

Abstract
NNN (1) is an esophageal carcinogen in rats. 2′-Hydroxylation of NNN is believed to be the major bioactivation pathway for NNN tumorigenicity. (S)-NNN is preferentially metabolized by 2′-hydroxylation in cultured rat esophagus, while there is no preference for 2′-hydroxylation vs. 5′-hydroxylation in the metabolism of (R)-NNN. 2′-Hydroxylation of NNN generates the reactive intermediate 4-oxo-4-(3-pyridyl)butanediazohydroxide (8), resulting in the formation of pyridyloxobutyl (POB)-DNA adducts. Based on these observations, we hypothesized that (S)-NNN treatment would produce higher levels of POB-DNA adducts than (R)-NNN in the rat esophagus. We tested this hypothesis by treating male F344 rats with 10 ppm of (R)-NNN, or (S)-NNN in the drinking water. After 1, 2, 5, 10, 16, or 20 weeks of treatment, POB-DNA adducts in esophageal, liver and lung DNA were quantified by HPLC-ESI-MS/MS. In rat esophagus, (S)-NNN treatment generated 3–5 times higher levels of POB-DNA adducts than (R)-NNN treatment, which supports our hypothesis. 7-[4-(3-pyridyl)-4-oxobut-1-yl]guanine (7-POB-Gua, 14) was the major adduct detected, followed by O2-[4-(3-pyridyl)-4-oxobut-1-yl]thymidine (O2-POB-dThd, 11) and O2-[4-(3-pyridyl)-4-oxobut-1-yl]cytosine (O2-[4-(3-pyridyl)-4-oxobut-1-yl]cytosine -POB-Cyt, 15). O6-[4-(3-pyridyl)-4-oxobut-1-yl]-2′-deoxyguanosine (O6-POB-dGuo, 10) was not detected. Total POB-DNA adduct levels in esophagus were 3–11 times higher than those in liver for (R)-NNN and 2–6 times higher for (S)-NNN. In contrast to esophagus and liver, (R)-NNN treatment produced more POB-DNA adducts than (S)-NNN treatment in rat lung, which suggested an important role for cytochrome P450 2A3 in NNN metabolism in rat lung. In both liver and lung, O2-POB-dThd was the predominant adduct and accumulated during the experiment. The results of this study demonstrate that individual POB-DNA adducts form and persist in esophagus, liver and lung of rats chronically treated with NNN enantiomers and demonstrate that (S)-NNN produces higher levels of POB-DNA adducts in esophagus than (R)-NNN, suggesting that (S)-NNN is more tumorigenic than (R)-NNN to the rat esophagus.
Introduction
Tobacco-specific nitrosamines are among the most important carcinogens in tobacco products (1, 2). N′-nitrosonornicotine (NNN, 1, Chart 1) and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK, 2, Chart 1) are the most carcinogenic of the tobacco-specific nitrosamines in laboratory animals and are found in virtually all tobacco products (3). NNN causes esophageal and nasal cavity tumors in rats, lung tumors in A/J mice, tracheal tumors in hamsters, and nasal cavity tumors in mink (1). When NNN was co-administered with NNK by swabbing in the rat oral cavity, oral tumors were induced (4) NNN is believed to be a causative agent for tumors of the esophagus, oral cavity, and nasal cavity in people who use tobacco products (2). NNN and NNK are classified as human carcinogens (Group 1) by the International Agency for Research on Cancer (3).
Chart 1.
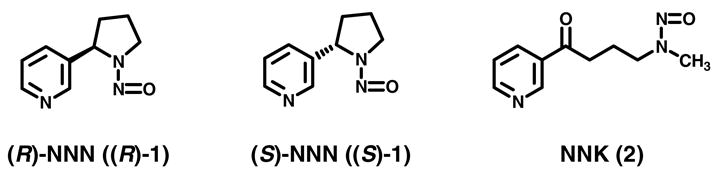
Structures of (R)-NNN, (S)-NNN and NNK,
NNN requires metabolic activation to form DNA adducts and express its mutagenicity and presumably its carcinogenicity (Scheme 1) (1). Both 2′-hydroxylation and 5′-hydroxylation, catalyzed by cytochrome P450 enzymes, lead to intermediates which form DNA adducts (1). 2′-Hydroxylation generates pyridyloxobutyl (POB)-diazohydroxide 8, the same intermediate that is formed by α-methyl hydroxylation of NNK. Intermediate 8 alkylates DNA to produce POB-DNA adducts such as O6-[4-(3-pyridyl)-4-oxobut-1-yl]-2′-deoxyguanosine (O6-POB-dGuo, 10), O2-[4-(3-pyridyl)-4-oxobut-1-yl]thymidine (O2-POB-dThd, 11), 7-[4-(3-pyridyl)-4-oxobut-1-yl]-2′-deoxyguanosine (7-POB-dGuo, 12), and O2-[4-(3-pyridyl)-4-oxobut-1-yl]-2′-deoxycytidine (O2-POB-dCyd, 13) (5–7) (Chart 2). Neutral thermal hydrolysis of unstable adducts 12 and 13 produces the stable adducts, 7-[4-(3-pyridyl)-4-oxobut-1-yl]guanine (7-POB-Gua, 14) and O2-[4-(3-pyridyl)-4-oxobut-1-yl]cytosine (O2-POB-Cyt, 15) (6–7) (Chart 2). DNA pyridyloxobutylation in rats treated with NNN or in organ cultures was demonstrated previously by analysis of 4-hydroxy-1-(3-pyridyl)-1-butanone (HPB, 7)-releasing DNA adducts (8–11). 5′-Hydroxylation of NNN leads to intermediate 9 which is also DNA reactive, yielding adducts 16 and 17 (Chart 2) (12). 2′-Hydroxylation is believed to be the major bioactivation pathway of NNN in rats based on previous studies in which it was the predominant metabolic pathway in both rat esophagus and nasal cavity, target tissues for NNN tumorigenicity, and POB-DNA adducts were detected in these tissues as HPB-releasing DNA adducts (9–11, 13–17) However, individual POB-DNA adducts of NNN have not been previously analyzed in vitro or in vivo.
