the cofactor may improve the catalytic efficiency of fVIIa in the extrinsic Xase complex (12). It has been noted that fVIIa makes extensive interactions with both the N- and C-terminal domains of sTF, and based on the elongated structure of the complex, it has been hypothesized that, similar to fVIIa, the cofactor would also interact with the natural substrates in a similar manner (10,12). In support of this proposal, recent fluorescence resonance energy transfer studies demonstrated that the active-site of fVIIa in complex with TF in the extrinsic Xase complex is also located far above the membrane surface (13). Thus, for effective recognition by fVIIa, both fIX and fX must also assemble into the activation complex in the same extended conformations in order to maintain the activation peptides of the substrates at a similar distance to that of the active-site of fVIIa above the membrane surface (2,12,14,15).
Like other vitamin K-dependent plasma serine proteases, the structure of fVIIa consists of a light and a heavy chain held together by a disulfide bond (16,17). The N-terminal light chain of fVIIa contains the Gla and two epidermal growth factor (EGF)-like domains and the C-terminal heavy chain contains the trypsin-like catalytic domain of the protease (16,17). Both the light and heavy chains of fVIIa have extensive interaction with both the membrane distal N-terminal and the membrane proximal C-terminal domains of TF in the extrinsic Xase complex (7,12,18) and most of these interactive-sites on both the cofactor and the protease have been mapped by mutagenesis studies (7,10,14,18). On the other hand, with the exception of the Gla-domain of fX, which is known to interact with the C-terminal domain of TF, no other recognition site on TF for the substrate has been characterized. Thus, it has been demonstrated that several basic residues on the C-terminal domain of TF (in particular Lys-165 and Lys-166) interact with the Gla-domain of fX (and also fIX), thereby stabilizing the substrate in an extended conformation on the membrane surface (10,19,20). While it is known that the first EGF domain (EGF-1) of fX is essential for the recognition and the assembly of the substrate into the extrinsic Xase complex (21), its interactive-site(s) on the fVII-TF complex has not been identified.
Molecular modeling based on the x-ray crystal structure of the fVIIa-sTF complex (Figure 1) has predicted that the membrane proximal TF residues Asn-199, Arg-200, and Lys-201 may have suitable height and topology on the membrane surface to interact with either the C-terminal residues of the Gla-domain or EGF-1 of fX in the extrinsic Xase complex (10,12). To test this hypothesis, we substituted all three residues individually with Ala in three different constructs and expressed them as soluble sTF2-219 forms in a bacterial periplasmic expression/purification vector system. All three mutants were purified to homogeneity and characterized with respect to their ability to function as cofactors in the enhancement of the amidolytic and proteolytic activity of fVIIa using Spectrozyme fVIIa, wild-type fX and two deletion derivatives of fX as substrates. All three TF mutants exhibited normal cofactor activity, thereby enhancing the amidolytic activity of fVIIa toward the chromogenic substrate with normal catalytic efficiency. On the other hand, the Arg-200 and Lys-201 mutants of TF each exhibited ~3-fold decreased cofactor activity with both wild-type and the mutant of fX in which the Gla-domain of the substrate was deleted (GD-fX). However, both mutants had normal cofactor activity with another mutant of fX in which both the Gla and EGF-1 (E2-fX) were deleted. Further kinetic analysis revealed that the decreased cofactor activities of the mutants are primarily due to impairments in the kcat of the activation reactions. These results suggest that both Arg-200 and Lys-201 of TF interact with the first EGF domain of fX to mediate the optimal docking of the activation peptide of the substrate into the active-site groove of fVIIa in the extrinsic Xase pathway.
Figure 1.
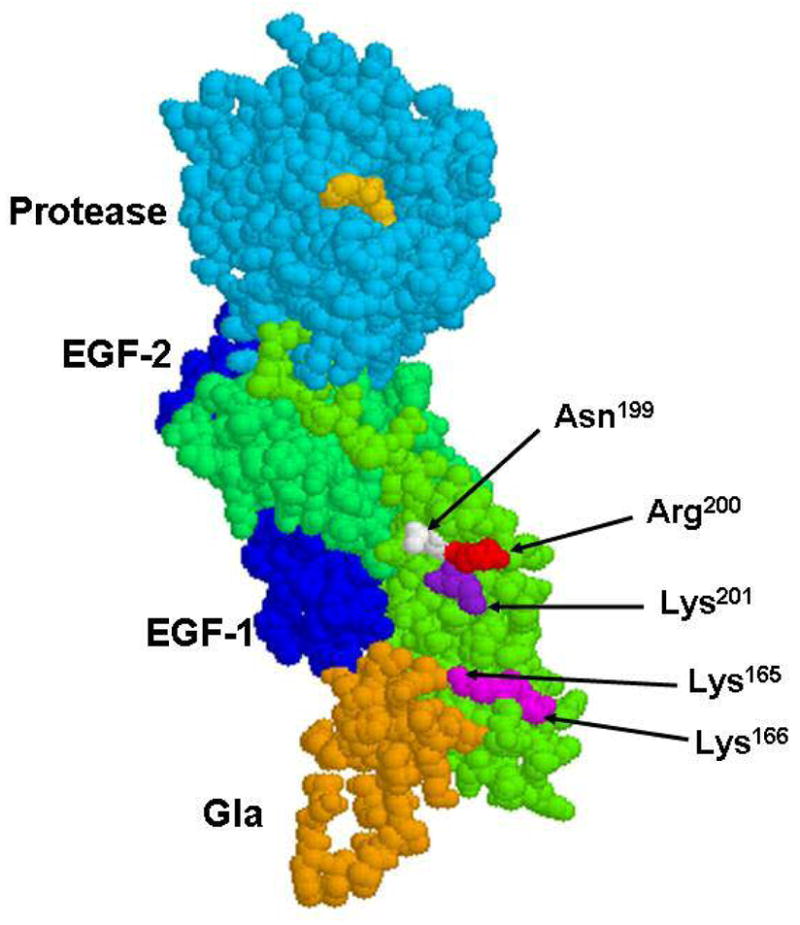
The space-filling model of the crystal structure of the fVIIa-sTF complex. The side chains of three residues Asn-199, Arg-200 and Lys-201 are shown by arrows. The side chains of Lys-165 and Lys-166 which are thought to interact with the Gla-domain of fVIIa (19,20) are also shown. The sTF residues are colored in green, the residues of the Gla-domain of fVIIa are colored in gold, EGF-1 and EGF-2 are in dark blue and the protease domain of the PPACK-inhibited fVIIa is shown in cyan. The inhibitor in the active-site of fVIIa is shown in yellow. The coordinates (Protein Data Bank accession code 1DAN) were used to prepare the figure (12).
