

Abstract
The iron(II) hydride dimers [LRFe(μ-H)2FeLR] (LMe = 2,4-bis(2,6-diisopropylphenylimino) pent-3-yl; LtBu = 2,2,6,6-tetramethyl-3,5-bis(2,6-diisopropylphenylimino)hept-4-yl) abstract hydrocarbyl groups from BR′3 (R′ = Et, Ph) to give LRFeR′ and LRFe(μ-H)2BR′2. Mechanistic studies with R = R′ = Me are consistent with a process in which the hyride dimer opens one Fe-H bond, and subsequent B-H bond formation is concerted with dissociation of an Fe-H unit. Cleavage of boron-carbon bonds is likely to proceed at least in part from transient quaternary borate anions. In a separate bond-breaking reaction, LMeFe(μ-H)2BEt2 reacts with N2H4 to eject H2 from the bridging hydrides and cleave the N-N bond in the diaminoborate complex LMeFe(μ-NH2)2BEt2. These novel bond-breaking reactions are facilitated by the low coordination number at the iron(II) center.
Keywords: Iron(II), B-C bonds, borane, N-N bonds, mechanism
Introduction
In addition to their classic roles as intermediates in homogeneous catalytic reactions of organometallic complexes,1 iron hydrides have been postulated to be reactive intermediates in the mechanisms of hydrogenase 2 and nitrogenase 3 enzymes. We have become especially interested in exploring the chemistry of unsaturated hydride complexes of the late transition metals because of the juxtaposition of the reactive M-H functionality and an open coordination site for substrate binding.
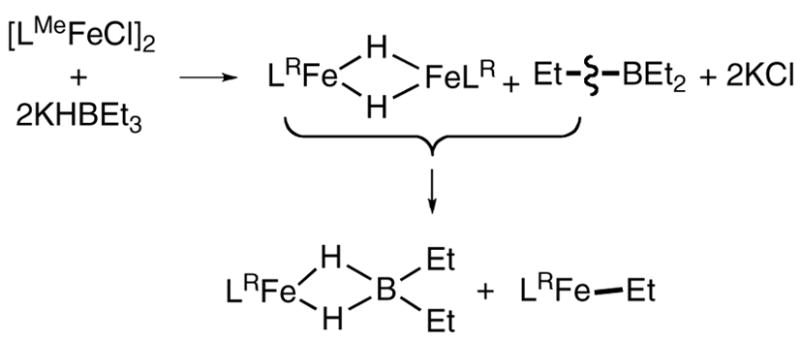
We have used bulky β-diketiminate ligands (Figure 1) to enable the synthesis of the only known three-and four-coordinate iron hydride complexes.4,5 These unsaturated iron hydrides, LRFe(μ-H)2FeLR, are prepared from reactions between iron(II) chloride complexes and potassium triethylborohydride (eq 1a and 1b).
Figure 1.
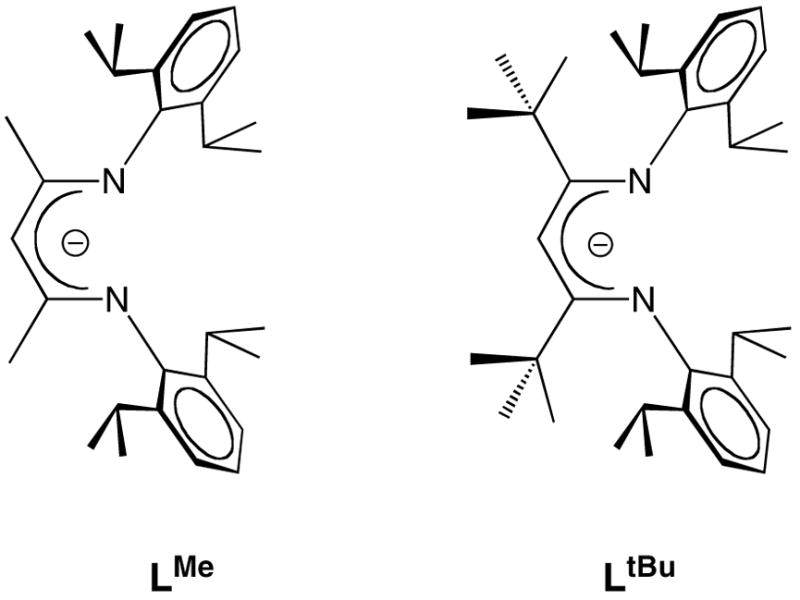
Diketiminate ligands LR, where R indicates the substituent on the N2C3 backbone.
| (1a) |
| (1b) |
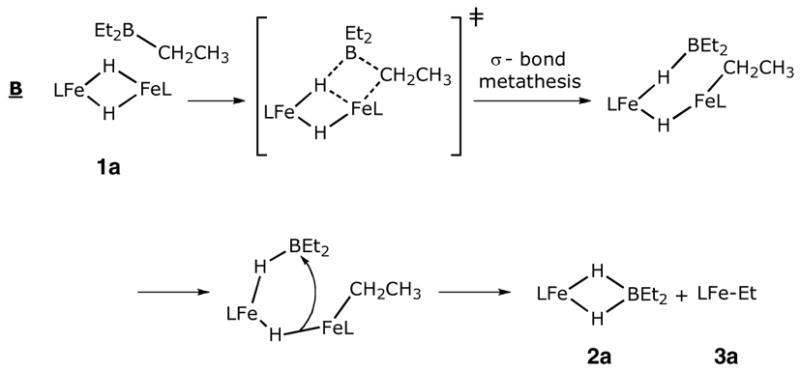
Although both iron hydride compounds exist as dimers in the solid state, they behave differently in solution: LtBuFe(μ-H)2FeLtBu dissociates into monomers in solution,4a while only dimer is observed in solutions of the LMeFe(μ-H)2FeLMe.4b One phenomenon caught our attention during the preparation of the iron hydride complexes: long reaction time (> 30 min at room temperature) or heating leads to the formation of red products instead of the brown iron hydride complexes. Here, we report that the iron hydride complex reacts with the byproduct BEt3 to give an iron dihydridoborate complex and an iron ethyl complex (Scheme 1).
Scheme 1.
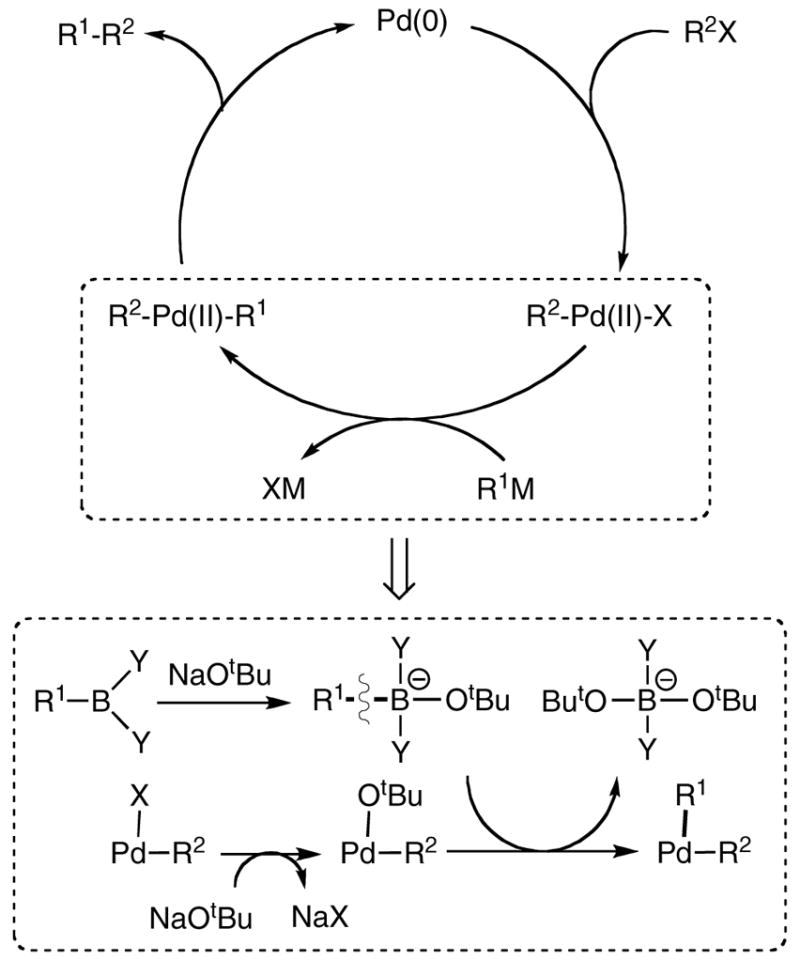
The reaction in Scheme 1 features migration of a boron-bound hydrocarbyl group. Boron-to-carbon migrations have precedent in the organic chemistry literature, where alkylboranes have been utilized for forming N-C or C-C bonds to nucleophiles that contain a potential leaving group (e.g. deprotonated α-haloesters or carbon monoxide).6 In these reactions, trialkylborane initially attacks the nucleophile to form a quaternary adduct, and one of the boron R groups subsequently migrates to the adjacent N or C, displacing a leaving group. The migration of hydrocarbyl groups from boron to a transition metal is also a key step of the Suzuki-Miyaura reaction, the transmetallation of the organoboron compound to form organopalladium species (Scheme 2).7 Interestingly, in both organic and organometallic precedents, quaternary anionic boron species are the actual alkyl/aryl group donor. For example, transmetallation from boron to palladium in the Sukuzi-Miyaura reaction is typically promoted by adding bases to form a quaternary borate anion (Scheme 2, bottom).6a, 8,9 Since the B-C bond cleavage in Scheme 1 requires no added base, and occurs in non-coordinating solvents, we were curious to learn whether this reaction is mechanistically distinct from the palladium reaction, possibly leading to insights on strategies for milder cross-coupling reactions.
Scheme 2.
Few boron alkylations of any sort are understood in great mechanistic detail.10 Below, we describe kinetic studies that give insight into the mechanism of this transformation. The hydridoborate product also undergoes an interesting reaction with hydrazine to cleave the N-N bond to yield a diaminoborate complex.