Chart 2.

Structures of POB-DNA adducts (10–15) and DNA adducts derived from 5′-hydroxylation of NNN (16 and 17). dR: 2′-deoxyribosyl.
NNN has a chiral center at the 2′-position and (S)-NNN is the predominant enantiomer in tobacco products (18). The comparative metabolism of (R)- and (S)-NNN was studied in vitro in cultured rat esophagus and in vivo in rats (19). Cultured rat esophagus metabolized (S)-NNN predominantly by 2′-hydroxylation, while it metabolized (R)-NNN similarly by 2′-hydroxylation and 5′-hydroxylation. In rats treated with tritium-labeled NNN enantiomers, metabolites formed by 2′-hydroxylation predominated in the urine of (S)-NNN-treated rats, whereas products of 5′-hydroxylation were more prevalent in the urine of rats treated with (R)-NNN (19). Based on these observations, we hypothesized that (S)-NNN treatment would give higher levels of POB-DNA adducts in the rat esophagus than (R)-NNN treatment. To explore this hypothesis and characterize POB-DNA adduct formation from NNN enantiomers, F344 rats were given 10 ppm of (R)-NNN or (S)-NNN in the drinking water for 20 weeks. At various time intervals, individual POB-DNA adducts were quantified in esophageal, liver and lung DNA by HPLC-ESI-MS/MS analysis (20–21).
Materials and methods
Caution
NNN is carcinogenic. It should be handled in a well-ventilated hood with extreme care, and with personal protective equipment.
Chemicals
(R)-NNN, (S)-NNN and standards of POB-DNA adducts were synthesized (18, 20, 22) Micrococcal nuclease (LS004797, 15 kU) and phosphodiesterase II (LS003603, 10 U) were obtained from Worthington Biochemical Corporation (Lakewood, NJ). Alkaline phosphatase (567752, 30 U/μL) was purchased from Roche Molecular Biochemicals (Indianapolis, IN). The reagents and enzymes for DNA isolation were purchased from Centra Systems (Minneapolis, MN). All other chemicals and solvents were acquired from Sigma-Aldrich Chemical Co. (Milwaukee, WI) or Fisher Scientific (Fairlawn, NJ).
Animal experiment
The study was approved by the University of Minnesota Institutional Animal Care and Use Committee. A total of 162 male F344 rats, 7 weeks old, were purchased from Charles River Laboratories (Kingston, NY). They were housed two animals per micro-isolator cage with corn cob bedding in the Research Animal Resources facility of the University of Minnesota under the following conditions: temperature 20–24 °C; relative humidity 50 ± 10%; 12 h light/dark cycle. They were given NIH-07 diet (Harlan, Madison, WI) and tap water ad libitum. The animals were allowed one week for acclimatization to the facility prior to carcinogen treatment. The rats were randomly divided into three groups of 54 rats as follows: (1) control; (2) (R)-NNN; (3) (S)-NNN. Mean body weights at the beginning of the experiment were: 170 ± 12 g for the control group; 172 ± 10 g for the (R)-NNN group; and 174 ± 7 g for the (S)-NNN group. Body weights were recorded once a month. The rats in the treatment groups received 10 ppm of the appropriate carcinogen in the drinking water, and the control rats were given tap water. Aqueous solutions of carcinogens were prepared weekly and stored at 4°C, conditions under which they are known to be stable. These solutions were placed in the plastic water bottles of the rat cages twice weekly and water consumption was recorded. Nine rats per group were sacrificed by CO2 overdose at 1, 2, 5, 10, 16, and 20 weeks. Tissues were harvested and stored at −80°C until DNA isolation.