Materials and Methods
Construction, Mutagenesis and Expression of Soluble Tissue Factor in Bacteria
Construction and expression of soluble TF lacking both the trans-membrane and cytoplasmic domains (sTF2-219) in the pINIH-pelB-neo bacterial periplasmic expression/purification vector system has been described (22). The Ala substitution mutants of sTF2-219 (N199A, R200A, and K201A) were prepared by PCR mutagenesis methods in the same vector system as described (22). After confirmation of the accuracy of the mutagenesis by DNA sequencing, the mutant constructs were transformed into the BL21 strain of E. coli and selection was carried out in kanamycin as described (9,22). Bacterial colonies harboring the wild-type and mutant sTF2-219 derivatives were grown in LB medium containing 50 μg/mL kanamycin in one liter flasks at 37 °C to a cell density of A600 = 0.6–1.0 and the expression was induced by addition of IPTG to a final concentration of 1 mM. The incubation was continued overnight (~12 h) at room temperature (~25 °C). The supernatant was collected by centrifugation at 4000g for 30 min and the periplasmic extract was prepared by hypotonic shock of bacterial pellet in 100 mL of H2O for 30 min and centrifugation at 10,000g for 15 min. The supernatant and the periplasmic extract were mixed and supplemented with 20 mM Tris-HCl (pH 7.5), 10 mM benzamidine, 0.02% NaN3, 5 mM CaCl2, and chromatographed on the HPC4 monoclonal antibody immobilized on Affi-Gel 10 (Bio-Rad) as described previously (9). The sTF2-219 concentrations were determined from the absorbance at 280 nm assuming a molecular weight of 27,000 and extinction coefficient (E1%1cm) of 14.8, and by an amidolytic activity assay as described (9). The Ser-195 to Ala substitution mutant of fX (fX S195A) was expressed and purified by a combination of immunoaffinity and anion exchange chromatography as described previously (23). The expression and characterization of the recombinant fX mutant lacking both the Gla and EGF-1 domains (E2-fX) has been described previously (24). A protein C mutant in which the Gla and hydrophobic stack domains of the zymogen (residues 1-45) was replaced with the corresponding domains of fX (PC fX-Gla) were prepared and expressed in human kidney 293 (HEK-293) as described (25). The mutant was purified by a combination of immunoaffinity and ion exchange chromatography using the monoclonal antibody HPC4 and an FPLC Mono Q column, respectively.
Human plasma proteins including factors VIIa, IX, X, Xa, and an fX mutant lacking the Gla-domain (GD-fX, cleaved by chymotrypsin after Tyr-44 as determined by N-terminal sequencing) were purchased from Haematologic Technologies Inc. (Essex Junction, VT). Phospholipid vesicles containing 80% phosphatidylcholine and 20% phosphatidylserine (PC/PS) were prepared as described (26). The chromogenic substrates Spectrozyme VIIa (SpVIIa, CH3SO2-D-CHA-But-Arg-pNA) and Spectrozyme FXa (SpFXa, MeO-CO-D-CHG-Gly-Arg-pNA) were purchased from American Diagnostica (Greenwich, CT). IPTG was purchased from Gold Bio Technology Inc. (St. Louis, MO).
Evaluation of the Kd(app) values
The affinity of the sTF2-219 mutants for binding to fVIIa was evaluated based on their ability to enhance the catalytic activity of fVIIa toward cleavage of SpVIIa as described (22). Briefly, increasing concentrations of the cofactor (0.78–100 nM) were incubated with fVIIa (5 nM) in 100 mM NaCl, 20 mM Tris-HCl (pH 7.5), containing 0.1 mg/mL bovine serum albumin, 0.1 % polyethylene glycol 8000, and 5 mM CaCl2 (TBS/Ca2+) in 96-well assay plates. After 1 min of incubation at room temperature, SpVIIa was added to a final concentration of 0.5 mM and the rate of hydrolysis was measured at 405 nm by a Kinetic Microplate Reader (Molecular Devices, Menlo Park, CA). The background activity of fVIIa in the absence of sTF2-219 was subtracted from the measured values. The maximal activity and Kd(app) values were calculated from the non-linear curve fitting of amidolytic activity data to the quadratic binding equation as described (22).
Factor X activation
The initial rate of fX, GD-fX and E2-fX activation by fVIIa in complex with wild-type or mutant sTF2-219 derivatives were studied in TBS/Ca2+ at room temperature as described (22). The generation of product was monitored by a two stage discontinuous assay. In the first stage, the time course of activation of each zymogen (0.015–5 μM) by fVIIa (0.2–100 nM) in complex with saturating concentrations of sTF2-219 mutants (200 nM) were monitored on PC/PS vesicles (0.3 mM) in 96-well assay plates. At each time point, aliquots of the activation reactions were terminated by adding 50 mM EDTA. In the second stage, the amidolytic activity of each sample was determined by the subsequent addition of SpFXa (final concentration of 0.2 mM). The absorbance at 405 nm was monitored over 5 min using a Vmax Kinetic Microplate Reader and the initial rates of chromogenic substrate hydrolysis (ΔA405/min) were converted to nanomolar of product by reference to a standard curve. The apparent Km and kcat values for substrate activation were calculated from Michaelis-Menten equation, and the catalytic efficiencies were expressed as the ratio of kcat/Km.
Competitive Binding Studies
The competitive effects of S195A fX and PC fX-Gla mutants on the wild-type zymogen activation by fVIIa in complex with the sTF2-219 derivatives were studied. In both cases, the activation of fX (150 nM) by a limiting fixed concentration of fVIIa (0.3 nM) in complex with each sTF2-219 derivative (100 nM) was monitored on PC/PS vesicles (0.05–0.3 mM) in TBS/Ca2+ in the presence of increasing concentrations of the competitors (0–2.5 μM). Following incubation for 3–6 min at room temperature, the reactions were terminated by adding 50 mM EDTA and the rate of fXa generation was determined as described above. The kinetic data were normalized to 100% maximal activity in the absence of the competitor and plotted as a function of increasing concentrations of the competitors. The apparent dissociation constants (Kd(app)) were determined by nonlinear regression analysis the kinetic data as described (27).
