

Abstract
Formalin-fixed, paraffin-embedded tissues are an invaluable tool for biomarker discovery and validation. As these archived specimens are not always compatible with modern genomic techniques such as gene expression arrays, we assessed the use of microRNA (miRNA) as an alternative means for the reliable molecular characterization of formalin-fixed, paraffin-embedded tissues. Expression profiling using two different microarray platforms and multiple mouse and human formalin-fixed, paraffin-embedded tissue types resulted in the correlation ratios of miRNA expression levels between frozen and fixed tissue pairs ranging from 0.82 to 0.99, depending on the cellular heterogeneity of the tissue type. The same miRNAs were identified as differentially expressed between tissues using both fixed and frozen specimens. While formalin fixation time had only marginal effects on microarray performance, extended storage times for tissue blocks (up to 11 years) resulted in a gradual loss of detection of miRNAs expressed at low levels. Method reproducibility and accuracy were also evaluated in two different tissues stored for different lengths of time. The technical variation between full process replicates, including independent RNA isolation methods, was approximately 5%, and the correlation of expression levels between microarray and real-time quantitative reverse transcriptase polymerase chain reaction was 0.98. Together, these data demonstrate that miRNA expression profiling is an accurate and robust method for the molecular analysis of archived clinical specimens, potentially extending the use of miRNAs as new diagnostic, prognostic, and treatment response biomarkers.
Microarray technology is a powerful approach that enables high-throughput and cost-efficient comparative analyses of global gene expression in normal and neoplastic tissues. Microarrays targeting DNA and mRNA have been used in classification of tumors, prediction of patients' response to chemotherapy, and for the discovery of new diagnostic, prognostic, and treatment response biomarkers.1,2,3,4,5 The potential of this technology is being realized in clinical applications with the recent introduction of the first microarray-based gene expression signature assay, MammaPrint, which uses a 70-gene signature with hundreds of control genes to predict the risk of distant metastases in breast cancer patients.5 While microarray technology has proven to be reproducible in a series of technical studies,6 before a microarray signature becomes a part of routine clinical practice, its utility must be validated using a large number of samples with known clinical outcomes, medical history, and histopathological findings. This large-scale analysis could be accomplished through accessing the wealth of human tissues that have been archived through the years as formalin-fixed, paraffin-embedded (FFPE) specimens. The formalin fixation process allows for permanent preservation of the architecture of tissues in optimal histological condition and easy long-term storage. Unfortunately, this process also compromises the yield, quality and integrity of nucleic acids through enzymatic and chemical degradation, extensive cross-linking with proteins and various chemical modifications.7,8 mRNA species are particularly affected with one study estimating that only 3% of the mRNA population from FFPE tissues is accessible for quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) analysis.9
Expression of various microRNAs (miRNAs) has been reported to be differentially altered across a variety of tumor types, suggesting their direct involvement in oncogenesis.10,11,12 miRNAs are small (19 to 24 nucleotides) regulatory RNA molecules, which, due to their size, are potentially more robust to FFPE-dependent degradation than mRNAs. Therefore miRNAs could become viable alternative analytes for expression profiling using FFPE tissues. Many research groups have been working toward enabling the use of microarray technology for miRNA expression studies. Novel labeling protocols have been developed to decrease the amount of required starting material, streamline the procedure, and improve the steps that can lead to quantitative bias.13,14 Modified microarray probes, such as locked nucleic acid chemistry, have also been introduced in an attempt to improve sensitivity and specificity and to allow detection of miRNAs differing by only one nucleotide.15 Using this platform, Xi et al recently reported that correlations of expression profiles between fixed and frozen mouse liver samples are higher with miRNAs than with mRNAs.16 The same authors also showed that miRNAs can efficiently be detected by qRT-PCR in archived colorectal cancer specimens up to 10 years old, suggesting that miRNAs are better analytes for expression analyses in archived samples.
In this study, we performed a thorough analysis of miRNA expression profiles in FFPE specimens using multiple tissue types and two different microarray profiling methods based on standard DNA probe chemistry. Initial evaluation using mouse brain, stomach, small intestine and kidney tissues indicated that the identity and quantity of miRNAs recovered from FFPE blocks are highly correlated to paired frozen samples. Human myometrium, B-cell lymphoma and colonic mucosa specimens also showed nearly identical miRNA profiles when flash-frozen tissues were compared with paired FFPE tissues fixed for 6 or 24 hours. FFPE blocks 1 to 3 years old showed highly comparable miRNA expression patterns, while detection of low-abundance miRNAs was reduced in tissue blocks stored for 7 years or longer. Our study also demonstrates that differential miRNA expression profiles as well as the magnitude of these differences are preserved in FFPE tissues and are very closely correlated with matched frozen samples. Finally, we show that the miRNA microarray method is reproducible and generates data highly concordant with real-time qRT-PCR. We conclude that miRNA microarray expression profiling and qRT-PCR are suitable platforms for retrospective molecular analysis and characterization of archived clinical samples provided that appropriate RNA isolation and microarray methodologies are used.