Quantitation of POB-DNA adducts by HPLC-ESI-MS/MS-SRM
Liver and lung DNA were isolated from three rats at each time point by following the modified Puregene® DNA isolation protocol (Gentra Systems), as previously described except that NaBH3CN was not used (23). Esophagi from nine rats in each group were divided into 3 pools of 3 rats for DNA isolation. DNA samples were subjected to HPLC-ESI-MS/MS-selected reaction monitoring (SRM) analysis for quantitation of POB-DNA adducts, as described previously (20). The characteristics of the method including limits of detection, accuracy, precision, and recoveries have been presented (20). In brief, 0.5–2 mg of each DNA sample, with addition of four deuterated internal standards, was subjected to neutral thermal hydrolysis (100 °C, 30 min), followed by enzymatic hydrolysis with micrococcal nuclease, phosphodiesterase II and alkaline phosphatase. The hydrolysate was partially purified by solid phase extraction (Strata-X cartridge, Phenomenex, CA). Then the sample was delivered by an Agilent 1100 capillary HPLC system (Agilent Technologies, Inc. Wilmington, DE) into a Finnigan TSQ Quantum Discovery Max triple quadrupole mass spectrometer (Thermo Electron, San Jose, CA). The ESI source was operated in the positive mode. POB-DNA adducts and corresponding internal standards were detected by monitoring the formation of the POB ion from the parent ion. The ion transitions were listed in Table 1. Contents of dGuo, dCyd and dThd were determined by HPLC (20). The amount of DNA is calculated from dGuo content by considering that 1 mg of DNA contains 3 μmol nucleotides (24), while dGuo accounts for 22% of total nucleotides in rat DNA, as previously determined in our laboratory. Adduct levels were expressed as fmol adduct/mg DNA. Adduct levels at the 20-week time point were also expressed as fmol per μmol appropriate nucleoside, i.e. fmol O2-POB-dThd/μmol dThd and fmol O2-POB-Cyt/μmol dCyd, and are listed in Table 3 in the Supporting Information. Levels of POB-DNA adducts were expressed as the mean ± S.D. of single analyses of three DNA samples per group.
Table 1.
Ion transitions for SRM of POB-DNA adducts and corresponding internal standards
| Adducts | Ion transitions | |
|---|---|---|
| Parent ion ([M + 1]+, m/z) | Daughter ion (m/z) | |
| O6-POB-dGuo(10) | 415.1 | 148.1 [POB]+ |
| [pyridine-D4]O6-POB-dGuo | 419.1 | 152.1 ([pyridine-D4]POB])+ and [Gua+H]+ |
| O2-POB-dThd(11) | 390.1 | 148.1 [POB]+ |
| [pyridine-D4](O2-POB-dThd | 394.1 | 152.1 ([pyridine-D4]POB])+ |
| 7-POB-Gua(14) | 299.1 | 148.1 [POB]+ |
| [pyridine-D4]7-POB-Gua | 303.1 | 152.1 ([pyridine-D4]POB])+ and [Gua+H]+ |
| O2-POB-Cyt(15) | 259.1 | 148.1 [POB]+ |
| [pyridine-D4]O2-POB-Cyt | 263.1 | 152.1 ([pyridine-D4]POB])+ |
Statistical analyses
The method of analysis for water consumption was a repeated measures approach using a generalized linear mixed model, which includes both fixed and random effects. The groups and time intervals are considered fixed effects and the rate of change over time for each cage is treated as a random effect. The variance/covariance structure for the data was estimated using restricted maximum likelihood (REML). Two-way analysis of variance (ANOVA) was used for comparisons of levels of total and individual POB-DNA adducts between groups ((R)-NNN and (S)-NNN) and time (1, 2, 5, 10, 16, 20 weeks). Since the individual adducts were obtained from the same tissue sample, a repeated measures ANOVA was chosen to compare these POB-DNA adducts within each group. Time was treated as a fixed effect and, again, REML was used to estimate the variance/covariance structure. Due to a highly skewed distribution, the POB-DNA adducts were analyzed on the natural log scale. Adjustments for multiple comparisons between groups or between POB-DNA adducts were made according to the Tukey method. A p-value <0.05 was considered to be statistically significant, except for comparisons of individual POB-DNA adducts within each group at each time point, for which a more restrictive p-value cutoff of less than 0.01 was used to judge statistical significance.
Results
Animal Experiment
This was designed according to a previous study (17), in which F344 rats treated with 5 ppm of NNN in the drinking water for 87 weeks developed an esophageal tumor incidence of 71%, except that we used NIH-07 diet instead of AIN-76A diet. In our preliminary study, POB-DNA adducts were readily detected in liver DNA isolated from rats treated with 5 ppm of racemic NNN in the drinking water for 8 weeks (data not shown). To ensure the detection of POB-DNA adducts in rats treated with each enantiomer of NNN, the dose of 10 ppm was chosen for the current experiment.
Overall, there were no significant differences in water consumption between the control rats and those treated with (R)-NNN or (S)-NNN, while there was a small but significant decrease in water consumption over time in each group (P < 0.05) (Figure 1A–C). There were no effects of carcinogen treatment on body weight (Figure 1D). All rats survived throughout the 20-week experiment.
Figure 1.
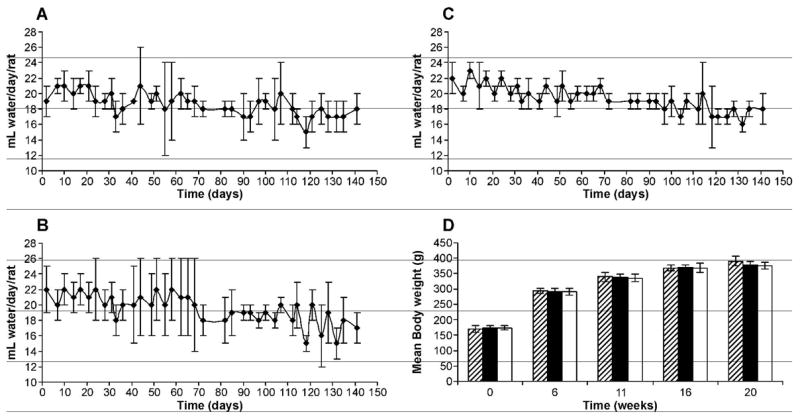
Water consumption by (A) control rats; (B) (R)-NNN-treated rats; (C) (S) NNN-treated rats; and (D) growth chart of rats in control (▨), (R)-NNN (■), and (S)-NNN (□) groups. Values are mean ± S.D. of measurements of all rats in each group.