RESULTS
Expression and Purification of sTF2-219 Derivatives
Both wild-type and mutant sTF2-219 derivatives were expressed in the periplasmic space of bacteria using the pINIII-pelB-neo expression/purification vector system as described under “Materials and Methods”. This vector system, incorporates a 12-residues epitope for the Ca2+-dependent monoclonal antibody HPC4 to the N-terminal of the expressed proteins, thereby facilitating their purification from the bacterial culture supernatants and periplasmic extracts by a single step immunoaffinity chromatography using immobilized HPC4 as described (9,22). SDS-PAGE analysis of the purified proteins under non-reducing conditions indicated that the isolated proteins were essentially homogenous (Figure 2). Previous results have indicated that the bacterial sTF2-219 with or without an N-terminal HPC4 epitope exhibits identical cofactor activity as the mammalian sTF2-219 (9). Thus, the cofactor activities of the sTF2-219 derivatives were evaluated without cleaving the HPC4 epitope from the N-termini of the recombinant proteins.
Figure 2.
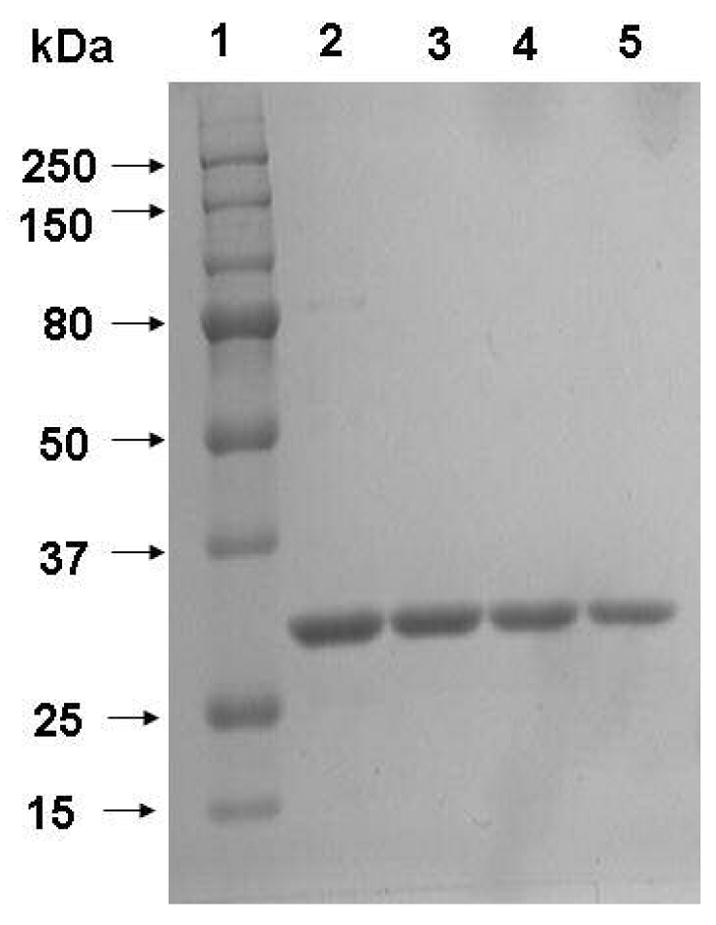
SDS-PAGE analysis of sTF2-219 mutants. Under non-reducing conditions: A: lane 1, molecular mass standards in kDa; lane 2, wild type sTF2-219; lane 3, N199A; lane 4, R200A; and lane 5, K201A.
Evaluation of the Kd(app) of sTF2-219 Mutants for binding to fVIIa
TF promotes the amidolytic activity of fVIIa toward tripeptidyl chromogenic substrates ~ 30–100-fold (2). This assay was employed to evaluate the affinity of sTF2-219 derivatives for fVIIa. All three sTF2-219 mutants exhibited normal cofactor activity, thus enhancing the catalytic efficiency of fVIIa toward SpVIIa with similar Vmax and Kd(app) of ~3 nM (Table 1), suggesting that mutagenesis of the residues under study did not adversely affect the folding of the mutant cofactors or their ability to allosterically render fVIIa catalytically competent.
Table 1.
The sTF2-219 mediated improvement in the amidolytic activity of fVIIa
| STF2-219 | Maximal velocity mOD/min | Kd(app) nM |
|---|---|---|
| WT | 9.2 ± 0.4 | 2.9 ± 0.7 |
| N199A | 8.0 ± 0.2 | 3.0 ± 0.5 |
| R200A | 7.7 ± 0.2 | 3.2 ± 0.6 |
| K201A | 7.3 ± 0.2 | 2.9 ± 0.7 |
The amidolytic function of 5 nM fVIIa toward SpVIIa in the presence of increasing concentrations of wild-type or mutant cofactors was determined in TBS/Ca2+ as described under “Materials and Methods”. All values are average of three measurements. The amidolytic activity of fVIIa in the absence of sTP2-219 was not significantly different from the background chromogenic substrate activity in the absence of the protease.
Proteolytic Function
The cofactor function of the sTF2-219 mutants in enhancing the proteolytic activity of fVIIa toward the natural substrate fX and its Gla and EGF-1 deletion derivatives was evaluated. Both wild-type and the N199A mutant of sTF2-219 exhibited similar cofactor activity, thus promoting the initial rate of the fVIIa activation of fX with similar catalytic efficiency (Figure 3). However, the catalytic efficiency of fVIIa in complex with both R200A and K201A mutants of the cofactor was impaired ~3-fold (Figure 3A). To determine whether either one of the basic sTF2-219 residues interacts with the Gla or with a site outside the Gla-domain of fX, the cofactor function of the mutants in promoting the fVIIa activation of GD-fX and E2-fX derivatives was evaluated. Like their decreased cofactor activity in the activation of wild-type fX, the cofactor activity of both R200A and K201A mutants was decreased ~3-fold when GD-fX was used as the substrate (Figure 3B), however, both cofactor mutants exhibited normal activity with E2-fX as the substrate (Figure 3C). These results suggested that the interactive-sites for both Arg-200 and Lys-201 of TF are located within the EGF-1 domain of fX in the extrinsic Xase complex. To understand the basis for the decreased cofactor activity of the mutants, the kinetic parameters for the cofactor-mediated fVIIa activation of wild-type fX were determined. As presented in Figure 4 and Table 2, the kcat for activation of the substrate by fVIIa in complex with both cofactor mutants was decreased with no change in the Km. A requirement for higher concentrations of the substrates excluded detailed kinetic analysis with the deletion mutants, however, initial rate studies at several sub-saturating concentrations of GD-fX suggested that the decreased cofactor activity of both Arg-200 and Lys-201 is also due to a decrease in kcat of the substrate activation by fVIIa (data not shown). These results suggest that the interaction of either Arg-200 or Lys-201 of TF with the EGF-1 domain of fX does not contribute to the binding energy of the initial complex formation, but rather both residues may play either an orientational or an allosteric role, thereby properly accommodating the activation peptide of the substrate into the active-site groove of fVIIa to achieve maximal catalysis. Further kinetic studies using fIX as the substrate of the extrinsic Xase complex yielded similar results, with both Arg-200 and Lys-201 mutant exhibiting an ~2-fold decrease in the kcat of the zymogen activation, with no significant change in Km(app) values (data not shown).