Materials and Methods
Tissue Samples
All human samples used in this study were collected as part of standard clinical care and were considered to be residual materials unnecessary for patient treatment. Patient identifiers were thoroughly removed from all samples before processing. Human normal liver tissue adjacent to tumor tissue (NAT) and normal spleen tissue were acquired from commercial suppliers by Asuragen's Tissue Acquisition Group in compliance with the regulations as outlined in Title 45 of the Code of Federal Regulations Part 46 and other regulatory guidance. Normal myometrium, small B-cell lymphoma, colonic mucosa, and colorectal carcinoma were surgically obtained from two donors for each tissue type by the Tissue Procurement Services at Ohio State University Medical Center, Columbus, OH. All of the specimens were collected within 4 weeks and processed within 30 minutes of surgical resection. Each specimen was divided into three 500-mg (5 cm3) pieces, one immediately snap-frozen in a cryovial using liquid nitrogen, and the remaining two pieces were fixed in 10% neutral-buffered formalin for 6 or 24 hours. Tissue sections from each block were stained with hematoxylin and eosin for verification of expected tissue type. Tissue sections at 20-μm thickness were used for isolation of total RNA. Detailed characteristics of each block, including pathology reports, can be found in Supplemental Table 1 at http://jmd.amjpathol.org. FFPE tissue was stored at ambient room temperature and frozen tissue was stored at −80°C. Archived FFPE specimens of myometrium stored from 7 to 11 years were also used for additional studies.
Surplus white mice (Harlan Sprague Dawley Inc., Indianapolis, IN) were humanely euthanized and the collected brain, stomach, small intestine, and kidney tissues were split into two halves. One half was immediately flash-frozen in liquid nitrogen and stored at −80°C, while the remaining half was placed in formalin (10% neutral-buffered formalin) and remained at room temperature for 6 hours. Fixed tissues were processed using standard histology techniques, embedded in paraffin, and stored at room temperature.
Total RNA Isolation
Total RNA from mouse and human FFPE tissues was isolated using RecoverAll Total Nucleic Acid Isolation Kit for FFPE Tissues (Ambion, Applied Biosciences, Austin, TX) according to the manufacturer's instructions. Total RNA from frozen tissues was extracted using mirVana miRNA Isolation Kit (Ambion) following the manufacturer's protocol.
Concentration and purity of total RNA samples were measured using the NanoDrop ND3.0 spectrophotometer (NanoDrop Technologies Inc, Wilmington, DE). RNA integrity was assessed with the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA) and the RNA 6000 LabChip kit (Agilent Technologies).
miRNA Expression Profiling
miRNA expression profiling of all mouse tissues was performed using a two-color microarray platform. Each test sample was compared against a unique reference sample, as described previously.17 Expression profiling of human myometrium, B-cell lymphoma, and colonic mucosa was performed as described previously by labeling 10 μg of total RNA with Cy5 fluorescent dye and hybridizing to one-color mirVana miRNA Bioarrays v1 containing 377 unique miRNA probes printed in duplicate (Ambion).12 Expression profiling of human liver NAT and spleen was performed as described above using 4 μg of total RNA and mirVana miRNA Bioarrays v2 platform (Ambion) containing a total of 662 unique miRNA probes printed in duplicate. The raw array data were normalized using the variance stabilization method.18
The microarray expression levels of selected miRNA species in liver NAT and spleen tissue blocks were confirmed via qRT-PCR using 10 ng of total RNA input and TaqMan microRNA assays (Applied Biosystems, Foster City, CA) as described.19
Results
Mouse Model Analysis
To assess the compatibility of FFPE samples with miRNA expression profiling, we first evaluated the correlation between expression profiles obtained from matched frozen and FFPE samples prepared from four different mouse tissues: brain, stomach, small intestine, and kidney. Four biological replicates of frozen and FFPE tissue pairs were prepared from four different mice, processed using a two-color microarray platform, and normalized as described previously.17 The normalized expression data for each of the 191 miRNAs interrogated can be found in Supplemental Table 2 at http://jmd.amjpathol.org. Unsupervised hierarchical clustering showed that miRNA expression profiles from FFPE samples are very similar to those obtained from frozen samples (Figure 1A). All samples derived from the same tissue, regardless of the preservation method, clustered together and away from samples from other tissue types.
Figure 1.
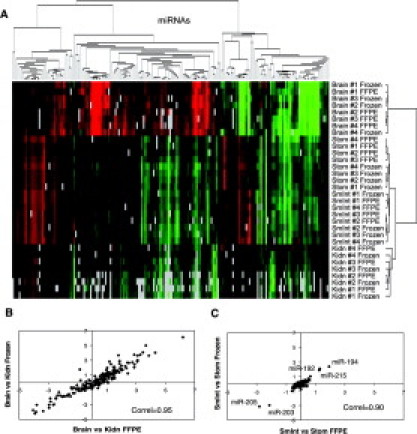
miRNA expression profiling in four mouse tissues. A: Unsupervised hierarchical clustering of miRNA expression in paired frozen and fixed mouse brain, stomach (Stom), small intestine (SmInt), and kidney (Kidn) using a two-color array platform. Expression levels are color-coded from green to red relative to the common reference hybridization sample used for each array, with red indicating enrichment in a given sample. Gray squares represent flagged miRNAs probes with low array signal below threshold (no data). B and C: Correlation of miRNA expression levels for paired fixed versus frozen mouse kidney, brain, small intestine, and stomach. Correlations were calculated using mean normalized expression levels in fixed and frozen samples (n = 4 donors per tissue).