Results and Discussion
Formation of the dihydridoborate complex from LRFe(μ-H)2FeLR and BEt3
Reaction of the iron hydride dimers LRFe(μ-H)2FeLR (LR = β-diketiminate ligands shown in Figure 1; 1a with R = Me and 1b with R = tBu) with one molar equivalent of BR3 (R = Et, Ph) in toluene at 80 °C for 22 h leads to an equimolar mixture of a four-coordinate hydridoborate complex, LRFe(μ-H)2BR′2 (R = Me, R′ = Et 2a or Ph 2b; R = tBu, R′ = Et 2c), and an iron alkyl complex LRFeR′ (R = Me, R′ = Et 3a or Ph 3b; R = tBu, R′ = Et 3c).11 These reactions are shown in Scheme 3. Reaction of 1a with B(OEt)3 and B(iBu)3 gave multiple unidentified products, and these reactions were not pursued further.
Scheme 3.
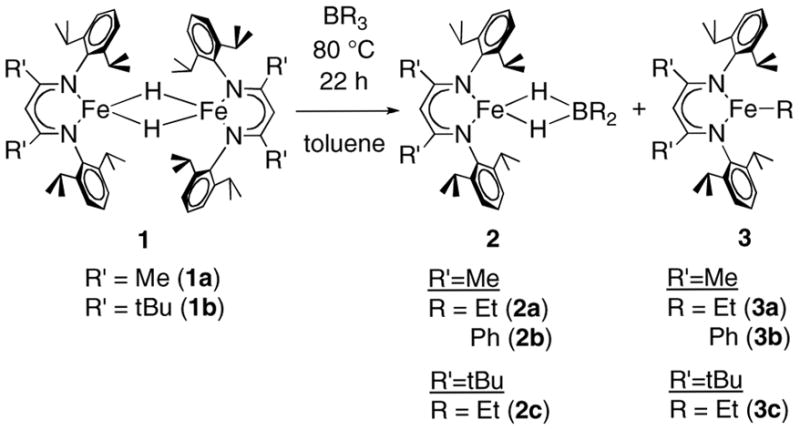
The alkyliron compounds 3a and 3c were identified by comparison to their known 1H NMR spectra (they can be prepared independently from [LRFeCl]n (n = 1 or 2) and Grignard reagents).12 The new complex LMeFePh (3b) was previously unknown, and so it was generated from the reaction of [LMeFe(μ-Cl)2FeLMe] with PhMgCl. Its spectroscopic features and X-ray crystal structure (see Experimental Section for spectroscopy and Supporting Information for solid-state structure) are analogous to those for the three-coordinate alkyl complexes. The 1H NMR spectrum of independently prepared 3b verifies that it is identical to one of the products from reaction of LMeFe(μ-Cl)FeLMe and BPh3.
The new (dihydridoborate)iron(II) complexes 2a, 2b, and 2c were isolated in 59–72% yield after fractional crystallization away from the iron(II) alkyl/aryl complexes. The reaction of 1a with BEt3 was very clean: 1a, 2a, and 3a are the only compounds observed in 1H NMR spectra of the reaction mixture. In the other reactions, there were a few unidentified paramagnetic peaks in the crude mixtures, representing side products that were removed by crystallization. Each dihydridoborate complex exhibits a magnetic moment of 4.0(1) to 5.7(1) μB in C6D6 solution, indicating a high-spin iron(II) center (S = 2). Each 1H NMR spectrum covers an extremely wide range from 100 ppm to −310 ppm, with large chemical shift dispersion as a result of the paramagnetic iron ion. The proton resonances were generally assigned from the relative integrations (see Experimental Section). The six-proton peaks for the BEt2 group and the LMe backbone could be distinguished because the 1H NMR spectra of LMeFe(μ-H)2BEt2 and LtBuFe(μ-H)2BEt2 differ only by the 6-proton resonance in the LMe compound and the 18-proton resonance in the LtBu compound. The bridging hydride protons are not observed because of fast relaxation.
A single crystal of each iron dihydridoborate complex was grown from pentane. The solid-state structure of 2a is shown in Figure 2 as an example, and others may be found in the Supporting Information. The bridging hydride ions were located and refined with an isotropic thermal parameter in each structure (the two hydrides are crystallographically distinct, except in the case of LMeFe(μ-H)2BEt2, where the hydride positions are related by a C2 axis). Including the two Fe-H bonds, each iron center has a pseudotetrahedral geometry with a Fe-H distance of about 1.7 Å (Table 1). Although the Fe···B distances are relatively short (ca. 2.24 Å), we do not postulate any direct Fe-B bonding interaction because the two atoms are held in proximity by the bridging hydride.13 The B-H distances range from 1.19(2) Å to 1.35(2) Å, which is comparable to the average M-H-B distance of 1.24 Å in the Cambridge Structural Database.
Figure 2.
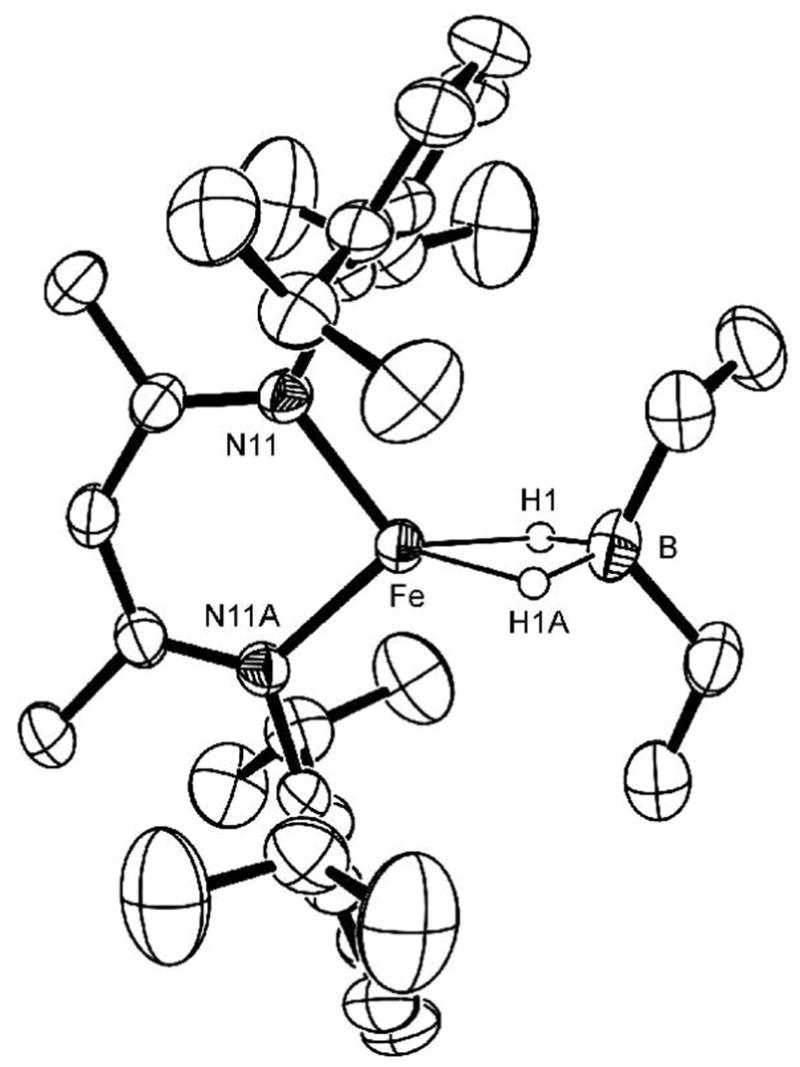
Thermal-ellipsoid plot of the molecular structure of LMeFe(μ-H)2BEt2 (2a). A crystallographic C2 axis passes through Fe and B. Other crystallographic data are in the Supporting Information.
Table 1.
Important bond distances and angles for dihydridoborate complexes in this work.
| Complex | Fe-H/Å | B-H/Å | Fe-B/Å |
|---|---|---|---|
| LMeFe(μ-H)2BEt2 (2a) | 1.74(2) | 1.35(2) | 2.237(3) |
| LMeFe(μ-H)2BPh2 (2b) | 1.72(2), 1.72(2) | 1.28(2), 1.19(2) | 2.239(2) |
| LtBuFe(μ-H)2BEt2 (2c) | 1.74(2), 1.73(2) | 1.26(2), 1.21(2) | 2.232(1) |
The formation of 2 and 3 is the result of an interesting B-C bond cleavage. Previous examples of B-C cleavage are most often from nucleophilic borates, rather than from boranes.14 In complexes with covalently bound hydridoborate ligands, BHnR4−n− (n = 1–4, R = H or alkyl) complexes are much less common than borohydride (BH4−) complexes.15 Known examples of MH2BR2 complexes typically come from double metathesis reactions of a metal chloride complex with an alkali metal salt of H2BR2 (R2 = H2, 16 C5H10, C8H14 17 and Et2 15b, 18). In some literature cases, the H2BEt2− group was present from impurities in commercial HBEt3−.15b, 1819 Here, the hydridoborate product is formed from cleavage of the B-C bond of a trialkylborane, and not from a dihydridoborate impurity in the starting material (see below).
Reaction of LMeFe(μ-H)2FeLMe with BR3: Rate Law and Activation Parameters
We used the reaction of LMeFe(μ-H)2FeLMe (1a) with BEt3 as the subject of kinetic studies because of the simple 1H NMR spectra of the starting material and the exceptionally clean reaction. Using an internal integration standard (LtBuFeCl) in a sealed capillary, we verified that the spectroscopic yields of products are 95% and 99% for LMeFe(μ-H)2BEt2 (2a) and LMeFeEt (3a) respectively. In the kinetics experiments, various amounts of 1a and BEt3 (>10 equiv relative to 1a) were mixed in C6D6 and held at various temperatures between 6.5 °C and 59.7 °C. Complexes 2a and 3a were the only products observed by 1H NMR spectroscopy. The integrations of the peaks at 13 ppm (1a), 17 ppm (2a), and −11 ppm (3a) were followed as a function of time, and invariably showed exponential decays. The rate constant was independent of [Fe] when a drop of liquid mercury was added to the reaction mixtures. Without the mercury, we observed a slight decrease in kobs from 3.97(7) × 10−3 s−1 to 2.51(2) × 10−3 s−1 with an increase of [1a] from 11.1 mM to 42.3 mM. With added Hg0, the derived kobs showed no clear trend while changing [1a] from 20 mM to 40 mM. Therefore, subsequent trials were run with a drop of mercury, which presumably amalgamates trace metallic iron.