Figure 3.
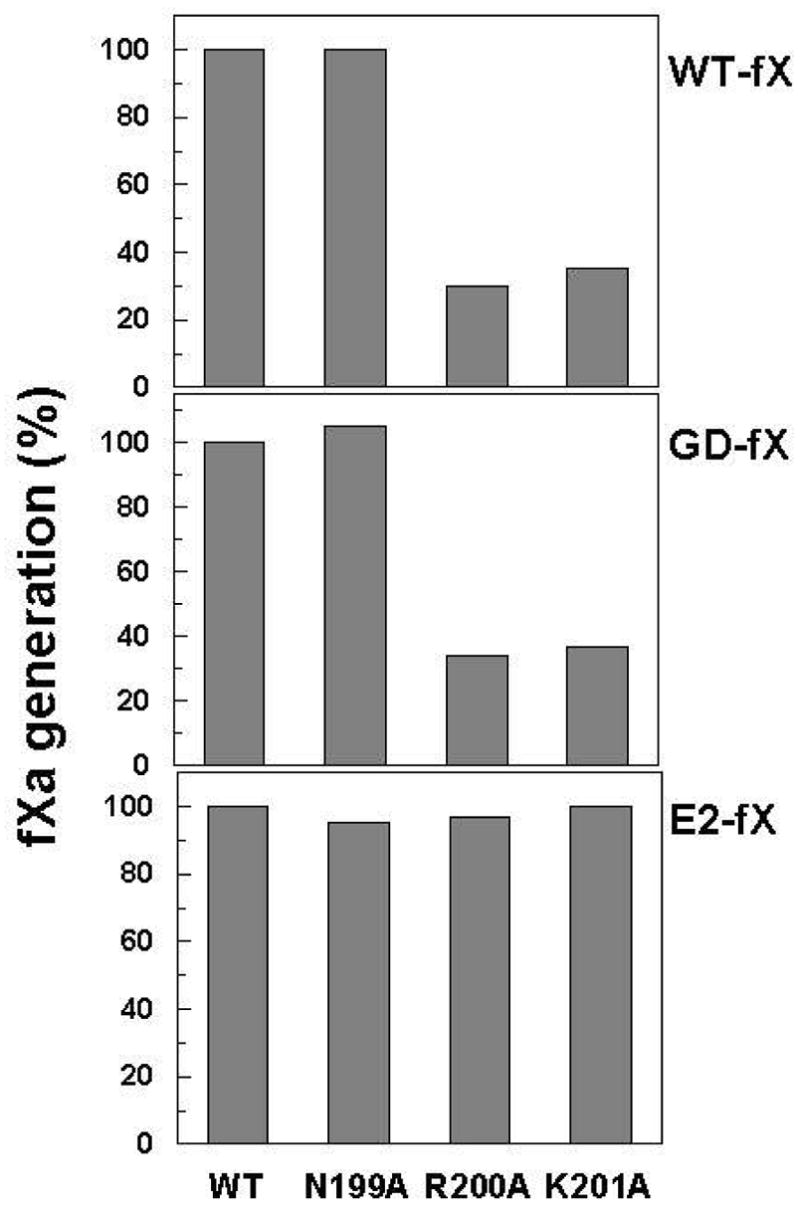
Cofactor effect of wild-type sTF2-219 and its mutants on the activation of fX derivatives by fVIIa. Top, the activation of wild-type fX (100 nM) by fVIIa (0.3 nM) in complex with sTF2-219 derivatives (200 nM) was monitored on PC/PS vesicles (0.3 mM) in TBS/Ca2+ at room temperature. The reactions were stopped by addition of 50 mM EDTA, and the rate of fXa generation was measured by an amidolytic activity assay described under “Materials and Methods”. Center, the same as A except that the activation of GD-fX (1 μM) was monitored by fVIIa (0.5 nM). C, the same as A except that the activation of E2-fX (1 μM) was monitored by fVIIa (100 nM). The normalized values are averages of at least three measurements.
Figure 4.
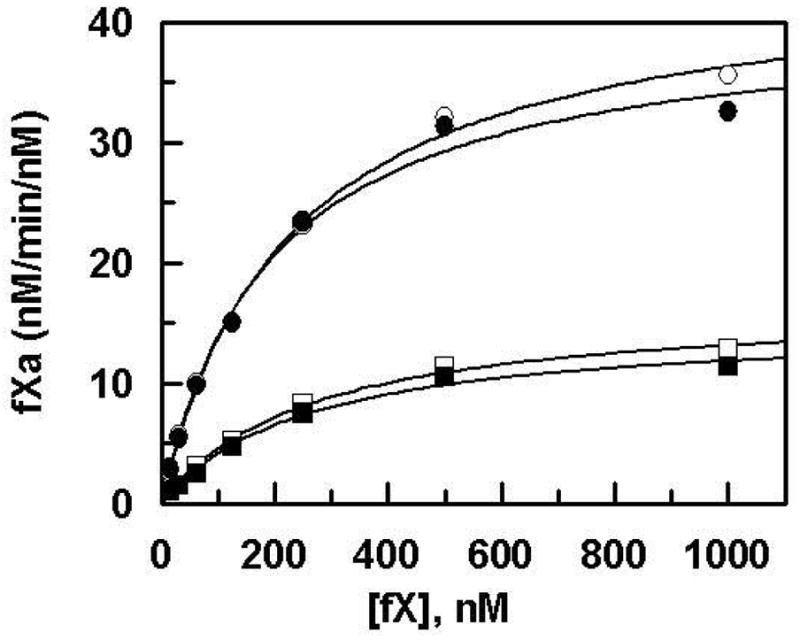
Cofactor effect of wild-type sTF2-219 and its mutants on the activation of fX by fVIIa. Saturating concentrations of wild-type sTF2-219 (○), N199A (●), R200A (□), and K201A (■) (250 nM each) were incubated with VIIa (0.2 nM) and PC/PS vesicles (0.3 mM) in TBS/Ca2+. The reaction was initiated by addition of different concentrations of fX shown on x-axis. Following 1–5 min activation, the reactions were stopped by addition of 50 mM EDTA, and the rate of fXa generation was measured by an amidolytic activity assay described under “Materials and Methods”. Solid lines are non-linear regression analysis of data according to the Michaelis-Menten equation.