The Pearson correlation coefficients for miRNA expression levels in each FFPE and frozen tissue pairs varied from 0.82 to 0.97 (Table 1). This correlation was above 0.91 for all tissue types when using the mean expression levels from the four biological replicates (Table 1). Furthermore, the same miRNAs were found differentially expressed between mouse tissues using either frozen or fixed samples. For tissues in which miRNA expression profiles were very different (eg, brain and kidney) the correlation of differential expression between FFPE and frozen tissue sets was >0.95 (Figure 1B). Even for tissues with very similar miRNA expression patterns, such as small intestine and stomach, the correlation of differential expression was still high (0.90; Figure 1C) and the same five miRNAs were found differentially expressed using either FFPE or frozen tissues (miR-192, -194, -203, -205 and -215; Figure 1C; see also Supplemental Table 2 at http://jmd.amjpathol.org). Overall these experiments indicate that miRNAs isolated from various recently fixed mouse tissues are compatible with downstream miRNA expression profiling methods and generate data similar to frozen reference tissues.
Table 1.
Pearson Correlation Coefficients for miRNA Expression Levels in Fixed Mouse Brain, Kidney, Small Intestine, and Stomach Relative to Matched Frozen Samples
| Correlation to frozen tissue |
|||||
|---|---|---|---|---|---|
| Sample ID | Set 1 | Set 2 | Set 3 | Set 4 | Mean* |
| Brain | 0.94 | 0.93 | 0.91 | 0.95 | 0.95 |
| Kidney | 0.88 | 0.94 | 0.92 | 0.83 | 0.96 |
| Small intestine | 0.96 | 0.97 | 0.96 | 0.95 | 0.97 |
| Stomach | 0.94 | 0.82 | 0.94 | 0.82 | 0.91 |
Calculated using mean normalized expression levels in fixed and frozen samples (n = 4 donors per tissue).
Evaluation of Human FFPE Tissue Blocks
To evaluate human specimens, we chose three tissues, myometrium, B-cell lymphoma and colon, with very different cell content and tissue block features (see Supplemental Table 1 at http://jmd.amjpathol.org). In addition, each biological replicate from two different donors was fixed for either 6 or 24 hours and compared with the corresponding paired frozen sample using a single-color microarray carrying 377 miRNA probes. Every sample generated quality array data that were subsequently normalized using the variance stabilization method (see Supplemental Table 3 at http://jmd.amjpathol.org). Unsupervised hierarchical clustering showed that the expression profiles between the frozen, fixed for 6 hours, and fixed for 24 hours myometrium and B-cell lymphoma tissues were nearly identical (data not shown). The Pearson correlation coefficients for the mean miRNA expression levels between fixed and frozen tissues were all greater than 0.97 (Table 2). As expected for tissues with higher heterogeneous cell content, both colon replicate sets showed lower correlation between fixed and frozen specimens with mean Pearson coefficient of 0.89 and 0.91 (Table 2). No significant differences were observed between FFPE specimens fixed for 6 or 24 hours for all three tissue types.
Table 2.
Pearson Correlation Coefficients for miRNA Expression Levels in Fixed Human Myometrium, B-Cell Lymphoma, and Colon Relative to Matched Frozen Tissues
| Correlation to frozen tissue |
|||
|---|---|---|---|
| Sample ID | Set 1 | Set 2 | Mean* |
| Myo_6 hours | 0.96 | 0.96 | 0.97 |
| Myo_24 hours | 0.97 | 0.99 | 0.99 |
| BCL_6 hours | 0.97 | 0.93 | 0.97 |
| BCL_24 hours | 0.96 | 0.98 | 0.98 |
| Colo_6 hours | 0.88 | 0.82 | 0.89 |
| Colo_24 hours | 0.86 | 0.90 | 0.91 |
Fixed tissues were immersed in formalin for either 6 or 24 hours.
Calculated using mean normalized expression levels in fixed and frozen samples (n = 2 donors per tissue).
Further analysis of the number of miRNA probes with signal above background showed that on average 60.3% of the miRNA sequences represented on the array were detected in frozen myometrium, 55% in B-cell lymphoma, and 51.5% in colon. In FFPE samples, the number of detected miRNAs was decreased by 5 to 8.6% independently of the fixation time. This observation suggests that the fixation process or the storage of the block at room temperature can result in partial loss of microarray signal intensity.
Effect of FFPE Block Storage Time
The human FFPE specimens described above were stored for 1 to 3 years before RNA isolation and array processing. To further determine the effect of prolonged block storage on microarray data quality we compared the 1-year-old FFPE and frozen myometrium replicates with independent blocks that had been fixed for the same amount of time (24 hours) and stored for 7 years (three biological replicates) or 11 years (four biological replicates). As expected, the 1-year-old FFPE specimens clustered together with their paired frozen samples (see Supplemental Figure 1 at http://jmd.amjpathol.org) and showed nearly identical miRNA expression profiles (mean Pearson correlation >0.99, Figure 2A). The older myometrium samples from different donors also clustered together, regardless of their age, but away from the 1-year-old samples. Their expression profiles, however, were still quite similar to the recently fixed samples as determined by the Pearson correlation of 0.89 and 0.84 for 7- and 11-year-old samples, respectively (Figure 2, B and C). A closer analysis of individual normalized miRNA probe signal (see Supplemental Table 4 at http://jmd.amjpathol.org) revealed that the expression levels of two putative miRNAs, miR-494 and miR-513, increased significantly with FFPE block age. In 1-year-old fixed myometrium, miR-494 and miR-513 were expressed at levels 3.1- and 1.8-fold higher than in matched frozen myometrium samples (Figure 2D). In 7-year-old myometrium samples, these fold changes further increased to 147- and 63-fold, while in 11-year-old samples they reached 464- and 161-fold, respectively.
Figure 2.