POB-DNA adducts in esophageal DNA from (R)-NNN or (S)-NNN-treated rats
POB-DNA adducts were not found in esophageal DNA of control rats and were readily detected in esophageal DNA of rats treated with either (R)-NNN or (S)-NNN (Figure 2A-C). Levels of POB-DNA adducts are summarized in Figure 3A, B (also see Table 1A and 2A in the Supporting Information). Three adducts, O2-POB-Thd, 7-POB-Gua and O2-POB-Cyt, were detected. Levels of O6-POB-dGuo were below the detection limit. 7-POB-Gua was the major adduct, accounting for 46–56% of total adduct levels in the (R)-NNN group and 36–46% in the (S)-NNN group. The corresponding percentages of O2-POB-dThd were 28–34% [(R)-NNN] and 31–42% [(S)-NNN]. Those of O2-POB-Cyt were 12–19% [(R)-NNN] and 19–29% [(S)-NNN]. At each time point, relative adduct levels in the (R)-NNN and (S)-NNN groups were in the same order: 7-POB-Gua > O2-POB-dThd > O2-POB-Cyt, except that the level of O2-POB-dThd exceeded that of 7-POB-Gua after twenty weeks of treatment with (S)-NNN (Figure 3A, B). In rats treated with (R)-NNN, there was a significant difference between levels of each individual adduct at weeks 1 and 2, and levels of 7-POB-Gua and O2-POB-Cyt were significantly different at weeks 16 and 20 (P < 0.01). In rats treated with (S)-NNN, a significant difference was observed between levels of 7-POB-Gua and O2-POB-Cyt at weeks 1, 5, and 16, and levels of O2-POB-dThd were significantly different from those of O2-POB-Cyt at weeks 5, 16 and 20 (P < 0.01). Comparisons among individual adduct levels are shown in Table 4A in the Supporting Information. Total adduct levels from (S)-NNN treatment reached a maximum after 10 weeks (1380 fmol/mg DNA), while total adduct levels from (R)-NNN treatment decreased with time (Figure 4A). Total adduct levels in the (S)-NNN group were 3–5 times higher than those in the (R)-NNN group throughout the experiment. Levels of each individual POB-DNA adduct were 2–8 times higher in the (S)-NNN group than in the (R)-NNN group. Levels of total adducts were significantly different between the (S)-NNN and (R)-NNN groups at all time points (P < 0.05).
Figure 2.
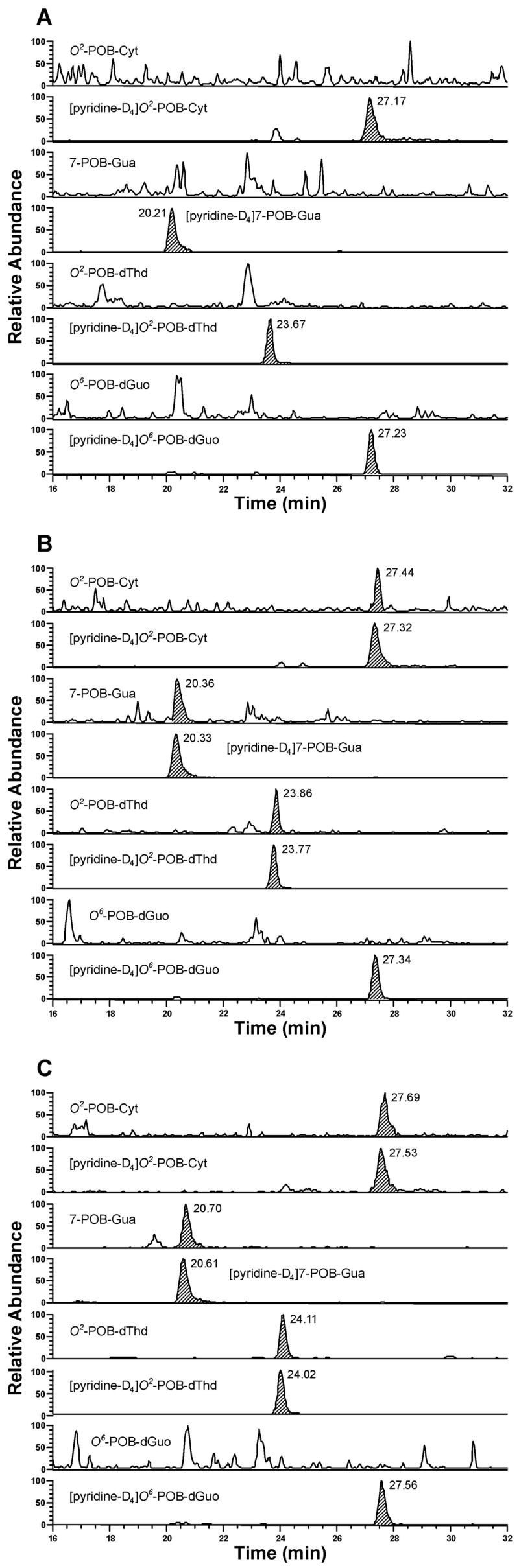
Typical SRM chromatograms obtained upon analysis of esophageal DNA isolated from (A) control rats; (B) (R)-NNN-treated rats; and (C) (S)-NNN-treated rats after 16 weeks of treatment. Individual POB-DNA adducts or internal standards were monitored as indicated on each channel.