Table 2.
Kinetic constants for the sTF2-219 mediated activation of fX by fVIIa
| STF2-219 | Km(app) (μM) | kcat (min−1) | kcat/Km(app) (μM−1 min−1) |
|---|---|---|---|
| WT | 0.23 ± 0.02 | 44.5 ± 0.1 | 193.5 ± 17.3 |
| N199A | 0.19 ± 0.02 | 40.6±1.8 | 213.7 ± 31.9 |
| R200A | 0.26 ± 0.02 | 16.6 ± 0.6 | 63.8 ± 7.2 |
| K201A | 0.26 ± 0.03 | 14.9 ± 0.8 | 57.3 ± 9.7 |
Enzyme assays were carried out at saturating concentrations of sTF2-219 derivatives over a range of substrate concentrations (0.015–1 pM) in the presence of 0.2 nM fVIIa and 0.3 mM PC/PS vesicles. Km(app) and kcat values were calculated from the initial activation rates of fX and GD-fX using Michaelis-Menten equation. All values are average of 2–3 experiments ± SE.
To provide further support for the hypothesis that the interaction of either Arg-200 or Lys-201 of sTF2-219 with EGF-1 of fX does not contribute to the specificity of the initial binding, the catalytically inactive S195A mutant of the zymogen fX was used as a competitive inhibitor of fX activation by fVIIa in complex with sTF2-219 derivatives. As shown in Figure 5A, S195A fX functioned as a competitive inhibitor of the extrinsic Xase complex with Kd(app) values (~200–300 nM), that is similar to Km(app) of the substrate activation by the activator complex, with both wild-type and mutant cofactors. These results suggest that a direct interaction of either Arg-200 or Lys-201 with fX makes no contribution to the specificity of the substrate assembly into the extrinsic Xase complex, but rather following the assembly of the substrate into the activation complex, the interaction of the basic residues of TF with EGF-1 of the substrate facilitates the optimal docking of the fX activation peptide into the active-site groove of the protease. Similar competitive kinetic studies using PC fX-Gla as the competitor of fX activation by the fVna-sTF2-219 complex revealed that the Gla-domain of fX primarily determines the recognition specificity of the complex assembly as evidenced by the ability of the mutant to function as a competitive inhibitor of the extrinsic Xase complex with Kd(app) values (~200–300 nM), that is again similar to Km(app) of the substrate activation by fVIIa in complex with both wild-type and mutant cofactors (Figure 5B).
Figure 5.
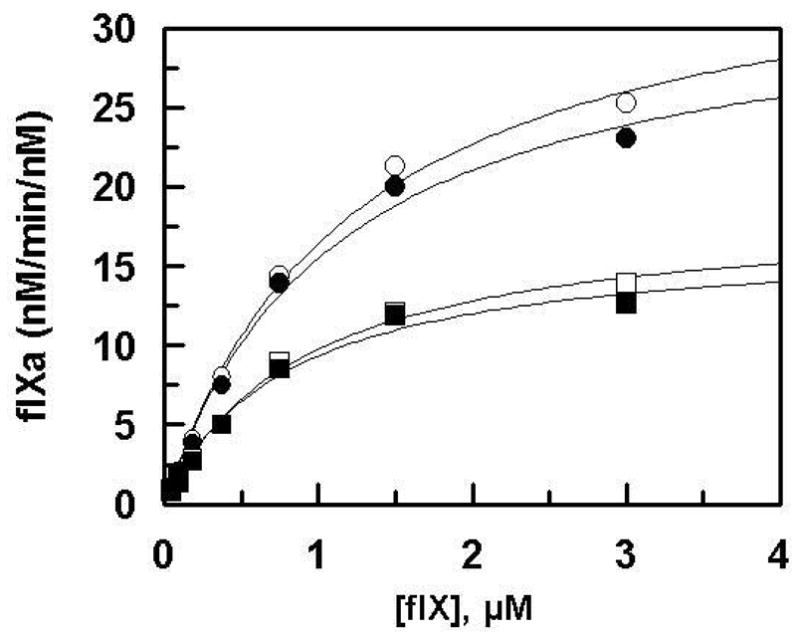
Cofactor effect of wild-type sTF2-219 and its mutants on the activation of fIX by fVIIa. Saturating concentrations of wild-type sTF2-219 (○), N199A (●), R200A (□), and K201A (■) (250 nM each) were incubated with VIIa (2 nM) and PC/PS vesicles (0.3 mM) in TBS/Ca2+. The reaction was initiated by addition of different concentrations of fX shown on x-axis. Following 15 min activation, the reactions were stopped by addition of 50 mM EDTA, and the rate of fXa generation was measured by an amidolytic activity assay. Solid lines are non-linear regression analysis of data according to the Michaelis-Menten equation. An apparent Km value of ~1 μM was obtained with all cofactor derivatives. The kcat values were: 33–36 nM/min/nM for wild-type and N199A, and 17–18 nM/min/nM for R200A and K201A.
Discussion
In this study we have demonstrated that the EGF-1 domain of fX has an interactive-site for TF that involves Arg-200 and Lys-201 of the cofactor. This proposal is based on the observation that the Ala substitution mutants of both cofactors exhibited impaired cofactor activity in the extrinsic Xase assays when either wild-type fX or GD-fX was used as the substrate, but normal cofactor activity when E2-fX (lacks both Gla and EGF-1) was used as the substrate. The lower cofactor activity of the mutants was not caused by the mis-folding of the mutant proteins since they both exhibited normal cofactor activity in the amidolytic activity assays, thus enhancing the catalytic efficiency of fVIIa toward the chromogenic substrate SpVIIa with Vmax and Kd(app) values similar to those observed for wild-type sTF2-219. Analysis of kinetic parameters suggested that a lower kcat for the activation of the protein substrate by fVIIa in complex with both sTF2-219 mutants is responsible for the defect with both cofactors since the Km(app) for fX activation remained similar for both wild-type and mutant cofactors. These results indicate that the interaction of either one of the TF residues with EGF-1 does not contribute to the initial binding step in substrate recognition, but rather to improvement in the rate of catalysis in the second step of the reaction. Further support for this hypothesis was provided by the observation that either the catalytically inactive S195A mutant of fX or the PC fX-Gla chimera functioned as competitive inhibitors of the extrinsic Xase complex with Kd(app) values that correlated well with the Km(app) values of fX activation by fVIIa in complex with both wild-type and mutant cofactors. These results are consistent with the previous hypothesis that TF-dependent exosite interactions determine the affinity and specificity of fX for the extrinsic Xase complex (28,29). Moreover, the results with the PC fX-Gla mutant further suggest that the Gla-domain of fX is primarily responsible for the binding specificity of the extrinsic Xase complex assembly on the membrane surface, which results in an improvement in the Km of the reaction. Thus, the interaction of TF with residues outside the Gla-domain of fX may primarily enhance the catalytic rate of the reaction as observed in this study. Analysis of the kinetic data obtained with the activation of fIX by fVIIa in complex with the both sTF2-219 mutants predicts a similar role for the cofactor in interaction with EGF-1 of fIX in the activation complex.