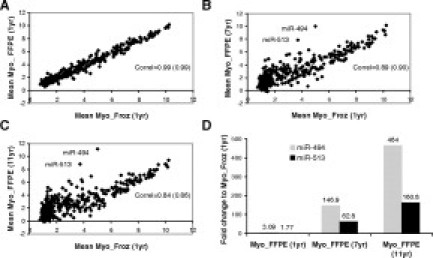
Correlation between fixed and frozen miRNA expression profiles in myometrium according to tissue age. A–C: Mean miRNA expression in 1-, 7-, and 11-year-old FFPE myometrium samples relative to frozen samples. The correlation value in brackets was calculated following removal of miR-494 and miR-513. D: Mean expression level of miR-494 and miR-513 in indicated fixed samples, relative to expression levels in frozen samples.
Without these two miRNAs, the correlation coefficients between the 7- or 11-year-old FFPE samples and frozen myometrium were still significantly lower than for the 1-year-old FFPE specimens (0.9 and 0.86, respectively; Figure 2, B and C). This difference mainly stemmed from a decrease in the number of probes detected above background over time of storage (Figure 3A). In frozen myometrium, approximately 60% of the miRNA sequences were detected as present, whereas following fixation only 53% miRNAs were detected above array threshold. Within the subsequent 10 years of storage, an additional 15% of miRNA present calls were lost, bringing the final percentage of miRNAs detected in 11-year-old myometrium to approximately 38%. This reduction was accompanied by a three- to fourfold decrease in overall foreground array signal intensity, with no significant change in background signal (Figure 3B). Further analysis of the percentage of miRNA detected in FFPE specimens according to expression level in the frozen reference samples showed that 100% of the miRNAs highly expressed in frozen tissues are detected in FFPE samples (Figure 3C, Myo_Froz (1yr) >6). In contrast, the detection of miRNAs expressed at medium or low levels in frozen samples is gradually lost over time, and can be as low as 30% for low abundance miRNAs in 11-year-old FFPE samples. Thus, miRNA expression profiles in archived samples are accurate providing their expression levels are robust enough (normalized expression level h > 6 in frozen samples).
Figure 3.

Variation of microarray platform performance according to FFPE tissue block age. A: Mean percentage of miRNAs detected above threshold for frozen (Myo_(1yr), n = 2), fixed 1-year-old (Myo_(1yr), n = 2), fixed 7-year-old (Myo_(7yr), n = 3), and fixed 11-year-old (Myo_(11yr), n = 4) myometrium tissues. The minimum threshold was defined as the 5% symmetric trimmed mean plus two standard deviations of the foreground minus background median intensities across all empty spots on individual arrays. B: Average foreground signals (excluding control spots) and background signals (including all spots) for microarrays described in A. C: Mean percentage of miRNAs detected above threshold in fixed myometrium samples according to expression levels in frozen samples.
Differential miRNA Expression in Archived Samples
A common objective for miRNA array analyses is identification of miRNAs that are differentially expressed between samples. Additionally, unsupervised techniques such as hierarchical clustering and principal component analysis can reveal similarities and differences between samples at the level of global expression signatures. We used principal component analysis to identify the major axes of variability among all 12 arrays of paired frozen and fixed myometrium and B-cell lymphoma (Figure 4A). The myometrium miRNA expression profiles from two separate donors were highly similar and clustered tightly together in the first two principal components, but did separate on the third principal component with a lower magnitude of variation (data not shown). There was a higher donor-to-donor variation in B-cell lymphoma samples; however, the fixed and frozen samples from the same donor had very comparable expression profiles (Table 2). A two-way variance analysis confirmed that there were no statistically significant differences between miRNA profiles of the same tissue preserved either as frozen or fixed for 6 or 24 hours and that all of the variation was solely associated with the tissue type (see Supplemental Table 5 at http://jmd.amjpathol.org).
Figure 4.
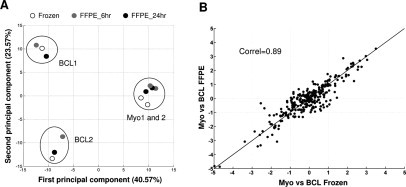
Differential miRNA expression for paired fixed versus frozen myometrium (n = 6 samples) and B-cell lymphoma (n = 6 samples). A: Principal component analysis of miRNA expression in fixed and paired frozen myometrium and B-cell lymphoma. B: Correlation between mean differential miRNA expression level (Δh) in fixed myometrium and B-cell lymphoma as compared to corresponding frozen tissue set. The indicated correlation coefficient includes probes that were detected in any of the eight analyzed samples.
The similarity of miRNA expression profiles in fixed and frozen tissue sets was supported by a high correlation (r = 0.89) between mean differential expression profiles in FFPE versus frozen myometrium and B-cell lymphoma (Figure 4B). We also verified that data analysis of differential expression between FFPE myometrium and B-cell lymphoma identifies the same miRNA candidates as analysis of matched frozen samples (Table 3). Nineteen miRNAs were found to exhibit sufficient statistical and signal differences to be considered differentially expressed miRNAs between myometrium and B-cell lymphoma (|Δh|4 >1.6 or fivefold change, P < 0.01). The overall correlation of differential expression level was >0.99 for this subset of miRNAs. Finally, we examined the preservation of log difference in miRNA expression between FFPE and frozen myometrium and B-cell lymphoma. We found that the mean log difference across all miRNA probes when comparing fold change within either frozen or FFPE tissue sets was 0.008 (−0.06 and 0.07 with 95% confidence). Together, these results demonstrate that the difference in expression between these two tissue types is essentially maintained with either method of tissue storage.
Table 3.