Figure 3.
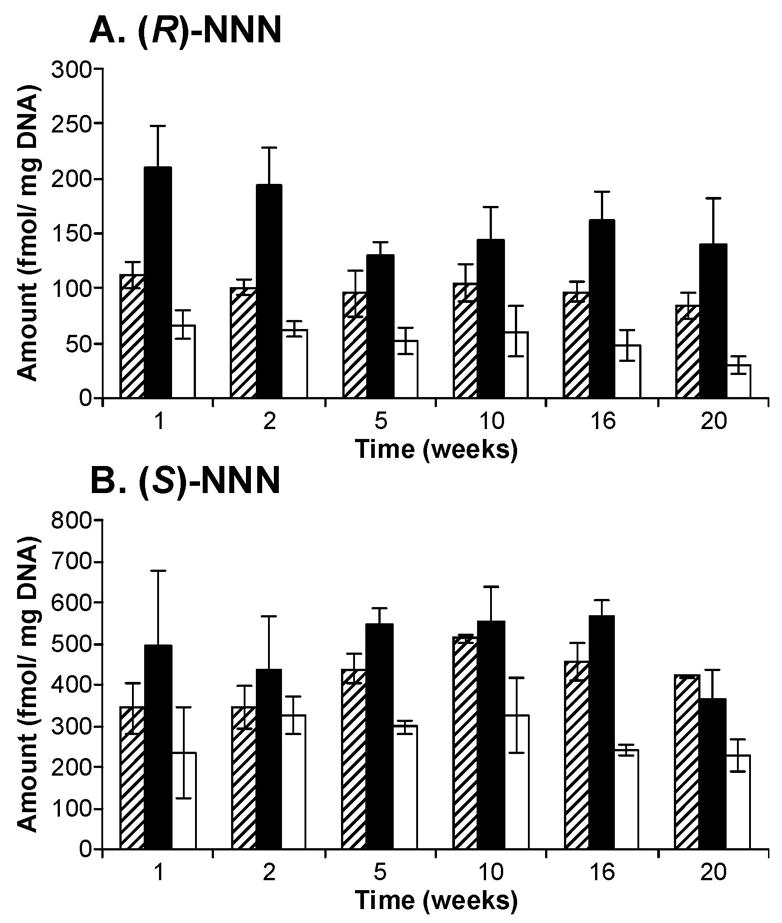
Levels of POB-DNA adducts in esophageal DNA isolated from rats treated with (A) (R)-NNN and (B) (S)-NNN. Symbol designations are: ▨, O2-POB-dThd; ■, 7-POB-Gua; □, O2-POB-Cyt. Each value is the mean ± S.D. of single analyses of DNA samples from 3 pools of 3 rats per group.
Figure 4.
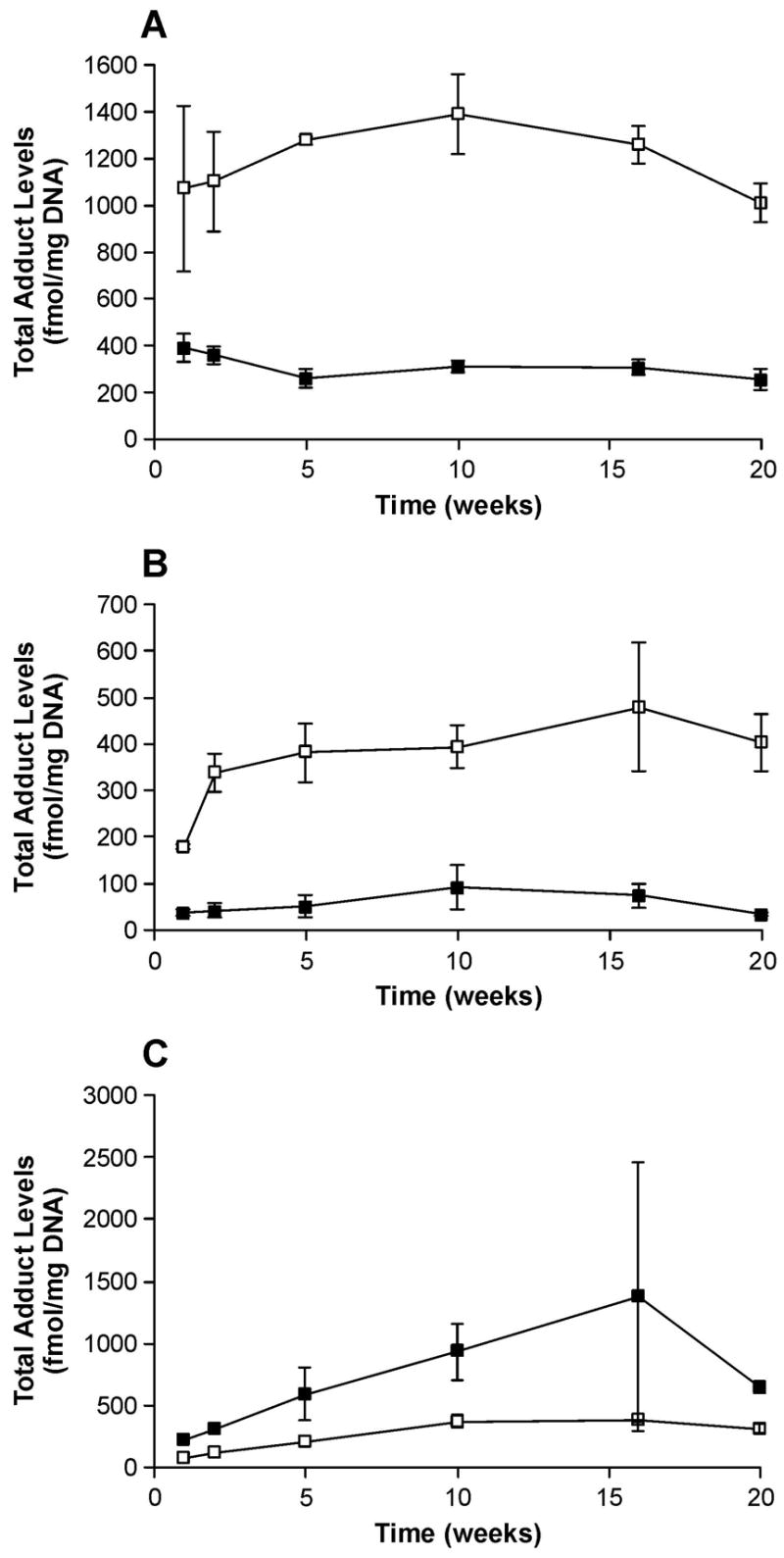
Plots of total adduct levels (fmol/mg DNA) versus time (weeks) in (A) esophageal, (B) liver and (C) lung DNA from (R)- and (S)-NNN-treated rats. Values of the total adduct levels are the sum of amounts of all POB-DNA adducts measured in esophageal, liver and lung DNA at each time point ± S. D. Symbol designations are: ■, total adduct levels from (R)-NNN treatment; □, total adduct levels from (S)-NNN treatment.
POB-DNA adducts in liver DNA from (R)-NNN or (S)-NNN-treated rats
Similar to esophageal DNA, no POB-DNA adducts were detected in liver DNA isolated from control rats and three adducts were detected in hepatic DNA from (R)-NNN and (S)-NNN-treated rats (Figure 5A-C). O6-POB-dGuo was not found in any liver DNA sample. Levels of POB-DNA adducts in liver DNA are summarized in Figure 6A, B (also see Table 1B and 2B in the Supporting Information). Total POB-DNA adduct formation was 4–12 times greater in the rats treated with (S)-NNN than in those given (R)-NNN. Levels of individual adducts were 3–9 times