Since fVIIa in the crystal structure of the fVIIa-sTF complex adopts an extended conformation and contacts both the N- and C-terminal domains of TF, it is anticipated that the substrates fIX and fX would also assemble into the complex in the same extended conformation in order to position their activation peptides into the catalytic groove of the protease in the activation complex. Thus, similar to fVIIa, both substrates can potentially interact with both domains of TF in the complex. In this context, a recognition site for fX on the membrane proximal C-terminal domain of TF consisting of residues Tyr-157, Lys-159, Ser-163, Gly-164, Lys-165 and Lys-166 and Tyr-185 has been identified (10,19,30). Noting the three dimensional locations of these residues in the x-ray crystal structure of the fVIIa-sTF complex (12) and the structural homology between fVIIa and fX, it is expected that all of these TF residues would interact with the Gla-domain of the substrate (10). In support of this hypothesis, mutagenesis of both Lys-165 and Lys-166 has resulted in mutants which have exhibited dramatically diminished cofactor activity in promoting the fVIIa activation of fX, but not GD-fX, suggesting that the acidic residues of the Gla-domain of fX constitute an exosite for interaction with these and perhaps other residues of the C-terminal TF mentioned above in the extrinsic Xase complex (20). No Gla-independent interactive-site on fX for TF had been reported prior to this study, though both the light (Gla, EGF-1 and EGF-2 domains) and the heavy (protease domain) chains of fVIIa are known to interact with TF in the extrinsic Xase complex, with the protease domain primarily interacting with the membrane distal N-terminal domain of TF (12,14,15,18). However, our previous mutagenesis of 23 charged residues of the N-terminal TF domain failed to identify a binding site for fX or fix on the cofactor (22). Thus, further studies will be required to determine whether TF has interactive-sites for either EGF-2 or the protease domain of the substrates in the activation complex. It should be noted that previous protein-protein docking studies using the structures of both the fVIIa-sTF complex and fXa have predicted that the protease domain of fX may also interact with the N-terminal domain of TF, however, no mutagenesis study has yet validated the molecular models (14,15).
Based on the x-ray crystal structure of the fVIIa-sTF complex, the side chains of both Arg-200 and Lys-201 of TF are solvent exposed and located at the same height as EGF-1 of fVIIa on the opposite face of the protease contact site (Figure 1). Although the structure of fVIIa-TF in complex with fX has not been resolved, nevertheless, noting the structural homology between fVIIa, fX and fXa, and molecular models built based on these structures (14,15), several acidic residues in EGF-1 of fX can interact with basic residues of TF in the extrinsic Xase complex. The structure of EGF-1 of fX consists of three disulfide bonded p sheets forming three loops (31). There are five acidic residues, with two being located outside (Asp-46 and Asp-48) and the other three within the first two disulfide loops of fX (Glu-51, Asp-63 and Glu-67). All of these acidic residues, with the exception of Glu-67, which is Glu-70 in fIX, have also been three-dimensionally conserved in EGF-1 of fIX. We hypothesize that one or more of these acidic residues can potentially interact with Arg-200 and Lys-201 of TF in both substrates. In support of this hypothesis, previous mutagenesis of Glu-51 of fX has impaired the catalytic efficiency of the fVIIa-TF complex toward the mutant substrate 2–3-fold (14). Further studies will be required to identify all residues of EGF-1 on fX that may interact with TF in the extrinsic Xase complex. We predict that the contributions of Arg-200 and Lys-201 of TF for interaction with EGF-1 of fX would be additive since a previous mutagenesis study substituting both of these residues with Ala reported an ~ 80% loss of the cofactor activity for the double mutant in a clotting assay (7).
In summary, we have demonstrated that the two basic residues Arg-200 and Lys-201 on TF interact with EGF-1 of fX (and most likely also flX) to improve the kcat of the substrate activation by fVIIa in the extrinsic Xase complex. The mechanism through which the interaction of these resides with EGF-1 of the substrate improves the rate of catalysis is not known. Nevertheless, such an interaction can play an orientational role, thus leading to the proper alignment of the activation peptide of the substrate in the catalytic pocket of fVIIa, or alternatively, the interaction can have an allosteric role, thus altering the conformation of the activation peptide and improving the stability of the protease-substrate transition-state in the activation complex.
Figure 6.