Expression Levels of Representative miRNAs Differentially Expressed by Fivefold or More (|Δh| >1.6, P < 0.01) in frozen versus fixed myometrium and B-cell lymphoma.
| MiRNA | Frozen (Δh*) | Fixed (Δh*) | P value (frozen) | P value (fixed) |
|---|---|---|---|---|
| hsa_miR_155 | −4.97 | −4.82 | 3.03E-06 | 3.65E-06 |
| hsa_miR_18 | −2.70 | −3.41 | 1.42E-02 | 5.04E-03 |
| ambi_miR_7073 | 1.93 | 1.70 | 2.45E-03 | 4.53E-03 |
| hsa_miR_143 | 1.96 | 2.09 | 6.34E-04 | 4.44E-04 |
| hsa_miR_100 | 2.03 | 2.18 | 5.41E-05 | 3.53E-05 |
| hsa_miR_196a | 2.07 | 1.67 | 2.06E-03 | 5.74E-03 |
| ambi_miR_7101 | 2.19 | 2.48 | 1.12E-02 | 6.49E-03 |
| hsa_miR_27b | 2.21 | 2.26 | 4.62E-05 | 3.98E-05 |
| hsa_miR_125b | 2.28 | 2.30 | 1.89E-04 | 1.78E-04 |
| hsa_miR_203 | 2.30 | 2.55 | 1.03E-02 | 6.51E-03 |
| hsa_miR_99a | 2.32 | 2.45 | 1.29E-02 | 1.02E-02 |
| hsa_miR_195 | 2.55 | 2.06 | 1.31E-03 | 3.75E-03 |
| hsa_miR_214 | 2.57 | 2.27 | 4.78E-05 | 9.70E-05 |
| hsa_miR_189 | 2.65 | 2.70 | 3.28E-03 | 2.98E-03 |
| hsa_miR_145 | 2.65 | 2.69 | 8.99E-04 | 8.35E-04 |
| hsa_miR_196b | 2.74 | 2.26 | 4.31E-04 | 1.20E-03 |
| hsa_miR_10b | 2.97 | 3.03 | 1.94E-03 | 1.76E-03 |
| hsa_miR_218 | 3.04 | 2.76 | 1.32E-04 | 2.25E-04 |
| hsa_miR_204 | 3.49 | 3.56 | 7.59E-04 | 6.87E-04 |
Mean differential miRNA expression level between myometrium and B-cell lymphoma (n = 2 donors per tissue type) using either frozen or fixed samples.
Method Reproducibility and Accuracy
To determine the reproducibility of miRNA gene expression measurements in archived tissues, we next performed three independent total RNA isolations from two individual FFPE blocks 8 or 12 years old (spleen and liver normal adjacent tissue; see Supplemental Table 1 at http://jmd.amjpathol.org). Each technical replicate was then labeled and hybridized individually on single color microarrays carrying 662 unique miRNA probes. Following data analysis, miRNA expression levels were found to be highly similar between tissue block replicates (see Supplemental Table 6 at http://jmd.amjpathol.org). The Pearson correlations between triplicate arrays were 0.95, 0.96, and 0.98 for liver and 0.97, 0.98, and 0.99 for spleen. The individual probes signal variation was also low. The mean %CV for miRNAs detected above background was 4.94% and 5.14% for spleen and liver, respectively, indicating high reproducibility for the entire process (RNA isolation and microarray platform).
To assess method accuracy, we compared microarray expression data with real-time qRT-PCR. We performed duplicate reverse transcription reactions followed by duplicate PCR on each of the technical replicate RNA samples using TaqMan microRNA assays specific for 14 miRNAs, including eight miRNAs that showed little to no change in expression levels between tissue types (miR-16, -17–5p, -24, -25, -103, -106a, -191, and let-7a), and six miRNAs that were differentially expressed to various extent (miR-122a, -155, -181a, -192, -194, and -223) (Figure 5A; see Supplemental Figure 2 at http://jmd.amjpathol.org). qRT-PCR data were highly reproducible between technical replicates with mean %CV of 1.28 and 0.99% for spleen and liver NAT, respectively. For each miRNA, the direction of change in miRNA expression between the two tissues was preserved relative to the microarray (Figure 5B). Overall, the concordance of differential expression levels between microarray and qRT-PCR platforms was excellent with a Pearson correlation of 0.98. Thus, with optimized RNA isolation, data normalization, and microarray methodologies, miRNA expression profiling in archived FFPE samples is a robust and accurate process appropriate for biomarker discovery and measurement of gene expression changes.
Figure 5.

Comparison between microarray and qRT-PCR platforms. A: Example of miRNAs differentially expressed in three independent liver NAT and spleen technical replicates as determined by microarray and qRT-PCR. For each RNA sample, duplicate reverse transcription reactions followed by duplicate PCR from each reverse transcription reaction was performed. B: Correlation between the microarray and qRT-PCR platforms for the mean differential expression of 14 miRNAs in liver NAT and spleen replicates.
Discussion
While traditional mRNA microarray platforms may lack appropriate performances for analysis of gene expression in fixed tissues, several reports suggest that miRNA expression profiling of archived human specimens can be used for biomarker discovery.20,21 However, no thorough analysis and systematic comparison with reference paired frozen samples have been performed. Previously, we identified and validated a superior-performing RNA extraction method allowing isolation of large amounts of quality RNA to perform robust miRNA expression analyses by qRT-PCR in FFPE tissues.19 Here, we used this method to evaluate miRNA expression profiling in 31 different FFPE tissue blocks up to 11 years old. Using nine different mouse and human tissue types, we demonstrate that the original miRNA composition of frozen tissue is essentially preserved after routine fixation in formalin and long-term storage. We show that independent sections, RNA isolations, and microarray processing produce reliable and reproducible miRNA expression measurements. We also determined that the difference in specimen-specific miRNA expression is larger than any technical variability associated with RNA isolation and profiling processes. This comprehensive study demonstrates the feasibility and utility of miRNA expression measurements in FFPE tissue specimens using optimized high-throughput microarray technologies.