higher from (S)-NNN treatment than from (R)-NNN treatment. Levels of total adducts were significantly different between the (S)-NNN and (R)-NNN groups at all time points (P < 0.05). O2-POB-dThd was the most abundant POB-DNA adduct in liver DNA at each time point, followed by 7-POB-Gua and O2-POB-Cyt. The percentages of O2-POB-dThd of total adduct levels increased during the period of carcinogen treatment, 43–100% in the (R)-NNN group and 50–78% in the (S)-NNN group (Table 2A, B in the Supporting Information). The corresponding figures for 7-POB-Gua were 0–50% [(R)-NNN] and 17–41% [(S)-NNN], while those for O2-POB-Cyt were 0–14% [(R)-NNN] and 4–12% [(S)-NNN]. In rats treated with (R)-NNN, there was a significant difference between levels of O2-POB-dThd and O2-POB-Cyt at weeks 1 and 10, and the level of O2-POB-dThd was significantly different from that of 7-POB-Gua at week 10 (P < 0.01). In rats treated with (S)-NNN, a significant difference was observed between levels of each individual adduct (P < 0.01), except for levels of O2-POB-dThd and 7-POB-Gua at week 2, and levels of 7-POB-Gua and O2-POB-Cyt at week 20. Comparisons among individual adduct levels are shown in Table 4B in the Supporting Information. Total adduct levels from both (R)-NNN and (S)-NNN treatment were maximal after 16 weeks (Figure 4B): 500 fmol/mg DNA [(S)-NNN] and 100 fmol/mg DNA [(R)-NNN].
Figure 5.