For consistency, the following use exclusively the rate constants derived from following the decrease of 1a (Table 2). The rate has a first-order dependence on [1a] (as evident from its exponential decay, and the constancy of the rate constant upon variation of [1a]) and a first-order dependence on [BEt3] (from the linear dependence of the pseudo-first-order rate constant on BEt3 concentration). Therefore, the rate law is
Table 2.
Effect of [BEt3] on observed rate constant
| Entry | Initial concentrations (mM)
|
Observed rate constant kobs (s−1) | |
|---|---|---|---|
| 1 | BEt3 | ||
| 1 | 6.4 | 64.8 | 2.2(4)×10−3 |
| 2 | 6.4 | 129.6 | 4.7(2)×10−3 |
| 3 | 6.4 | 259.0 | 9.4(2)×10−3 |
| (2) |
The second-order rate constant at room temperature (21.5 °C) was 4.9(1) × 10−4 M−1s−1. Activation parameters were calculated from an Eyring plot of second-order rate constants from 6.5 °C to 59.7 °C, giving ΔH‡ = 21.8 ± 0.8 kcal·mol−1 and ΔS‡ = − 1 ± 2 eu.20
Table 4 shows that varying the steric demands of the diketiminate ligand and BR3 gives rate constants of the same magnitude, which slightly increase with a larger borane (BPh3) or diketiminate ligand (LtBu). Therefore, there is only a small steric influence on the reaction rate.
Table 4.
Rate constants with variation of R groups on the diketiminate and boranea
| Iron hydride reagent | BR3 reagent | Pseudo first-order rate constant kobs/s−1 |
|---|---|---|
| LMeFe(μ-H)2FeLMe (1a) | BEt3 | 3.09(2) × 10−3 |
| LMeFe(μ-H)2FeLMe (1a) | BPh3 | 8.91(5) × 10−3 |
| LtBuFe(μ-H)2FeLtBu (1b) | BEt3 | 4.8(2) × 10−3 |
[BR3] = 0.419 M, [LRFe(μ-H)2FeLR] = 26 mM, T = 40 °C.
Finally, the reaction of 1a with BEt3 was repeated in different polar solvents (75% o-difluorobenzene/25% C6D6 or THF-d8). We observed a small increase in rate constant using THF, but no increase in a more polar mixture of o-difluorobenzene and benzene (Table 5). The similarities in rate with a more polar solvent mixture suggests that charged species are not formed in the rate-limiting transition state (see below). The rate increase in THF may be from coordination to THF to intermediates, but the nature of this interaction was not queried further.
Table 5.
Solvent effect on rate constanta
B-C Bond Cleavage: Mechanistic Hypotheses
Several reasonable mechanisms can be proposed for the B-C bond cleavage reaction. We do not consider mechanisms in which BR3 undergoes β-hydride elimination because the reaction is equally facile with BPh3, for which β-hydride elimination is impossible. Moreover, independent experiments show that BEt3 and 1-hexene do not react when heated for 2 h at 80 °C, indicating that borane alkyl groups do not undergo substantial β-hydride elimination under the conditions of the reaction in Scheme 1.
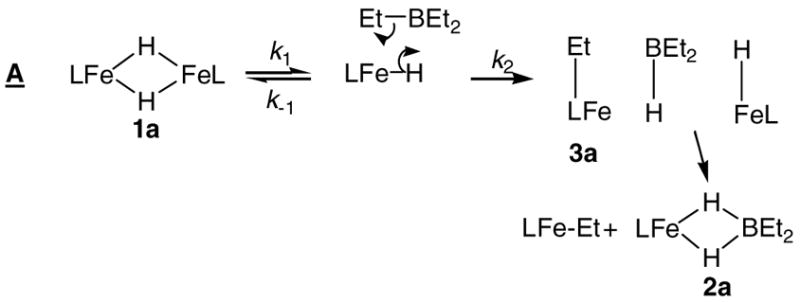
The mechanisms considered here are grouped into categories: dissociation of the iron hydride dimer into monomers before reaction with borane (A), concerted Fe-H and B-C cleavage with B-H formation (B, C, D), and formation and disproportionation of a transient iron triethylborohydride species (E, F).
Dissociation of 1a into monomer
Mechanism A (Scheme 4) illustrates the potentialdissociation of the iron hydridedimer as part of the mechanism. A mechanism of this type was strongly implicated in the reaction of L tBuFe(μ-H)2FeLtBu with 3-hexyne, where a zero-order dependence on alkyne concentration indicated an intramolecular rate-limiting step prior to any interaction with alkyne.4 However, in the reaction with borane described here, hydride dissociation can be ruled out because it does not predict the observed rate law. Mechanism A would lead to a zero-order dependence on [BEt3] if k1 (dimer dissociation) were rate-limiting, or a half order dependence on [1a] if k2 (attack of borane) were rate-limiting.
Scheme 4.
| (8) |
| (9) |
The time course of the concentration of 1a is inconsistent with a half-order dependence of the rate on [1a].
Concerted mechanisms
In mechanism B, 1a reacts with BEt3 in an initial rate-limiting step. While the empty boron p-orbital interacts with the bridging hydride, the nucleophilic boron-bound alkyl group simultaneously approaches the iron atom of 1a. This resembles the “σ-bond metathesis” mechanism that explains C-H activation reactions by d0 metal centers.24 This pathway leads to the observed products after dissociation of a bridging hydride, and is the microscopic reverse of the reaction of a ruthenium alkyl complex with catecholborane through a σ-bond metathesis mechanism.25
Mechanism C, on the other hand, begins with Fe-H bond breaking. After dissociating one of the Fe-H bonds, the “open” isomer leads to a six-membered transition state for a pericyclic reaction. As long as the initial Fe-H opening is rapid and reversible, mechanism C agrees with the experimental rate law, rate = k[1a][BEt3].
Mechanism D (Scheme 7) starts with rate-limiting abstraction of “H−” from 1a by the borane, breaking two of the Fe-H bonds and forming free HBEt3−. Previous researchers have observed alkyl group transfers from a borate to a metal center, which is more common compared to group transfer from borane species.14 In analogy, mechanism D concludes with transfer of an ethyl group from HBEt3− to iron. The ion pair undergoes a σ-bond metathesis to give the final products. Whether the hydride abstraction (D1) or the recombination of the ion pair (D2) is rate limiting, the predicted rate law is the same as mechanisms B and C: a first order dependence on both [1a] and [BEt3], in concord with the observed rate law.
Scheme 7.
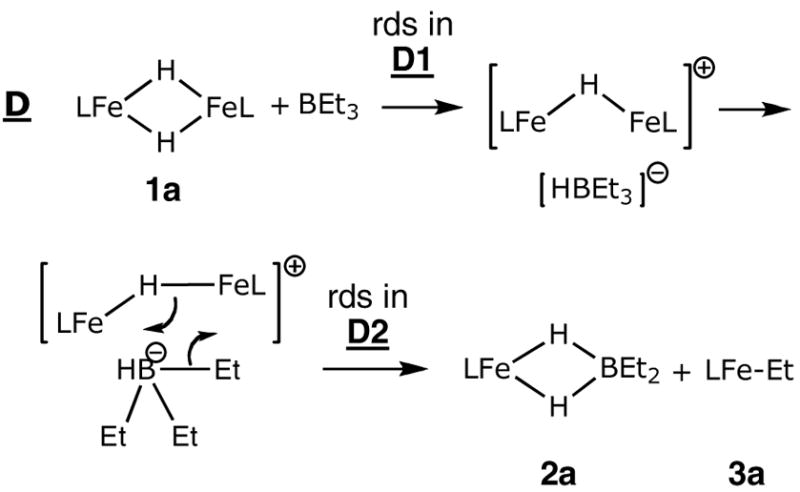
Notably, each of these mechanisms suffers from inconsistencies with other mechanistic data. First, the entropy of activation near zero is difficult to rationalize in the light of the bimolecular transition states in these mechanisms. In order to outweigh the expected large negative entropy change from bringing two molecules together, it is necessary to have a roughly equivalent release of entropy prior to the rate-limiting step. This situation is provided only by mechanisms C and D2, where the observed rate constant kobs is actually a product of the equilibrium constant for cleavage one or two Fe-H bond(s) in 1a (KFeH) and the rate constant for the elementary step consisting of the pericyclic reaction, kperi; kobs = KFeHkperi. Therefore ΔS‡obs = ΔSFeH + ΔS‡peri. We cannot determine either of these individual entropy values quantitatively, but it is reasonable to postulate that the entropy increase from partly opening the crowded dimer structure of 1a (ΔSFeH) could roughly equal the entropy decrease of bringing in the borane or borate to reach the transition state for the pericyclic reaction (ΔS‡peri). Molecular models of the reactants suggest that only an intermediate generated by cleavage of the Fe2H2 ring (as in mechanism C and D2) would have sufficient space for approach of the borane or borate B-C bond.
However, it is difficult to rationalize the similar observed reaction rate between BEt3 and BPh3 if mechanism C is followed. In the six-membered ring transition state, one of the R groups from the borate sits between the BR2 fragment and the LMeFe fragment. Considering the huge hindrance from the β-diketiminate ligand, this conformation is expected to have different steric effects for ethyl and phenyl groups. This argument, as well as the near-zero entropy of activation, makes mechanism C less plausible.
In mechanisms D, we note that an ionic intermediate is formed in or before the rate-limiting transition state. The rate of disappearance of 1a has very little dependence on solvent polarity, though polar solvents are expected to stabilize transition states in which charge is developing. Therefore, the solvent effect is inconsistent with mechanism D.