A pilot study using freshly fixed mouse brain, kidney, small intestine, and stomach demonstrated that the FFPE-derived miRNA expression data are highly comparable with the data obtained from corresponding frozen specimens. A similar study was performed using remnant archived human specimens that were specifically chosen to represent different tissue types with variable cellular content and heterogeneity. While myometrium is almost entirely made of smooth muscle cells (99%), B-cell lymphoma consists primarily of monoclonal lymphocytes (95%), and colon mucosal epithelial cells constitute about 60% of colon tissue cellular makeup.22 Independent of the tissue type, we observed that miRNA expression profiles in tissues fixed in formalin for 6 hours are nearly identical to those fixed for 24 hours. The “percent present call” metric, used to determine the total number of genes detected relative to the total number of probe sets on an array, can be a useful measure of the sample and/or platform performance. The analysis of miRNA percent present calls in FFPE myometrium, B-cell lymphoma, and colon revealed a loss of up to 17% miRNAs in comparison to the paired frozen tissue. Failure to capture a larger percentage of miRNAs in FFPE samples may be a consequence of a lower miRNA fraction recovery or excessive RNA modification during FFPE tissue storage. More than 90% of the miRNA sequences detected in the flash-frozen myometrium and B-cell lymphoma specimens were also detected in their matching FFPE samples, and the Pearson correlation values between average miRNA expression signals in FFPE and frozen tissue sets were high (Table 2). For colon, only 62% of the miRNA sequences detected in the frozen specimens were also found present in fixed specimens, and therefore the correlation was lower (Table 2). This observation is likely a consequence of cellular heterogeneity of colon accentuated by the histological and functional subdivision of this organ.
Based on high tissue homogeneity, we chose myometrium as the model system to assess the effect of extended storage time on the quality of miRNA expression profiling. In our study, the percentage of miRNAs detected decreased by a total of 22% within an 11-year-long storage of fixed myometrium tissue relative to frozen myometrium (Figure 3A). This loss was the result of a decreased global foreground array signal (Figure 3B) leading to a lower signal to noise ratio over time. This observation suggests that the higher extent of RNA degradation in older FFPE blocks resulted in recovery of short nonspecific RNA fragments that competed with and reduced the efficiency of miRNA labeling. Therefore, this loss in performance impacted primarily miRNAs expressed at low to medium levels in myometrium (h < 6 in frozen reference sample), leaving the high signal probes relatively unaffected (Figure 3C). Consistent with this observation, a gradual increase in the probe signal intensity of miR-494 and -513 was detected in aging myometrium tissue blocks, peaking at 460- and 161-fold in the 11-year-old samples respective to frozen tissue. This unexpected increase in abundance could be indicative of degradation of precursor miRNA or mRNA species that produced fragments that were non-specifically labeled and hybridized to miR-494 and -513 probes on the array.
Recently, we reported specific miRNA expression patterns that differentiate frozen pancreatic cancer specimens from chronic pancreatitis and normal pancreas samples.12 A similar study was subsequently published by Bloomston et al using FFPE tissue blocks.20 There were 46 common miRNAs found differentially expressed between pancreatic cancer and reference samples across both studies, out of which 42 (91.3%) showed concordant direction of fold change, while four (8.7%) were discordant. This correlation is surprisingly high considering that different tissue sets, RNA isolation methods, miRNA labeling protocols, microarray platforms, data normalization strategies, and reference samples (normal pancreas from healthy donors versus normal adjacent tissue) were used. In the present study, we thoroughly assessed the ability of miRNA microarrays to identify miRNAs differentially expressed between samples by using paired frozen and fixed biological replicates and unsupervised techniques, such as hierarchical clustering and principal component analysis. Myometrium RNA sample preparations from different donors generated similar levels of miRNA expression and the tight clustering of the samples is likely a consequence of high homogeneity of the tissue. There was a higher donor-to-donor variation in B-cell lymphoma samples, which likely result from the difference in cellular composition between the two tissue specimens, as shown in the pathology report (see Supplemental Table 1 at http://jmd.amjpathol.org).
A total of 108 miRNAs differentially expressed (|Δh| >1 or 2.7-fold change) between myometrium and B-cell lymphoma were detected in RNA from frozen and fixed tissue sets; 79 in frozen tissue set in contrast to 77 in the FFPE tissue set. The majority of those miRNAs (48/108, 44.4%) were common between frozen and fixed myometrium and B-cell lymphoma. Of the total number of miRNAs detected, 28.7% (31/108) were unique to the flash-frozen tissue set and represent miRNAs whose signal intensity decreased as a result of the fixation process, perhaps due to increased array background. miRNAs observed only in the FFPE tissue set (29/108, 26.8%) exemplify those genes whose signal intensity increased following the fixation process, likely as a result of nonspecific binding of fragmented RNA species due to degradation. Regardless of these small discrepancies, expression levels of the top 19 differentially expressed miRNAs (|Δh| > 1.6, P < 0.01) between the FFPE and frozen myometrium and B-cell lymphoma were highly concordant, with a Pearson correlation value of 0.99 (Table 3). This result was further supported by a paired t-test analysis across matched samples for each miRNA probe where we tested the hypothesis that differential expression is equivalent between tissue storage methods. The resulting P value was 0.819, indicating that this hypothesis cannot be rejected by the data. However, it should be noted that within the limitations of this study we cannot eliminate the possibility that for a larger sample set, there may be a statistically significant difference associated with different preservation conditions.