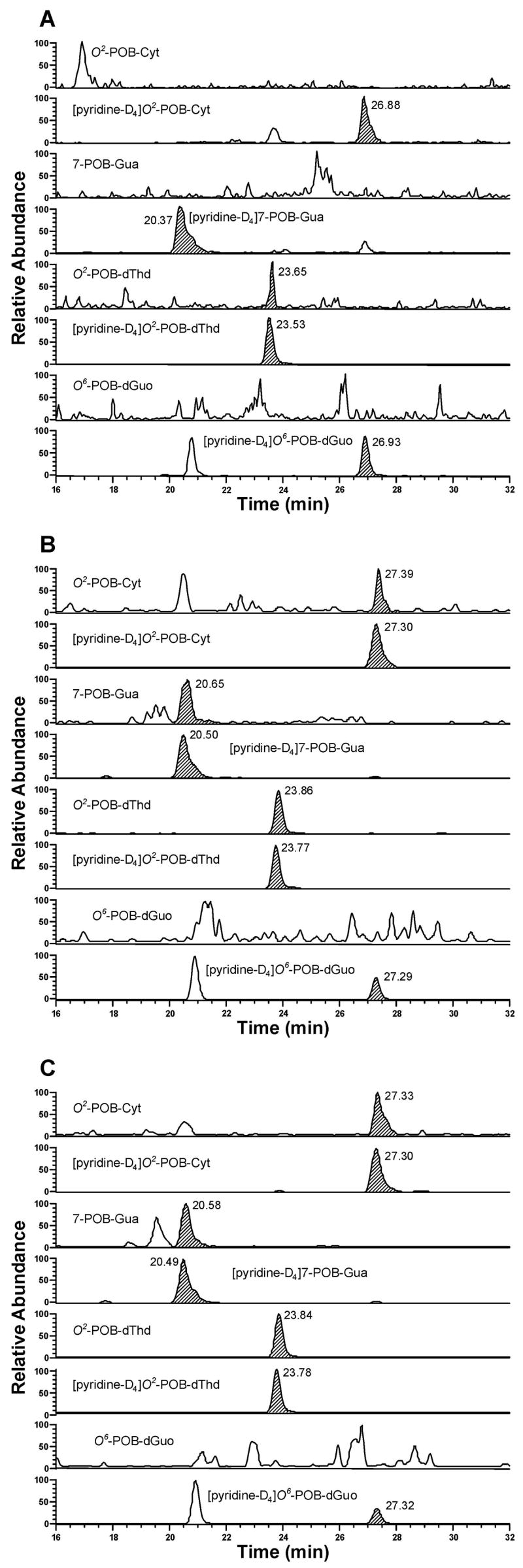
Typical SRM chromatograms obtained upon analysis of liver DNA from (A) control rats; (B) (R)-NNN-treated rats; and (C) (S)-NNN-treated rats after 10 weeks of treatment. Individual POB-DNA adducts or internal standards were monitored as indicated on each channel. In Figure 5A, the peak eluting at the position of O2-POB-dThd, the area of which is 0.2 % of [pyridine-D4]O2 -POB-dThd, originates from the internal standard.
Figure 6.
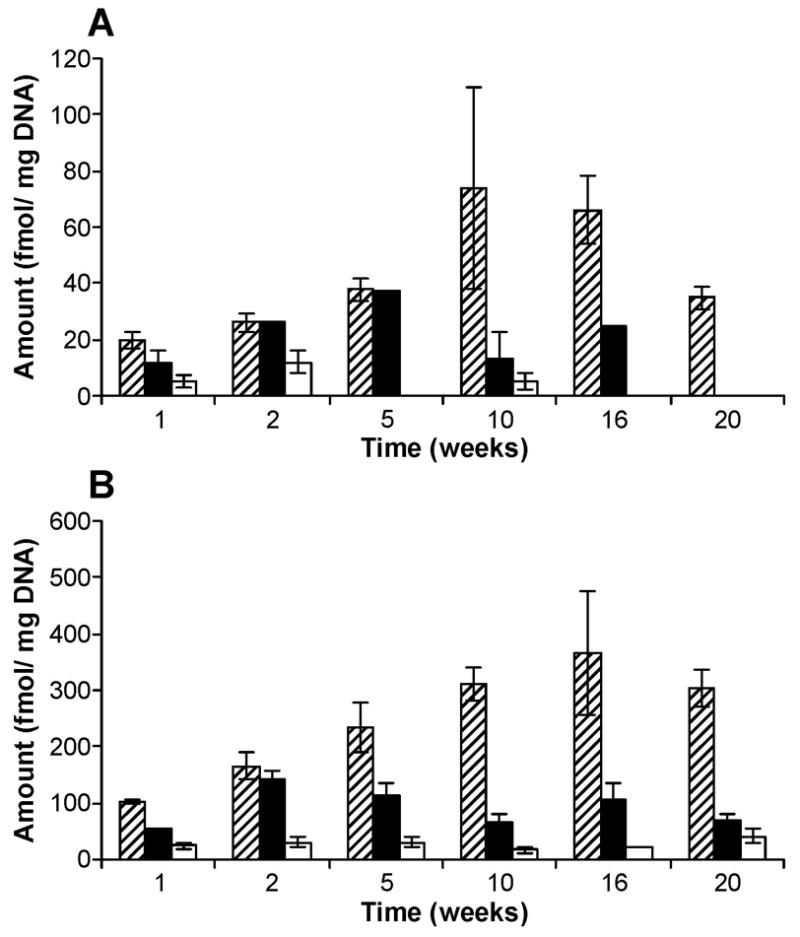
Levels of POB-DNA adducts in liver DNA isolated from rats treated with (A) (R)-NNN and (B) (S)-NNN. Symbol designations are: ▨, O2-POB-dThd; □, 7-POB-Gua; ■, O2-POB-Cyt. Each value is the mean ± S.D. of single analyses of DNA samples from three rats per group.