Borohydride disproportionation mechanisms
Commercial solutions of Super-Hydride (LiHBEt3) have been reported to undergo disproportionation to LiBEt4 and Li[BH2Et2] at room temperature over long time periods (eq 3).15b,18b,18c, 26 The resulting Li[BH2Et2] can lead to H2BEt2 complexes by metathesis with a metal chloride. In addition, solutions of LiHBEt3·THF are known to react with BEt3 to give LiBEt4 and (HBEt2)2, and subsequently Li[BH2Et2] 18c,26 (eq 3, 4).
| (3) |
| (4) |
However, the spurious presence of diethylborohydride anions is not likely to be involved in the reaction of 1a with BR3 for the following reasons. First, our KHBEt3 was prepared in-house from KH and BEt3 in toluene (eq 4) and stored as a solid. The 11B NMR spectrum of this isolated KHBEt3 in C6D6 has a single peak at −16 ppm, with no signs of dihydridoborate or BEt3 impurities. The 11B NMR spectrum of KHBEt3 did not show any change when heated to 80 °C for 2 h in benzene-d6 (mimicking the conditions of our kinetic experiments). An even more convincing piece of evidence is our ability to synthesize LMeFeH2BEt2 from 1a and BEt3, a reaction in which KHBEt3 is not present. Therefore, the presence of potassium diethylborohydride in our reaction is unlikely, and there is no necessity for hydridoborate salts in for the reaction to occur.
It is still necessary to consider the transient formation of trialkylborohydride species later in the pathway. For example, the first step of the reaction might resemble the reaction of alkali metal hydrides with triethylborane27 (eq 5).
| (5) |
The analogous reaction at iron would form a transient iron triethylborohydride complex, LMeFe(HBEt3), either through monomer (mechanism E) or through a partially open dimer (mechanism F). Because LMeFe(HBEt3) is the presumed intermediate in the reaction of LMeFeCl and KHBEt3 to form LMeFe(μ-H)2FeLMe + KCl + BEt3 (eq 1a, above), steps preceding the formation of this intermediate are assumed to be reversible (Scheme 8)
Scheme 8.

In Scheme 8, the transient iron triethylborohydride species lies in the center. Since LMeFe(HBEt3) is not observed by NMR spectroscopy during any reaction, this intermediate must be consumed very quickly (kd, k−2 and k′−2 are large). Assuming that LMeFe(HBEt3) is an intermediate on the way to [LMeFe(μ-H)2FeLMe]2 in eq 1, then k−2 and k−1 in mechanism E or k′ −1 in mechanism F must be larger than kd (or else 1a could not be isolated in the reaction of [LMeFeCl]2 and KHBEt3). Therefore k1 or k2 (in E) or k′1 (in F) is rate-limiting. In other words, the rate determining step (RDS) must happen before the formation of LMeFe(HBEt3).
E if RDS is k1,
| (6a) |
if RDS is k2,
| (6b) |
Mechanism E predicts a zero-order dependence on [BEt3] if k1 is the RDS or a half-order dependence on [1a] if k2 is the RDS (eq 6). Neither situation agrees with the observed rate law.
In F, as shown in Scheme 8, the hydride dimer first reacts with one molecule of BEt3, going through transition state a* to give LMeFeHBEt3 and LMeFeH. The LMeFeH monomer rapidly reacts with another molecule of BEt3 to form a second equivalent of LMeFeHBEt3. The two LMeFeHBEt3 species then undergo disproportionation to give the final products. If k′1 is rate-limiting the rate law is F
| (7) |
The first order dependence predicted for [1a] and [BEt3] is consistent with the experimental rate law. Moreover, the transition state a* involves the dissociation of one Fe-H bond and the formation of one B-H bond. As such, it bears a resemblance to the transition state for an interchange mechanism for substitution at a transition-metal complex (the intermediate stage between associative and dissociative substitution reactions). For Ia or Id mechanisms, values of ΔS‡ near zero are common.28
Mechanism F is also consistent with other mechanistic data. With the opening of one iron-hydride bond, the β-diketiminate has more space to avoid steric interactions with the borane R group. This explains the similar observed reaction rate for BEt3 and BPh3. Finally, there are no ionic intermediates prior to the rate-determining-step, which agrees with the lack of a solvent polarity effect during the reaction. Based on the above analysis, mechanism F is the one most consistent with all experimental data.
Because the key disproportionation step lies after the rate-limiting step, kinetic data are not useful in elucidating its characteristics. Therefore, we endeavored to independently synthesize LMeFe+BEt−4, the putative intermediate in the reaction.29 First, reaction of [LMeFeCl]2 and NaBEt4 in Et2O leads cleanly to the formation of LMeFeEt, as observed by 1H NMR spectroscopy. Also, LMeFeEt does not react with BEt3 up to 120 °C. This is a thermodynamic phenomenon rather than a kinetic barrier, because heating LMeFePh to 80 °C for 3 h with one equiv of BEt3 gives a mixture of LMeFePh and LMeFeEt. These experiments strongly suggest that transient LMeFe+BR4− species are unstable with respect to dissociation of borane, as required in the last step of mechanism F. In benzene, the ionic species may exist as a contact ion pair or as a tight ion pair, analogous to one that was previously observed.30
In summary, the available kinetic and mechanistic evidence are most consistent with mechanism F, in which a transient ring-opened isomer of 1a reacts with BR3 to give LFeHBR3, which converts into the iron hydridoborate and iron alkyl products through the ionic intermediate LFeBR4.
LMeFe(μ-H)2BEt2 reactivity with hydrazine: N-N bond cleavage
Exploration of the reactivity of the dihydridoborate complex LMeFe(μ-H)2BEt2 (2a) has shown an interesting series of reactions upon the addition of hydrazine. At room temperature, complex 2a reacts rapidly with H2NNH2 to give a hydrazine adduct, LMeFe(η1-H2NNH2)(μ-H)2BEt2 (4), isolated in 90% yield. Titration of hydrazine into a solution of 2a gave solutions with 1H NMR spectra similar to 2a but variable chemical shifts, showing that the bound hydrazine is in rapid exchange with free hydrazine. Proton NMR resonances for the N-bound protons are not seen because of their proximity to the paramagnetic metal center.
The binding stoichiometry in solution can be determined by Job plot analysis, where small aliquots of hydrazine are added to 2a and the 1H NMR spectrum is recorded after each addition. The chemical shift of peaks near −260 ppm and 19 ppm vary depending on the ratio of H2NNH2 and 2a (Figure 4). The intersection of the initial line (deficiency of hydrazine) and final line (deficiency of 2a) shows that the two molecules bind in a 1:1 manner.
Figure 4.
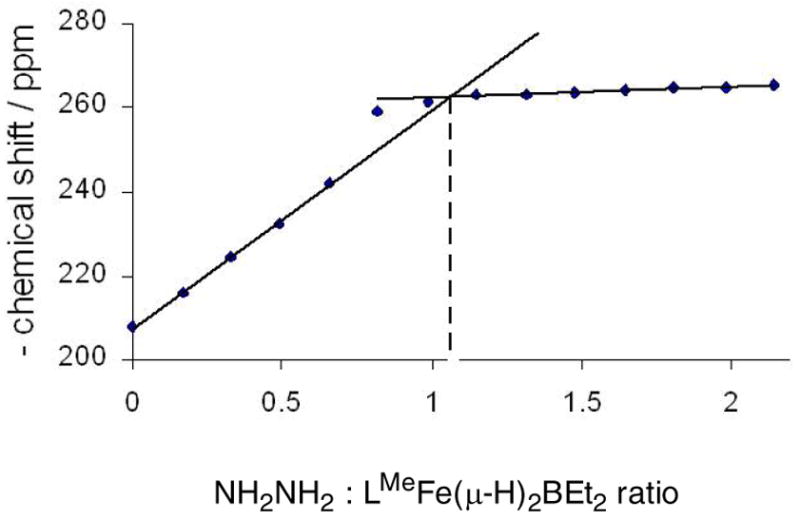
Titration curve and Job plot, using the chemical shift of the 1H NMR resonance near −260 ppm.
Figure 5 displays the X-ray crystal structure of 4. The hydrazine ligand binds to the iron atom in a η1 mode and points towards the diketiminate. The bridging hydrogen atoms were again located in Fourier difference maps, and their positions were refined. The geometry of the five-coordinate iron atom is intermediate between ideal square-pyramidal and trigonal-bipyramidal with τ = 0.4331. With the higher coordination number at iron, the Fe-H distances increase to 1.82(2) Å and 1.91(2) Å, which are about 0.1 Å longer than those in 1. Accordingly, the H-B distances decrease by 0.12 Å to 1.22(2) Å, comparable to the average value for literature hydridoborate complexes (see Supporting Information). The FTIR spectrum of 4 shows two bands at 3368 cm−1 and 3333 cm−1, which shifted to 2519 cm−1 and 2493 cm−1 when N2D4 was used (calculated from harmonic oscillator: 2460 cm−1 and 2434 cm−1). Therefore, these are assigned as N-H (N-D) stretching vibrations. 32
Figure 5.
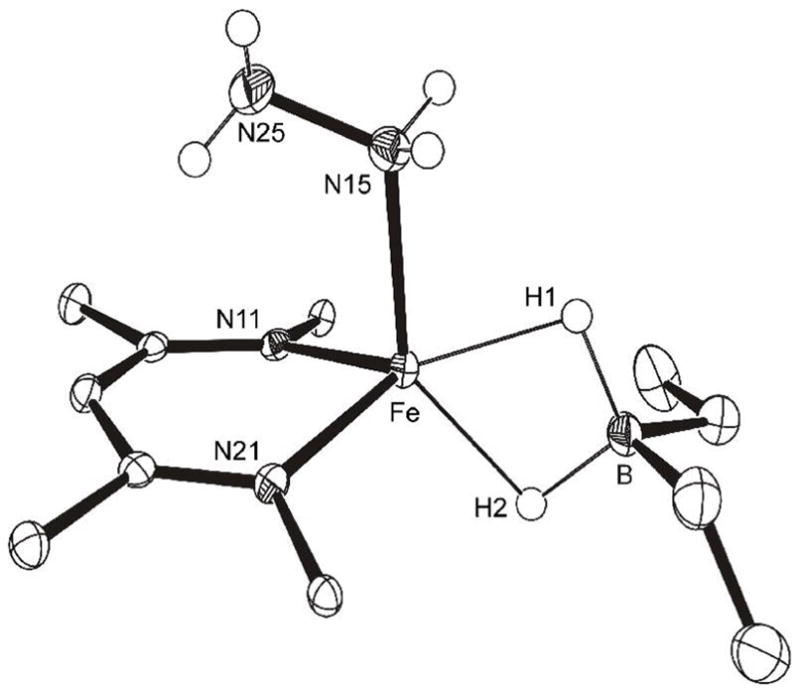
ORTEP diagram of the molecular structure of LMeFe(η1-H2NNH2)(μ-H)2BEt2 (4). Aryl groups and C-H are omitted for clarity. Important bond distances (Å) and angles (°): Fe–H1 1.91(2), Fe–H2 1.82(2), Fe–N15 2.214(2), B–H1 1.22(2), B–H2 1.22(2), N15–N25 1.441(2), Fe–B 2.350(2), H1-Fe-H2 61.2(9), H1-B-H2 103(1).