Similar to other gene expression measurement platforms, miRNA expression profiling in FFPE tissues must be reproducible and accurate before it can be used routinely in discovery studies and potential diagnostic applications. We showed here that the technical variability associated with independent RNA isolations, sample processing and array hybridization of two FFPE tissues, liver NAT and spleen, is acceptable (%CV approximately 5%) in comparison to the differences resulting from biological variability between specimens. Furthermore, a subset of differentially expressed miRNAs identified through comparative analysis of microarray data were verified by qRT-PCR, and the Pearson correlation coefficient between platforms was 0.98. On this subset of miRNAs, the intraplatform variability was better with qRT-PCR than with microarray, with %CV of 0.99 and 1.28% versus 3.0 and 5.9% for liver and spleen, respectively. This discordance can be explained by the lower sensitivity and linear dynamic range of the microarray platform, which result in compression of microarray data for low intensity signals. As discussed above, degradation of longer RNA species in archived FFPE blocks can also further reduce detection of low abundance miRNAs by affecting the labeling efficiency of the miRNA population. In contrast, more sensitive and gene specific methods such as qRT-PCR should be less susceptible to RNA degradation, and qRT-PCR has been shown to accurately quantify miRNA levels in FFPE blocks up to 12 years old.19 For example, miR-122a and -192 were found overexpressed in liver NAT relative to spleen using both platforms in the present study. However, these miRNAs were below the microarray detection threshold in spleen (see Supplemental Table 6 at http://jmd.amjpathol.org). Removing these two-probe signals from our reproducibility analysis significantly reduced the mean microarray %CV from 5.9 to 2.4%.
Overall, our data demonstrate the reproducibility and accuracy of miRNA expression profiling in FFPE tissues and validate its use for molecular characterization of various tissue types up to 11 years old. The understanding of miRNA microarray performances using FFPE tissues is extremely valuable to appropriately design retrospective studies using well-annotated archived clinical samples and extend the application of miRNA to new diagnostic, prognostic, and treatment response biomarkers.
Acknowledgements
We thank Emily Zeringer and Richard Conrad for the preparation of the mouse FFPE specimens.
Footnotes
Supported in part by National Cancer Institute grant 1R44CA118785.
Supplemental material for this article can be found at http://ajp.amjpathol.org.
J.S. is affiliated with Luminex Corp. A.E.S., T.D., J.S., M.D., and E.L. are employees of Asuragen, Inc. None of the authors declare any financial conflicts of interest with companies whose products were evaluated in this manuscript.
Supplementary data
References
- 1.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, Moore T, Hudson J, Jr, Lu L, Lewis DB, Tibshirani R, Sherlock G, Chan WC, Greiner TC, Weisenburger DD, Armitage JO, Warnke R, Levy R, Wilson W, Grever MR, Byrd JC, Botstein D, Brown PO, Staudt LM. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 2.Ghadimi BM, Grade M, Difilippantonio MJ, Varma S, Simon R, Montagna C, Fuzesi L, Langer C, Becker H, Liersch T, Ried T. Effectiveness of gene expression profiling for response prediction of rectal adenocarcinomas to preoperative chemoradiotherapy. J Clin Oncol. 2005;23:1826–1838. doi: 10.1200/JCO.2005.00.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takahashi H, Nemoto T, Yoshida T, Honda H, Hasegawa T. Cancer diagnosis marker extraction for soft tissue sarcomas based on gene expression profiling data by using projective adaptive resonance theory (PART) filtering method. BMC Bioinformatics. 2006;7:399. doi: 10.1186/1471-2105-7-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, Lin L, Chen G, Gharib TG, Thomas DG, Lizyness ML, Kuick R, Hayasaka S, Taylor JM, Iannettoni MD, Orringer MB, Hanash S. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8:816–824. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- 5.Glas AM, Floore A, Delahaye LJ, Witteveen AT, Pover RC, Bakx N, Lahti-Domenici JS, Bruinsma TJ, Warmoes MO, Bernards R, Wessels LF, Van't Veer LJ. Converting a breast cancer microarray signature into a high-throughput diagnostic test. BMC Genomics. 