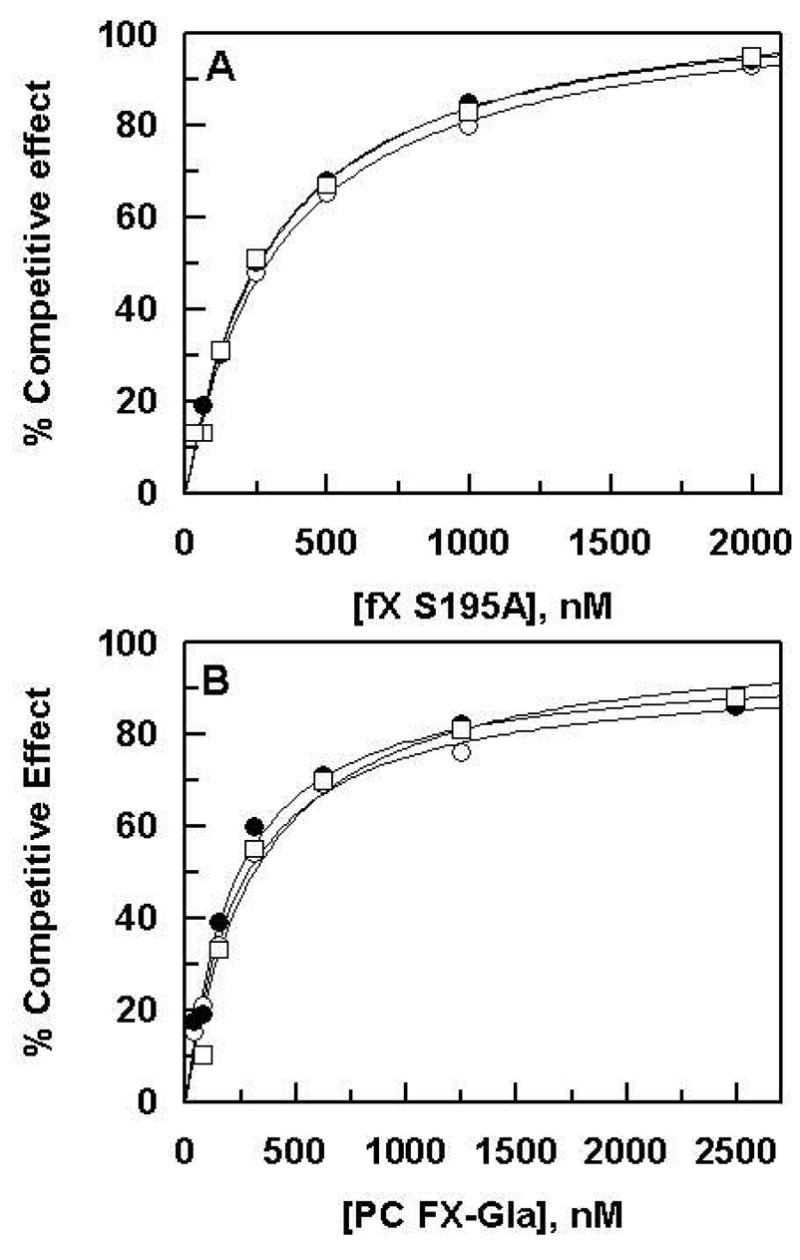
Competitive effect of fX SI95A and PC fX-Gla on wild-type fX activation by fVIIa in complex with sTF2-219 derivatives. A, fX (150 nM) was incubated with fVIIa (0.3 nM) in complex with PC/PS vesicles (0.05 mM) and sTF2-219 (100 nM) in TBS/Ca2+ in the presence of increasing concentrations of fX S195A (0.03–2 μM). Following incubation for 3–5 min at room temperature, the reactions were terminated by adding 50 mM EDTA and the rate of fXa generation was determined as described under “Materials and Methods”. B, the same as A except that PC fX-Gla was used as the competitor in the activation reactions. The symbols in both panels are: wild-type sTF2-219 (○), R200A (●), and K201A (□). The Kd(app) values, obtained from the nonlinear regression analysis of data (average of 2–3 experiments), were ~200–300 ± 20–50 nM for both competitors with all sTF2-219 derivatives.
Acknowledgments
We would like to thank Audrey Rezaie for her proofreading of this manuscript.
The research discussed herein was supported by a grant awarded by the National Heart, Lung, and Blood Institute of the National Institutes of Health (R01 HL68571 to ARR)
Abbreviations
- TF
tissue factor
- sTF
soluble tissue factor
- sTF2-219
soluble tissue factor containing residues 2-219 of extracellular domain
- fVIIa
activated factor VII
- fIX
factor IX
- fX
factor X
- fXa
activated factor X
- Gla
γ-carboxyglutamic acid
- GD-fXa
Gla-domainless factor X, factor X from which the amino-terminal residues 1-44 have been removed by chymotrypsin digestion
- fX S1950A
factor X derivative in which the catalytic residue Ser-195 (in the chymotrypsin numbering system of Bode et al. (1) has been substituted with an Ala
- EGF
epidermal growth factor
- E2-fX
deletion mutant of fX in which both the Gla and first EGF domain have been deleted by recombinant DNA methods
- PC fX-Gla
protein C mutant containing the Gla and hydrophobic stack domains of fX from residues 1–45
- PC/PS
phospholipid vesicles containing 80% phosphatidylcholine and 20% phosphatidylserine
References
- 1.Bode W, Mayr L, Baumann U, Huber R, Stone SR, Hofsteenge J. The refined 1.9 Å crystal structure of human α-thrombin: interaction with D-Phe-Pro-Arg chlorometheylketone and significance of the Tyr-Pro-Pro-Trp insertion segment. EMBO J. 1989;8:3467–3475. doi: 10.1002/j.1460-2075.1989.tb08511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morrissey JH, Neuenschwander PF, Huang Q, McCallum CD, Bixia S, Johnson AE. Factor VIIa-tissue factor: Functional importance of protein-membrane interactions. Thromb Haemostas. 1997;78:112–116. [PubMed] [Google Scholar]
- 3.Jackson CM, Nemerson Y. Blood coagulation. Ann Rev Biochem. 1980;49:765–811. doi: 10.1146/annurev.bi.49.070180.004001. [DOI] [PubMed] [Google Scholar]
- 4.Morrissey JH, Fakhrai H, Edgington TS. Molecular cloning of the cDNA for tissue factor, the cellular receptor for the initiation of the coagulation protease cascade. Cell. 1987;50:129–135. doi: 10.1016/0092-8674(87)90669-6. [DOI] [PubMed] [Google Scholar]
- 5.Harlos K, Martin DMA, O’Brien DP, Jones EY, Stuart DL, Polikarpov I, Miller A, Tuddnham EGD, Boys CWG. Crystal structure of the extracellular region of human tissue factor. Nature. 1994;370:662–666. doi: 10.1038/370662a0. [DOI] [PubMed] [Google Scholar]
- 6.Muller YA, Ultsch MH, Kelley RF, de Vos AM. Structure of the extracellular domain of human tissue factor: Location of the factor Vila binding site. Biochemistry. 1994;33:10864–10870. doi: 10.1021/bi00202a003. [DOI] [PubMed] [Google Scholar]
- 7.Ruf W, Schullek JR, Stone MJ, Edgington TS. Mutational mapping of functional residues in tissue factor: Identification of factor VII recognition determinants in both structural modules of the predicted cytokine receptor homology domain. Biochemistry. 1994;33:1565–1572. doi: 10.1021/bi00172a037. [DOI] [PubMed] [Google Scholar]
- 8.Kelly CR, Schullek JR, Ruf W, Edgington TS. Tissue factor residue Asp44 regulates catalytic function of the bound proteinase factor VIIa. Biochem J. 1996;315:145–151. doi: 10.1042/bj3150145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rezaie AR, Fiore MM, Neuenschwander PF, Esmon CT, Morrissey JH. Expression and purification of a soluble tissue factor fusion protein with an epitope for an unusual calcium-dependent antibody. Prot Exp Purif. 1992;3:453–460. doi: 10.1016/1046-5928(92)90062-2. [DOI] [PubMed] [Google Scholar]
- 10.Kirchofer D, Lipari MT, Moran P, Eigenbrot C, Kelley RF. The tissue factor region that interacts with substrates factor IX and factor X. Biochemistry. 2000;39:7380–7387. doi: 10.1021/bi000182+. [DOI] [PubMed] [Google Scholar]
- 11.Silverberg SA, Nemerson Y, Zur M. Kinetics of the activation of bovine coagulation factor X by components of the extrinsic pathway: kinetic behavior of two-chin factor VII in the presence and absence of tissue factor. J Biol Chem. 1977;252:8481–8488. [PubMed] [Google Scholar]