Well-characterized complexes of iron with N2 and its partial reduction products N2H2 and N2H4 are rare. A search of the Cambridge Structural Database showed only eight examples of Fe-N2H4 species.32,33 The Fe-N distance in our complex is 2.214(2) Å, which is comparable to the average value of 2.14(3) Å for the known iron-hydrazine adducts. The N-N distance in 4 also agrees well with the average literature value of 1.448(3) Å for bound N2H4.
After heating a solution of 4 in toluene at 60 °C for 22 h, the new complex LMeFe(μ-NH2)2BEt2 (5) can be isolated in 57% yield (Scheme 9). The solid-state structure of 5 is shown in Figure 6. Interestingly, the N-N bond has been cleaved (N···N = 2.459(3) Å) to yield a diaminoborate complex. The IR spectrum shows a single N-H stretching band at 3393 cm−1, which shifts to 2494 cm−1 in the compound synthesized from N2D4. Most complexes containing M(μ-NH2)2M fragments exhibit two N-H stretching bands between 3100–3300 cm−1.34
Scheme 9.
Figure 6.
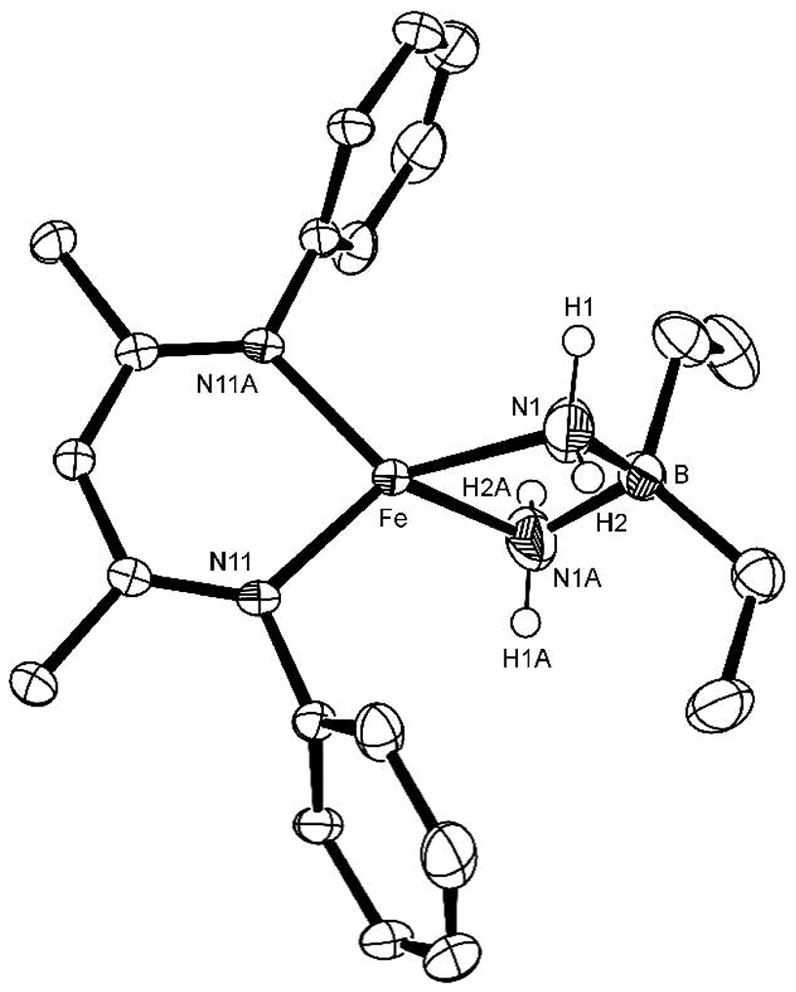
ORTEP diagram of the molecular structure of LMeFe(μ-NH2)2BEt2 (5) (aryl isopropyl groups and hydrogens are omitted for clarity). There is a crystallographic C2 axis through Fe and B. Important bond distances (Å) and angles (°): Fe-N1 2.064(2), Fe-N1A 2.064(1), B-N1 1.571(8), B-N1A 1.577(8), N1-N1A: 2.4589(31), N1-Fe-N1A: 73.12(9), N1-B-N1A: 102.5(3).
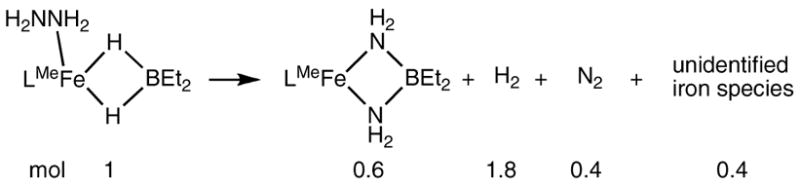
The observation of one N-H stretching band suggests a possible alternative formulation for 5, LMeFe(μ-NH)2BEt2. However, this formally iron(IV) complex seems less likely for several reasons. First, all examples of tetrahedral iron(IV) in the literature are low-spin and supported by strongly donating terminal nitride or imide ligands.35 The solution magnetic moment of 5 is 4.8(1) μB, which is more consistent with a high-spin Fe(II) complex. Second, each of the four N-bound H atoms was clearly visible in Fourier maps from the X-ray diffraction experiment and was refined.36 Finally, the alternative formulation implies that 2 equiv of H2 are produced in the reaction. Using gas chromatography, we determined that only 1.67 ± 0.03 equiv of H2 is produced (see Supporting Information for details), inconsistent with the diimidoborate formulation.
We have no definitive explanation for the observation of H2 in excess of the amount predicted by the stoichiometry of the reaction in Scheme 9. No substantial amount of H2 was produced in control experiments where 2a was heated with hydrazine. However, it is important to note that the spectroscopic yield of 5 is only 60% (from an NMR experiment with internal standard). A substantial amount of a black precipitate is formed along with 5. It seems possible that the remaining 0.4 equiv of hydrazine is disproportionated to H2 and N2 by the iron byproducts, with complete disproportionation giving the limiting stoichiometry in Scheme 10. The rough agreement with the observed H2 production supports this speculation.
Scheme 10.
Because 4 has hydrogens in the hydride bridges as well as the bound hydrazine, a final question regards the source of the N-H protons in 5. Importantly, the synthesis of 5 from N2D4 gave a product with no N-H peak and a new N-D peak in the infrared spectrum. This observation indicates that all N-H protons in 5 derive from hydrazine, and that the bridging hydrides in 4 are released as H2 upon heating. Although the unknown nature of some products dissuades us from further mechanistic studies, one can speculate a mechanism where loss of H2 from 4 generates unsaturation at iron and boron. This would enable coordination of the free lone pair on the bound hydrazine, followed by N-N cleavage to give 5. However, we stress that other mechanisms are also consistent with the limited data.
A limited number of literature examples report the cleavage of hydrazine N-N bonds with metal-containing clusters, often through formation of bridging NH or NH2.37 There are even fewer complexes that cleanly perform this reaction with two or less metals.38 Here, the Fe-N interactions are reinforced by the low-coordinate environment, which creates strong bonds to electronegative groups, as we have demonstrated previously.12
Conclusions
We have discovered an interesting series of borane alkylations/arylations of iron hydride complexes, which give iron hydridoborate products through attack of a ring-opened iron hydride species on a borane. In this reaction, the hydride ligand acts as a nucleophile, apparently activating the borane toward transfer of the R group to iron. A combination of kinetics studies has been unusually informative with respect to the mechanism, elucidating the nature of the rate-determining transition state. In addition, the hydridoborate product of the reaction reacts to cleave the N-N bond of hydrazine, releasing the two bridging hydrides as H2. The B-C and N-N cleavage reactions demonstrate the ability of low-coordinate iron complexes to perform difficult bond transformations under mild conditions.
Experimental Section
General Considerations
All manipulations were performed unde r a nitrogen atmosphere using standard Schlenk techniques or in an M. Braun glovebox maintained at or below 1 ppm of O2 and H2O. Glassware was dried at 150 °C overnight. 1H NMR spectra were recorded on a Bruker Avance 500 spectrometer (500 MHz) at 22 °C and referenced internally to residual protiated solvent (C6D5H at 7.15 ppm). Resonances are broad singlets unless otherwise specified. Infrared spectra (450–4000 cm−1) were recorded on KBr pellet samples in a Shimadzu FTIR spectrophotometer (FTIR-8400S). A total of 32 scans at 2 cm−1 resolution were collected in each case. Electronic spectra were recorded between 280 nm and 1000 nm on a Cary 50 UV-visible spectrophotometer, using screw-cap quartz cuvettes of 1 cm optical path length. Elemental analyses were determined by Desert Analytics (Tucson, AZ). Pentane, tetrahydrofuran (THF), and toluene were purified by passage through activated alumina and “deoxygenizer” columns obtained from Glass Contour Co. (Laguna Beach, CA). Deuterated benzene and THF were dried over CaH2, then over Na/benzophenone, and then vacuum transferred into a storage container. Before use, an aliquot of each solvent was tested with a drop of sodium benzophenone ketyl in THF solution. Celite was dried overnight at 200 °C under vacuum. Hydrazine, purchased from Aldrich, was dried over KH and vacuum transferred before use. KH, purchased for Aldrich as suspensions in oil, was washed with pentane three times and dried. BEt3 (1.0 M in hexane) was purchased from Aldrich and used as it arrived without any further purification. The preparation and properties of [L MeFeCl]2, [L MeFeH]2 (1) and KHBEt3 were previously reported.39
LMeFe(μ-H)2BEt2 (2a) from 1
A sample of 1 (139 mg, 0.147 mmol) was dissolved in 15 mL of toluene. Triethylborane (0.29 mL of a 1.0 M solution in hexane) was added via syringe to the bright red solution. The solution was heated at 70 °C for 14 h. Volatile materials were removed under vacuum and the residue was extracted with pentane (15 mL), filtered and concentrated to 5 mL. Crystallization from pentane at −35 °C gave bright red needles (68 mg, 72%).