2006;7:278. doi: 10.1186/1471-2164-7-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi L, Reid LH, Jones WD, Shippy R, Warrington JA, Baker SC, Collins PJ, de Longueville F, Kawasaki ES, Lee KY, Luo Y, Sun YA, Willey JC, Setterquist RA, Fischer GM, Tong W, Dragan YP, Dix DJ, Frueh FW, Goodsaid FM, Herman D, Jensen RV, Johnson CD, Lobenhofer EK, Puri RK, Schrf U, Thierry-Mieg J, Wang C, Wilson M, Wolber PK, Zhang L, Amur S, Bao W, Barbacioru CC, Lucas AB, Bertholet V, Boysen C, Bromley B, Brown D, Brunner A, Canales R, Cao XM, Cebula TA, Chen JJ, Cheng J, Chu TM, Chudin E, Corson J, Corton JC, Croner LJ, Davies C, Davison TS, Delenstarr G, Deng X, Dorris D, Eklund AC, Fan XH, Fang H, Fulmer-Smentek S, Fuscoe JC, Gallagher K, Ge W, Guo L, Guo X, Hager J, Haje PK, Han J, Han T, Harbottle HC, Harris SC, Hatchwell E, Hauser CA, Hester S, Hong H, Hurban P, Jackson SA, Ji H, Knight CR, Kuo WP, LeClerc JE, Levy S, Li QZ, Liu C, Liu Y, Lombardi MJ, Ma Y, Magnuson SR, Maqsodi B, McDaniel T, Mei N, Myklebost O, Ning B, Novoradovskaya N, Orr MS, Osborn TW, Papallo A, Patterson TA, Perkins RG, Peters EH, Peterson R, Philips KL, Pine PS, Pusztai L, Qian F, Ren H, Rosen M, Rosenzweig BA, Samaha RR, Schena M, Schroth GP, Shchegrova S, Smith DD, Staedtler F, Su Z, Sun H, Szallasi Z, Tezak Z, Thierry-Mieg D, Thompson KL, Tikhonova I, Turpaz Y, Vallanat B, Van C, Walker SJ, Wang SJ, Wang Y, Wolfinger R, Wong A, Wu J, Xiao C, Xie Q, Xu J, Yang W, Zhang L, Zhong S, Zong Y, Slikker W., Jr The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nature Biotechnol. 2006;24:1151–1161. doi: 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lewis F, Maughan NJ, Smith V, Hillan K, Quirke P. Unlocking the archive–gene expression in paraffin-embedded tissue. J Pathol. 2001;195:66–71. doi: 10.1002/1096-9896(200109)195:1<66::AID-PATH921>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 8.Masuda N, Ohnishi T, Kawamoto S, Monden M, Okubo K. Analysis of chemical modification of RNA from formalin-fixed samples and optimization of molecular biology applications for such samples. Nucleic Acids Res. 1999;27:4436–4443. doi: 10.1093/nar/27.22.4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Godfrey TE, Kim SH, Chavira M, Ruff DW, Warren RS, Gray JW, Jensen RH. Quantitative mRNA expression analysis from formalin-fixed, paraffin-embedded tissues using 5′ nuclease quantitative reverse transcription-polymerase chain reaction. J Mol Diagn. 2000;2:84–91. doi: 10.1016/S1525-1578(10)60621-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Esquela-Kerscher A, Slack FJ. Oncomirs—microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 11.Sevignani C, Calin GA, Siracusa LD, Croce CM. Mammalian microRNAs: a small world for fine-tuning gene expression. Mamm Genome. 2006;17:189–202. doi: 10.1007/s00335-005-0066-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szafranska AE, Davison TS, John J, Cannon T, Sipos B, Maghnouj A, Labourier E, Hahn SA. MicroRNA expression alterations are linked to tumorigenesis and non-neoplastic processes in pancreatic ductal adenocarcinoma. Oncogene. 2007;26:4442–4452. doi: 10.1038/sj.onc.1210228. [DOI] [PubMed] [Google Scholar]
- 13.Wang H, Ach RA, Curry B. Direct and sensitive miRNA profiling from low-input total RNA. RNA. 2007;13:151–159. doi: 10.1261/rna.234507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nelson PT, Baldwin DA, Scearce LM, Oberholtzer JC, Tobias JW, Mourelatos Z. Microarray-based, high-throughput gene expression profiling of microRNAs. Nat Methods. 2004;1:155–161. doi: 10.1038/nmeth717. [DOI] [PubMed] [Google Scholar]
- 15.Castoldi M, Schmidt S, Benes V, Noerholm M, Kulozik AE, Hentze MW, Muckenthaler MU. A sensitive array for microRNA expression profiling (miChip) based on locked nucleic acids (LNA) RNA. 2006;12:913–920. doi: 10.1261/rna.2332406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xi Y, Nakajima G, Gavin E, Morris CG, Kudo K, Hayashi K, Ju J. Systematic analysis of microRNA expression of RNA extracted from fresh frozen and formalin-fixed paraffin-embedded samples. RNA. 2007;13:1668–1674. doi: 10.1261/rna.642907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shingara J, Keiger K, Shelton J, Laosinchai-Wolf W, Powers P, Conrad R, Brown D, Labourier E. An optimized isolation and labeling platform for accurate microRNA expression profiling. RNA. 2005;11:1461–1470. doi: 10.1261/rna.2610405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huber W, von Heydebreck A, Sultmann H, Poustka A, Vingron M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics. 2002;18(Suppl 1):S96–S104. doi: 10.1093/bioinformatics/18.suppl_1.s96. [DOI] [PubMed] [Google Scholar]
- 19.Doleshal M, Magotra AA, Choudhury B, Cannon BD, Labourier E, Szafranska AE. Evaluation and validation of total RNA extraction methods for microRNA expression analyses in formalin-fixed, paraffin-embedded tissues. J Mol Diagn. 2008;10(3):203–211. doi: 10.2353/jmoldx.2008.070153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bloomston M, Frankel WL, Petrocca F, Volinia S, Alder H, Hagan JP, Liu CG, Bhatt D, Taccioli C, Croce CM. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA. 2007;297:1901–1908. doi: 10.1001/jama.297.17.1901. [DOI] [PubMed] [Google Scholar]
- 21.Nelson PT, Baldwin DA, Kloosterman WP, Kauppinen S, Plasterk RH, Mourelatos Z. RAKE and LNA-ISH reveal microRNA expression and localization in archival human brain. RNA. 2006;12:187–191. doi: 10.1261/rna.2258506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cotran R, Kumar V, Collins T, Robbins SL. Robbins Pathologic Basis of Disease. ed 6. W.B. Saunders Company; Philadelphia: 1999. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.