- 12.Banner DW, D’Arcy A, Chene C, Winkler FK, Guha A, Konigsberg WH, Nemerson Y, Kirchofer D. The crystal structure of the complex of blood coagulation factor VIIa with soluble tissue factor. Nature. 1996;380:41–46. doi: 10.1038/380041a0. [DOI] [PubMed] [Google Scholar]
- 13.McCallum CD, Su B, Neuenschwander PF, Morrissey JH, Johnson AE. Tissue factor positions and maintains the factor VIIa active site far above the membrane surface even in the absence of the factor VIIa Gla domain. J Biol Chem. 1997;272:30160–30166. doi: 10.1074/jbc.272.48.30160. [DOI] [PubMed] [Google Scholar]
- 14.Norledge BV, Petrovan RJ, Ruf W, Olson AJ. The tissue factor/factor VIIa/factor Xa complex: A model built by docking and site-directed mutagenesis. Proteins. 2003;53:640–648. doi: 10.1002/prot.10445. [DOI] [PubMed] [Google Scholar]
- 15.Venkateswarlu D, Duke RE, Perera L, Darden TA, Pedersen LG. An all-atom solution-equilibrated model of human extrinsic blood coagulation complex (sTF-VIIa-Xa): a protein-protein docking and molecular dynamics refinement study. J Thromb Haemost. 2003;1:2577–2588. doi: 10.1111/j.1538-7836.2003.00421.x. [DOI] [PubMed] [Google Scholar]
- 16.Furie B, Furie BC. The molecular basis of blood coagulation. Cell. 1988;53:505–518. doi: 10.1016/0092-8674(88)90567-3. [DOI] [PubMed] [Google Scholar]
- 17.Stenflo J. Structure-function relationships of epidermal growth factor modules in vitamin K-dependent clotting factors. Blood. 1991;78:1637–1651. [PubMed] [Google Scholar]
- 18.Dickinson CD, Kelly CR, Ruf W. Identification of surface residues mediating tissue factor binding and catalytic function of the serine protease factor VIIa. Proc Natl Acad Sci (USA) 1996;93:14379–14384. doi: 10.1073/pnas.93.25.14379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruf W, Miles DJ, Rehemtulla A, Edgington TS. Cofactor residues lysine 165 and 166 are critical for protein substrate recognition by the tissue factor-factor Vila protease complex. J Biol Chem. 1992;267:6375–6381. [PubMed] [Google Scholar]
- 20.Huang Q, Neuenschwander PF, Rezaie AR, Morrissey JH. Substrate recognition by tissue factor-factor VIIa. J Biol Chem. 1996;271:21752–21757. doi: 10.1074/jbc.271.36.21752. [DOI] [PubMed] [Google Scholar]
- 21.Zhong D, Bajaj MS, Schmidt AE, Bajaj SP. The N-terminal epidermal growth factor-like domain in factor IX and factor X represents an important recognition motif for binding to tissue factor. J Biol Chem. 2002;277:3622–3631. doi: 10.1074/jbc.M111202200. [DOI] [PubMed] [Google Scholar]
- 22.Kittur FS, Manithody C, Morrissey JH, Rezaie AR. The cofactor function of the N-terminal domain of tissue factor. J Biol Chem. 2004;279:39745–39749. doi: 10.1074/jbc.M406628200. [DOI] [PubMed] [Google Scholar]
- 23.Rezaie AR, Manithody C, Yang L. Identification of factor Xa residues critical for interaction with protein Z-dependent protease inhibitor. J Biol Chem. 2005;280:32722–32728. doi: 10.1074/jbc.M505517200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rezaie AR, Neuenschwander PF, Morrissey JH, Esmon CT. Analysis of the functions of the first epidermal growth factor-like domain of factor X. J Biol Chem. 1993;268:8176–8180. [PubMed] [Google Scholar]
- 25.Yang L, Manithody C, Rezaie AR. Contribution of basic residues of the 70–80-loop to heparin binding and anticoagulant function of activated protein C. Biochemistry. 2002;41:6149–6157. doi: 10.1021/bi015899r. [DOI] [PubMed] [Google Scholar]
- 26.Smirnov MD, Esmon CT. Phosphatidylethanolamine incorporation into vesicles selectively enhances factor Va inactivation by activated protein C. J Biol Chem. 1994;269:816–819. [PubMed] [Google Scholar]
- 27.Rezaie AR, Kittur FS. The critical role of the 185–189-loop in the factor Xa interaction with Na+ and factor Va in the prothrombinase complex. J Biol Chem. 2004;279:48262–48269. doi: 10.1074/jbc.M409964200. [DOI] [PubMed] [Google Scholar]
- 28.Shobe J, Dickinson CD, Edgington TS, Ruf W. Macromolecular substrate affinity for the tissue factor-factor VIIa complex is independent of scissile bond docking. J Biol Chem. 1999;274:24171–24175. doi: 10.1074/jbc.274.34.24171. [DOI] [PubMed] [Google Scholar]
- 29.Baugh RJ, Dickinson CD, Ruf W, Krishnaswamy S. Exosite Interactions determine the affinity of factor X for the extrinsic Xase complex. J Biol Chem. 2000;275:28826–28833. doi: 10.1074/jbc.M005266200. [DOI] [PubMed] [Google Scholar]
- 30.Ruf W, Miles DJ, Rehemtulla A, Edgington TS. Tissue factor residues 157-167 are required for efficient proteolytic activation of factor X and factor Vn. J Biol Chem. 1992;267:22206–22210. [PubMed] [Google Scholar]
- 31.Ullner M, Selander M, Persson E, Stenflo J, Drakenberg T, Teleman O. Three-dimensional structure of the Apo form of the N-terminal EGF-like module of blood coagulation factor X as determined by NMR spectroscopy and simulated folding. Biochemistry. 1992;31:5974–5983. doi: 10.1021/bi00141a004. [DOI] [PubMed] [Google Scholar]