LMeFe(μ-H)2BEt2 (2a) from FeCl2
The compound was typically synthesized in one flask from LMeLi and Fe(THF)1.5Cl2 as follows. A Schlenk flask was loaded with a mixture of LMeLi (1.69 g, 3.98 mmol) and Fe(THF)1.5Cl2 (0.94 g, 4.0 mmol) in toluene (50 mL). The mixture was stirred at 80 °C for 22 h. All solvent was removed under vacuum and a solution of KHBEt3 (0.56 g, 4.0 mmol) in toluene (50 mL) was added. The mixture was heated at 80 °C for 4 h. Volatile materials were removed under vacuum and the residue was extracted with pentane (100 mL), filtered and concentrated to 20 mL. Crystallization at −35 °C gave bright red needles (777 mg, 72%). 1H NMR (400 MHz, C6D6): 81 (6H, BEt2 CH3), 46 (6H, backbone CH3), 17 (4H), −4 (12H, iPr CH3), −36 (12H, iPr CH3), −40 (4H), −50 (2H, p-H), −208 (4H) ppm. (Peaks integrated as 4H could be BEt2 CH2, m-H or iPr methine. Peaks for the backbone H and bridging H were not observed.) UV-vis (pentane): 299 (ε= 27.3 mM−1cm−1), 398 (ε = 15.0 mM−1cm−1), 554 (ε = 2.9 mM−1cm−1) nm. μeff (C6D6, 25 °C): 5.1(1) μ B. Elem. Anal. Calcd for C33H53N2BFe: C, 72.80; H, 9.81; N, 5.15. Found: C, 73.52; H, 9.52; N, 5.62.
LMeFe(μ-H)2BPh2 (2b)
1 (127 mg, 0.13 mmol) and triphenylborane (32 mg, 0.13 mmol) were dissolved in 20 mL of toluene. The solution was heated at 80 °C for 18 h. Volatile materials were removed under vacuum and the residue was extracted with toluene (15 mL), filtered and concentrated to 5 mL. Crystallization from pentane at −26 °C gave red blocks (56 mg, 67%). 1H NMR (500 MHz, C6D6): 78 (6H, backbone CH3), 32 (4H), 17 (4H+4H), −4 (12H, iPr CH3), −8 (2H), −40 (12H, iPr CH3), −46 (4H), −52 (2H) ppm. (Peaks integrated as 4H could be iPr methine, aryl m-H, phenyl o-H or phenyl m-H. Peaks integrated as 2H could be aryl p-H or phenyl p-H. Peaks for the backbone H and bridging H were not observed.) UV-vis (toluene): 291 (ε = 14.6 mM−1cm−1), 398 (ε = 6.3 mM−1cm−1), 567 (ε = 1.2 mM−1cm−1) nm. μeff (C6D6, 25 °C): 4.0(1) μ B. Elem. Anal. Calcd for C41H53BFeN2: C, 77.12; H, 8.05; N, 4.39. Found: C 76.88, H 7.85, N 4.38,.
LtBuFe(μ-H)2BEt2 (2c)
A Schlenk flask was loaded with a mixture of LtBuFeCl (722 mg, 1.21 mmol) and KHBEt3 (167 mg, 1.21 mmol) in toluene (50 mL). The mixture was stirred at 80 °C for 18 h. Volatile materials were removed under vacuum and the residue was extracted with pentane (50 mL), filtered and concentrated to 10 mL. Crystallization at −26 °C gave dark red blocks (224 mg, 59%). 1H NMR (500 MHz, C6D6): 71 (6H, BEt2 CH3), 42 (1H, backbone), 35 (18H, backbone CH3), 16 (4H), −13 (12H, iPr CH3), −61 (12H, iPr CH3), −66 (4H), −89 (2H, p-H), −313 (4H) ppm. (Peaks integrated as 4H could be BEt2 CH2, m-H or iPr methine. Peaks for the bridging H were not observed.) UV-vis (pentane): 333 (ε = 9.42 mM−1cm−1), 414 (ε = 6.82 mM−1cm−1), 597 (ε = 1.27 mM−1cm−1) nm. μeff (C6D6, 25 °C): 5.7(1) μ B. Elem. Anal. Calcd for C39H65N2BFe: C, 74.52; H, 10.42; N, 4.46. Found: C, 74.60; H 10.20,; N, 4.34.
Complexes 3a and 3c have been reported in ref 12.
LMeFePh (3b)
The compound was typically synthesized in one flask from LMeLi and FeCl2(THF)1.5 as follows. A resealable flask was loaded with a mixture of LMeLi (592 mg, 1.4 mmol) and FeCl2(THF)1.5 (329 mg, 1.4 mmol) in toluene (20 mL). The mixture was stirred at 80 °C for 22 h. Phenylmagnesium chloride (2.0 M in THF, 0.7 mL, 1.4 mmol) was added, and the mixture was stirred for 4 h at room temperature. Solvent was removed under vacuum. The residue was extracted with pentane (20 mL), filtered through Celite, concentrated to 3 mL, and cooled to −45 °C to give bright yellow crystals (487 mg, 63% yield). 1H NMR (C6D6, 21 °C): 163 (2H, phenyl m-CH), 126 (1H, phenyl p-CH), 47 (6H, backbone CH3), 28 (1H, backbone CH), −10 (4H, m-CH), −21 (12H, iPr CH3), −82 (2H, p-CH), −118 (4H, iPr CH), −125 (12H, iPr CH3). (Phenyl o-CH was not observed. Peak assignments were based on integrations and 1H NMR spectra of other known LMeFeR complexes.) UV-vis (pentane): 325 (ε = 15.6 mM−1cm−1), 368 (ε = 11.4 mM−1cm−1), 494 (ε = 1.0 mM−1cm−1) nm. μeff (C6D6, 25 °C): 5.2(1) μ B. Elem. Anal. Calcd for C35H46N2Fe: C 76.35, H 8.42, N 5.09. Found: C, 76.06; H, 8.20; N, 4.99.
LMeFe(H2NNH2)(μ-H)2BEt2 (4)
LMeFe(μ-H)2BEt2 (221 mg, 0.407 mmol) was dissolved in diethyl ether (10 mL). Hydrazine (13 μL, 0.41 mmol) was added via syringe to the bright red solution. The solution was shaken, causing an immediate color change from bright red to orange-pink. The solution was concentrated to 2 mL and cooled to −35 °C to give bright pink-yellow needles of 3 (212 mg, 90%). 1H NMR (400 MHz, C6D6): 24 (6H), 19 (4H), 3 (12H, iPr CH3), 1 (6H), −15 (12H, iPr CH3), −24 (4H), −40 (2H, p-H), −62 (1H, backbone C-H), −260 (4H) ppm. (Peaks integrated as 4H could be BEt2 CH2, m-H or iPr methine. Peaks integrated as 6H could be BEt2 CH3 or backbone CH3. Peaks for the bridging H and hydrazine H were not observed.) IR (KBr pellet): 3368 cm−1, 3338 cm−1 (N-H). UV-vis (pentane): 298 (ε = 13.4 mM−1cm−1), 397 (ε = 7.5 mM−1cm−1), 552 (ε = 1.5 mM−1cm−1) nm. μeff (C6D6, 25 °C): 3.8(1) μB. Elem. Anal. Calcd for C33H57N4BFe: C, 68.75; H, 9.97; N, 9.72. Found: C, 68.01; H, 8.83; N, 5.70. The bound hydrazine could be removed by extended pumping under vacuum (i.e. 50% hydrazine was removed after 26 h at RT), and we suspect that this is the reason for the low nitrogen analysis.
LMeFe(μ-NH2)2BEt2 (5)
A sample of LMeFe(μ-H)2BEt2 (201 mg, 0.371 mmol) was dissolved in toluene (15 mL). Hydrazine (14 μL, 0.45 mmol) was added via syringe to the bright red solution. The solution was shaken and then heated at 60 °C for 22 h. Volatile materials were removed under vacuum and the residue was extracted with pentane (15 mL), filtered and concentrated to 2 mL. Crystallization from pentane at −35 °C gave brown blocks (121.6 mg, 57%). 1H NMR (500 MHz, C6D6): 57 (4H), 33 (6H), 19 (4H), 2 (12H, iPr CH3), −20 (12H, iPr CH3), −35 (4H), −42 (1H, backbone C-H), −49 (2H, p-H), −71 (6H) ppm. (Peaks integrated as 4H could be BEt2 CH2, m-H or iPr methine. Peaks integrated as 6H could be BEt2 CH3 or backbone CH3. Peaks for the bridging NH2 groups were not observed.) IR (KBr pellet): 3393 cm−1 (N-H). UV-vis (pentane): 331 (ε = 16.0 mM−1cm−1). μeff (C6D6, 25 °C): 4.8(1) mu;B. Elem. Anal. Calcd for C33H55N4BFe: C, 68.99; H, 9.65; N, 9.75. Found: C, 68.68; H, 9.45; N, 9.35.
Kinetic experiments: effect of BEt3
[LMeFeH]2, (16.4 mg, 17.3 mmol), was dissolved in C6D6 (2.0 mL). A J. Young NMR tube, which was pre-dried at 180°C, was loaded with 0.4 mL of the above iron hydride complex solution and 0.14 mL of BEt3 (1.0 M in hexane) C6D6 solution (with various BEt3 concentrations - see Supporting Information). The NMR tube was placed into the NMR probe, which was equilibrated at the appropriate temperature (typically 319.2 K). After the sample had equilibrated for 2 min, 1H NMR spectra were recorded at preset times using an automated program. After Fourier transform, phasing, calibrating and integrating each spectrum using MestRec, a plot of normalized [1] (y) vs. reaction time (x) was generated. Then these experimental data were fitted into the general, integrated equation: y0+A1*exp(−x/t1), where y0, A1, t1 are variables, t1 being the reciprocal of the first order rate constant. Origin 6.1 was employed for the data fitting. The error bars on the rate constant came from the least-squares fit to the data.
Kinetic experiments: effect of [LMeFeH]2
BEt3 (1.34, 1.0 M hexane solution) was mixed with C6D6 (1.86 mL) and used as a stock solution. A pre-dried J. Young NMR tube was charged with different amounts of 1 and BEt3 (0.3 mL of standard solution), with a drop of metallic mercury (see text).
Kinetic experiments: activation parameters
In the range 280–300 K, temperatures in the NMR probe were calibrated using 100% methanol; in the range 300–380 K, 100% ethylene glycol was used for calibrations (Bruker Instruments, VT-Calibration manual). 1a (73.7 mg, 77.7 mmol) was dissolved in C6D6 (1.86 mL) then BEt3 (1.34 mL, 1.0 M hexane solution) was added to make the reaction solution. For each temperature, 0.3 mL of the reaction solution was injected into a J. Young NMR tube with one drop of Hg0. The reaction solution was stored at −35 °C when it was not in use.
Supplementary Material
Synthetic, analytical, and kinetic details (PDF), and crystallographic data (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
Figure 3.
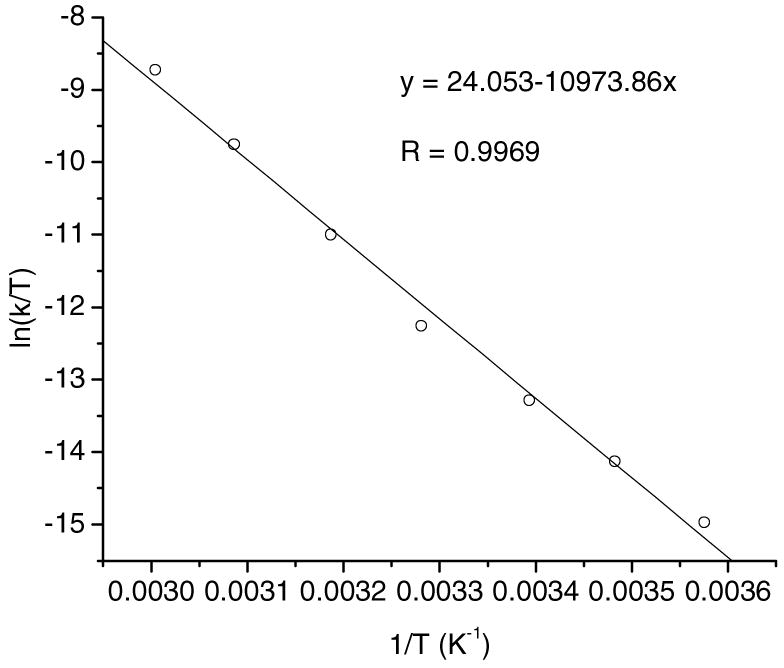
Eyring plot for the reaction of 1a + BEt3 to give 2a and 3a.
Scheme 5.
Scheme 6.
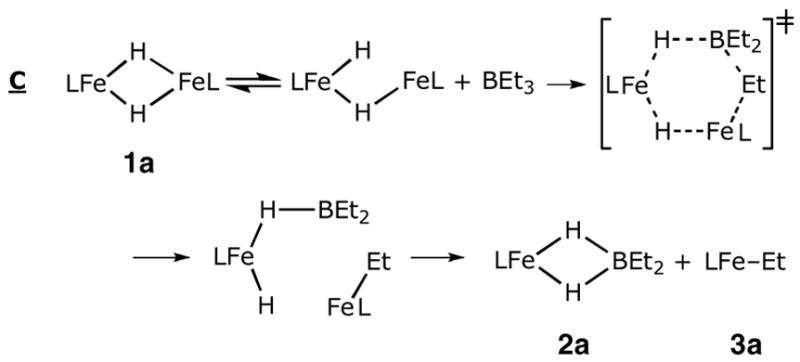
Table 3.
Effect of [1a] on observed rate constanta
| Entry | Initial concentrations (mM)
|
Observed rate constant kobs (s−1) | |
|---|---|---|---|
| 1a | BEt3 | ||
| 1 | 11.6 | 418.8 | 6.58(4)×10−3 |
| 2 | 20.8 | 418.8 | 4.59(2)×10−3 |
| 3 | 30.8 | 418.8 | 4.52(4)×10−3 |
| 4 | 41.6 | 418.8 | 4.27(8)×10−3 |
For the effect of changes in [1a] in the absence of mercury, see SI.
Acknowledgments
This work was supported by the NSF (CHE-0134658 to P.L.H.), the NIH (GM-065313 to P.L.H.), and the University of Rochester (Weissberger Fellowship to Y.Y.). P.L.H. acknowledges an A. P. Sloan Fellowship. We thank Jeremy Smith for initial synthetic experiments, William Jones and Thomas Cundari for helpful discussions, Pingwu Du and Richard Eisenberg for assistance with GC equipment, and Christine Flaschenriem for collecting crystallographic data.
References and Notes
- 1.(a) Dedieu A, editor. Transition Metal Hydrides. VCH; New York: 1992. [Google Scholar]; (b) Peruzzini M, Poli R, editors. Recent Advances in Hydride Chemistry. Elsevier; Amsterdam: 2001. [Google Scholar]
- 2.Maroney MJ. Hydrogen Metabolism and Hydrogenase. In: Bertini I, Gray HB, Stiefel EI, Valentine JS, editors. Biological Inorganic Chemistry. University Science Books; Sausalito, CA: 2007. pp. 443–452. [Google Scholar]
- 3.a) Igarashi RY, Laryukhin M, Dos Santos PC, Lee H-I, Dean DR, Seefeldt LC, Hoffman BM. J Am Chem Soc. 2005;127:6231–6241. doi: 10.1021/ja043596p. [DOI] [PubMed] [Google Scholar]; (b) Lukoyanov D, Barney BM, Dean DR, Seefeldt LC, Hoffman BM. Proc Natl Acad Sci USA. 2007;104:1451–1455. doi: 10.1073/pnas.0610975104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Smith JM, Lachicotte RJ, Holland PL. J Am Chem Soc. 2003;125:15752–15753. doi: 10.1021/ja038152s. [DOI] [PubMed] [Google Scholar]; (b) Vela J, Smith JM, Yu Y, Ketterer NA, Flaschenriem CJ, Lachicotte RJ, Holland PL. J Am Chem Soc. 2005;127:7857–7870. doi: 10.1021/ja042672l. [DOI] [PubMed] [Google Scholar]
- 5.A four-coordinate iron(II)-hydride complex has been implicated in the formation of a cyclohexadienyliron(II) complex: Brown SD, Peters JC. J Am Chem Soc. 2004;126:4538–4539. doi: 10.1021/ja0399122.This insertion is analogous to the benzene reactivity of an isolated iron(II) silyl complex: Turculet L, Feldman JD, Tilley TD. Organometallics. 2003;22:4627–4629.
- 6.Carey RA, Sundberg RJ. Adavanced Organic Chemistry, Part B: Reactions and Synthesis. 4. Kluwer Academic/Plenum Publishers; New York: 2001. pp. 549–563. [Google Scholar]
- 7.a) Miyaura N, Suzuki A. Chem Rev. 1995;95:2457–83. [Google Scholar]; (b) Chemler SR, Trauner D, Danishefsky SJ. Angew Chem, Int Ed. 2001;40:4544–4568. doi: 10.1002/1521-3773(20011217)40:24<4544::aid-anie4544>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]; (c) Hayashi H, Yamasaki K. Chem Rev. 2003;103:2829–2844. doi: 10.1021/cr020022z. [DOI] [PubMed] [Google Scholar]
- 8.(a) Onak T. Organoborane Chemistry. Academic; New York: 1975. [Google Scholar]; (b) Mikhailov BM, Bubnov YN. Organoboron Compounds in Organic Synthesis. Hanvood Academic Publishers; Amsterdam: 1983. [Google Scholar]; (c) Pelter A, Smith K, Brown HC. Borane Reagents. Academic; New York: 1988. [Google Scholar]
- 9.BEt4− has been used as an ethyl source for the synthesis of organometallics like tetraethyllead: Honeycutt JB, Riddle JM. J Am Chem Soc. 1961;83:369–373.Fischer R, Rapsomanikis S, Andreae MR. Anal Chem. 1993;65:763–766. doi: 10.1021/ac00054a019.Rapsomanikis S. Analyst. 2004;119:1429–1439.
- 10.(a) Miyaura N. J Organomet Chem. 2002;653:54–57. [Google Scholar]; (b) Zhao P, Incarvito CD, Hartwig JF. J Am Chem Soc. 2007;129:1876–1877. doi: 10.1021/ja068587q. [DOI] [PubMed] [Google Scholar]
- 11.Identical product mixtures are obtained when [LRFeCl]n (n = 1 or 2) is stirred with KBHEt3 for more than 15 h.
- 9.Vela J, Vaddadi S, Cundari TR, Smith JM, Gregory EA, Lachicotte RJ, Flaschenriem CJ, Holland PL. Organometallics. 2004;23:5226–5239. [Google Scholar]
- 13.Mehn MP, Brown SD, Paine TK, Brennessel WW, Cramer CJ, Peters JC, Que L., Jr Dalton. 2006:1347–1351. doi: 10.1039/b509580h. [DOI] [PubMed] [Google Scholar]
- 14.Examples of stoichiometric B-C bond cleavage by transition metals: Sacconi L, Dapporto P, Stoppioni P. Inorg Chem. 1976;15:325–329.Siegmann K, Pregosin PS, Venanzi LM. Organometallics. 1989;8:2659–2664.Thaler EG, Caulton KG. Organometallics. 1990;9:1871–1876.Aresta M, Quaranta E, Tommasi I, Derien S, Dunach E. Organometallics. 1995;14:3349–3356.Spence RE, Piers WE, Sun Y, Parvez M, MacGillivray LR, Zaworotko MJ. Organometallics. 1998;17:2459–2469.Lee LWM, Piers WE, Elsegood MRJ, Clegg W, Parvez M. Organometallics. 1999;18:2947–2949.Pleènik CE, Liu FC, Liu S, Liu J, Meyers EA, Shore SG. Organometallics. 2001;20:3599–3606.Schebler PJ, Mandimutsira BS, Riordan CG, Liable-Sands LM, Incarvito CD, Rheingold AL. J Am Chem Soc. 2001;123:331–332. doi: 10.1021/ja002638g.Hayes PG, Piers WE, Parvez M. Organometallics. 2005;24:1173–1183.
- 15.(a) Marks TJ, Kolb JR. Chem Rev. 1977;77:263–93. [Google Scholar]; (b) Ghilardi CA, Innocenti P, Midollini S, Orlandini A. J Organomet Chem. 1982;231:C78–C80. [Google Scholar]; (c) Ghilardi CA, Innocenti P, Midollini S, Orlandini A. J Chem Soc, Dalton Trans. 1985:605–9. [Google Scholar]; (d) Jia G, Lough AJ, Morris RH. J Organomet Chem. 1993;461:147–56. [Google Scholar]
- 16.Kandiah M, McGrady GS, Decken A, Sirsch P. Inorg Chem. 2005;44:8650–8652. doi: 10.1021/ic051541y. [DOI] [PubMed] [Google Scholar]
- 17.(a) Liu FC, Plecnik CE, Liu S, Liu J, Meyers EA, Shore SG. J Organomet Chem. 2001;627:109–120. [Google Scholar]; (b) Liu FC, Chen JH, Chen SC, Chen KY, Lee GH, Peng SM. J Organomet Chem. 2005;690:291–300. [Google Scholar]
- 18.(a) Fryzuk MD, Lloyd BR, Clentsmith GKB, Rettig SJ. J Am Chem Soc. 1994;116:3804–3812. [Google Scholar]; (b) Galler JL, Goodchild S, Gould J, McDonald R, Sella A. Polyhedron. 2004;23:253–262. [Google Scholar]; (c) Crestani MG, Munoz-Hernandez M, Arevalo A, Acosta-Ramirez A, García JJ. J Am Chem Soc. 2005;127:18066–18073. doi: 10.1021/ja056000m. [DOI] [PubMed] [Google Scholar]
- 19.Selected examples of hydridoborate complexes formed from transition metal hydride complexes: Baker RT, Ovenall DW, Harlow RL, Westcott SA, Taylor NJ, Marder TB. Organometallics. 1990;9:3028–3030.Baker RT, Calabrese JC, Westcott SA, Marder TB. J Am Chem Soc. 1995;117:8777–8784.Essalah K, Barthelat JC, Montiel V, Lachaize S, Donnadieu B, Chaudret B, Sabo-Etienne S. J Organomet Chem. 2003;680:182–187.Westcott SA, Marder TB, Baker RT, Harlow RL, Calabrese JC, Lam KC, Lin Z. Polyhedron. 2004;23:2665–2677.
- 20.Activation parameters derived from experiments without Hg0 gave similar values of ΔH‡ = 21.6 ± 0.7 kcal/mol and ΔS‡ = 2.3 ± 0.6 eu. This suggests that the reaction profile is the same in the presence of Hg0.
- 21.Nath J, Tripathi AD. J Chem Soc, Faraday Trans. 1984:1517–1524. [Google Scholar]
- 22.Laurence C, Nicolet P, Dalati MT, Abboud JM, Notario R. J Phys Chem. 1994;98:5807–5816. [Google Scholar]
- 23.Szulejko JE, Mcmahon TB. J Am Chem Soc. 1993;115:7839–7848. [Google Scholar]
- 24.(a) Thompson ME, Baxter SM, Bulls AR, Burger BJ, Nolan MC, Santarsiero BD, Schaefer WP, Bercaw JE. J Am Chem Soc. 1987;109:203–19. [Google Scholar]; (b) Cundari TR. J Am Chem Soc. 1994;116:340–7. [Google Scholar]
- 25.Hartwig JF, Bhandari S, Rablen PR. J Am Chem Soc. 1994;116:1839–1844. [Google Scholar]
- 26.Smith G, Cole-Hamilton DJ, Thornton-Pett M, Hursthouse MB. J Chem Soc, Dalton Trans. 1983:2501. [Google Scholar]
- 27.Brown HC, Krishnamurthy S, Hubbard JL. J Am Chem Soc. 1978;100:3343–3349. [Google Scholar]
- 28.Wilkins RG. Kinetics and Mechanism of Reactions of Transition Metal Complexes. VCH; New York: 1991. [Google Scholar]
- 29.LtBuFe(OEt2)+ BArF4− has been reported: Gregory EA, Lachicotte RJ, Holland PL. Organometallics. 2005;24:1803–1805.
- 30.Sciarone TJJ, Meetsma A, Hessen B, Teuben JH. Chem Commun. 2002:1580–1581. doi: 10.1039/b204454d. The equilibrium between LMeFe(benzyl) + B(C6F5)3 and LMeFe+[BnB(C6F5)3] − lies to the right. [DOI] [PubMed] [Google Scholar]
- 31.This parameter is 1 for trigonal bipyramidal, and 0 for square pyramidal. Addison AW, Rao TN, Reedijk J, Van Rijn J, Verschoor GC. J Chem Soc, Dalton Trans. 1984:1349–1356.
- 32.In the literature, the number of observed N-H stretching vibrations in Fe-N2H4 species varies from two to five. Sellmann D, Kreutzer P, Huttner G, Frank A. Z Naturforsch, B: Chem Sci. 1978;33B:1341–1346.Casey MT, Guinan P, Canavan A, McCann M, Cardin C, Kelly NB. Polyhedron. 1991;10:483–489.Sellmann D, Soglowek W, Knoch F, Ritter G, Dengler J. Inorg Chem. 1992;31:3711–3717.Sellmann D, Shaban SY, Heinemann FW. Eur J Inorg Chem. 2004:4591–4601.
- 33.(a) Sellmann D, Blum N, Heinemann FW. Z Naturforsch B. 2001;56:581–588. [Google Scholar]; (b) Sellmann D, Friedrich H, Knoch F. Z Naturforsch B. 1994;49:660–664. [Google Scholar]; (c) Goedken VL, Peng SM, Molin-Norris JA, Park YA. J Am Chem Soc. 1976;98:8391–400. doi: 10.1021/ja00442a014. [DOI] [PubMed] [Google Scholar]; (d) Rath SP, Olmstead MM, Balch AL. Inorg Chem. 2004;43:6357–6365. doi: 10.1021/ic049581+. [DOI] [PubMed] [Google Scholar]
- 34.(a) Janik JF, Duesler EN, Paine RT. Inorg Chem. 1987;26:4341–4345. [Google Scholar]; (b) Park S, Rheingold AL, Roundhill DM. Organometallics. 1991;10:615–623. [Google Scholar]; (c) Alcock NW, Bergamini P, Kemp TJ, Pringle PG, Sostero S, Traverso O. Inorg Chem. 1991;30:1594–1598. [Google Scholar]; (d) Kormos BL, Jegier JA, Ewbank PC, Pernisz U, Young VG, Jr, Cramer CJ, Gladfelter WL. J Am Chem Soc. 2005;127:1493–1503. doi: 10.1021/ja045149f. [DOI] [PubMed] [Google Scholar]; (e) Stanciu C, Hino SS, Stender M, Richards AF, Olmstead MM, Power PP. Inorg Chem. 2005;44:2774–2780. doi: 10.1021/ic048362l. [DOI] [PubMed] [Google Scholar]
- 35.(a) Verma AK, Nazif TN, Achim C, Lee SC. J Am Chem Soc. 2000;122:11013–11014. [Google Scholar]; (b) Betley TA, Peters JC. J Am Chem Soc. 2004;126:6252–6254. doi: 10.1021/ja048713v. [DOI] [PubMed] [Google Scholar]; (c) Thomas CM, Mankad NP, Peters JC. J Am Chem Soc. 2006;128:4956–4957. doi: 10.1021/ja0604358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.In our refinements, the positional parameters and thermal parameter for each hydrogen atom was constrained with respect to the nitrogen atom. If the occupancy of these hydrogen atoms was set to one-half (to consider the possibility of two disordered NH bridges), the thermal parameters for the hydrogen atoms were plainly smaller than those for the parent nitrogen atom, suggesting that insufficient electron density was being modeled, and that a full H atom was required in each position.
- 37.Selected N-N bond cleavage by metal clusters: Coucouvanis D. J Biol Inorg Chem. 1996;1:594–600.Shan H, Yang Y, James AJ, Sharp PR. Science. 1997;275:1460–1462.Verma AK, Lee SC. J Am Chem Soc. 1999;121:10838–10839.Seino H, Masumori T, Hidai M, Mizobe Y. Organometallics. 2003;22:3424–3431.Nakajima Y, Suzuki H. Organometallics. 2003;22:959–969.Nakajima Y, Inagaki A, Suzuki H. Organometallics. 2004;23:4040–4046.Takei I, Dohki K, Kobayashi K, Suzuki T, Hidai M. Inorg Chem. 2005;44:3768–3770. doi: 10.1021/ic0500560.
- 38.N-N bond cleavage by mononuclear complexes: Schrock RR, Glassman TE, Vale MG, Kol M. J Am Chem Soc. 1993;115:1760–1772.Vale MG, Schrock RR. Inorg Chem. 1993;32:2767–2772.Ohki Y, Takikawa Y, Hatanaka T, Tatsumi K. Organometallics. 2006;25:3111–3113.By di-nuclear complexes: Schollhammer P, Petillon FY, Poder-Guillou S, Saillard JY, Talarmin J, Muir KW. Chem Commun. 1996:2633–2634.Schollhammer P, Guenin E, Petillon FY, Talarmin J, Muir KW, Yufit DS. Organometallics. 1998;17:1922–1924.Petillon FY, Schollhammer P, Talarmin J, Muir KW. Inorganic Chemistry. 1999;38:1954–1955. doi: 10.1021/ic9813359.Lin CJ, Hwang WS, Chiang MY. J Organomet Chem. 2001;640:85–92.Shaver MP, Fryzuk MD. J Am Chem Soc. 2005;127:500–501. doi: 10.1021/ja043732q.
- 39.Eckert NA, Smith JM, Lachicotte RJ, Holland PL. Inorg Chem. 2004;43:3306–3321. doi: 10.1021/ic035483x. [DOI] [PubMed] [Google Scholar]; Smith JM, Lachicotte RJ, Holland PL. J Am Chem Soc. 2003;125:15752–15753. doi: 10.1021/ja038152s. [DOI] [PubMed] [Google Scholar]; Vela J, Smith JM, Yu Y, Ketterer NA, Flaschenriem CJ, Lachicotte RJ, Holland PL. J Am Chem Soc. 2005;127:7857–7870. doi: 10.1021/ja042672l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Synthetic, analytical, and kinetic details (PDF), and crystallographic data (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.