POB-DNA adducts in lung DNA from (R)-NNN or (S)-NNN-treated rats
POB-DNA adducts were not detected in lung DNA isolated from control rats, and three POB-DNA adducts were found in lung DNA isolated from the treated groups (Figure 7A–C). Levels of POB-DNA adducts in lung DNA are summarized in Figure 8A, B (also see Table 1C and 2C in the Supporting Information). In contrast to esophagus and liver, (R)-NNN treatment produced 2–4 times more total POB-DNA adducts than (S)-NNN in lung (Figure 4C). For each POB-DNA adduct, levels were 2–4 times higher in the (R)-NNN-treated rats than in those given (S)-NNN. Levels of total adducts were significantly different between the (S)-NNN and (R)-NNN groups at all time points except for 16 weeks (P < 0.05). O2-POB-dThd was the predominant adduct in lung DNA, accounting for 59–76% of total adducts in the rats treated with (R)-NNN and 63–76% in the (S)-NNN group. In both groups, the percentage of O2-POB-dThd of total adducts increased slightly over time, while the percentage of 7-POB-Gua of total adducts decreased. 7-POB-Gua accounted for 15–27% of total adducts in the rats treated with (R)-NNN and 16–25% in the (S)-NNN-treated rats. The corresponding figures for O2-POB-Cyt were 5–14% [(R)-NNN] and 5–13% [(S)-NNN]. At each time point, the relative levels of adducts in lung DNA of the (R)-NNN and (S)-NNN-treated rats shared the same trend: O2-POB-dThd > 7-POB-Gua > O2-POB-Cyt. In rats treated with (R)-NNN, there was a significant difference between levels of each individual adduct (P < 0.01), except for levels of O2-POB-Cyt and 7-POB-Gua at week 20. In rats treated with (S)-NNN, a significant difference was observed between levels of each individual adduct (P < 0.01) (Table 4C in the Supporting Information). Total adduct levels reached their maxima after 16 weeks (Figure 4C): 1370 fmol/mg DNA [(R)-NNN] and 380 fmol/mg DNA [(S)-NNN].
Figure 7.
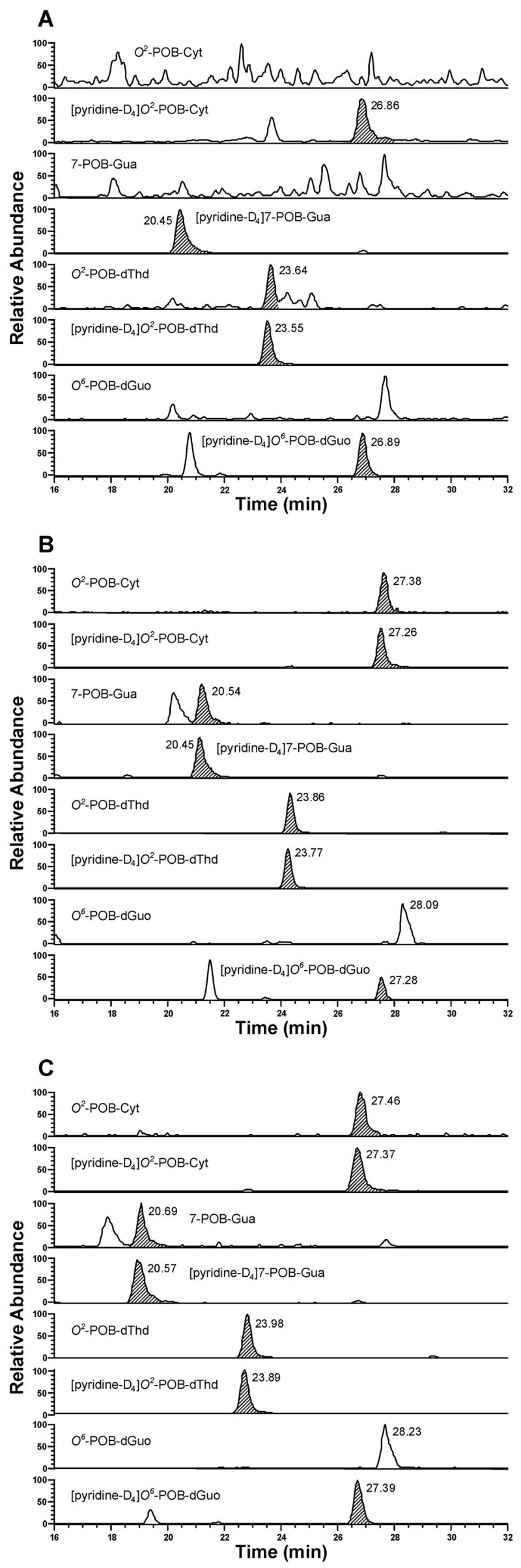
Typical SRM chromatograms obtained upon analysis of lung DNA from (A) control rats; (B) (R)-NNN-treated rats; and (C) (S)-NNN-treated rats after 16 weeks of treatment. Individual POB-DNA adducts or internal standards were monitored as indicated on each channel. In Figure 7A, the peak eluting at the position of O2-POB-dThd, the area of which is 0.2 % of [pyridine-D4]O2-POB-dThd, originates from the internal standard.
Figure 8.
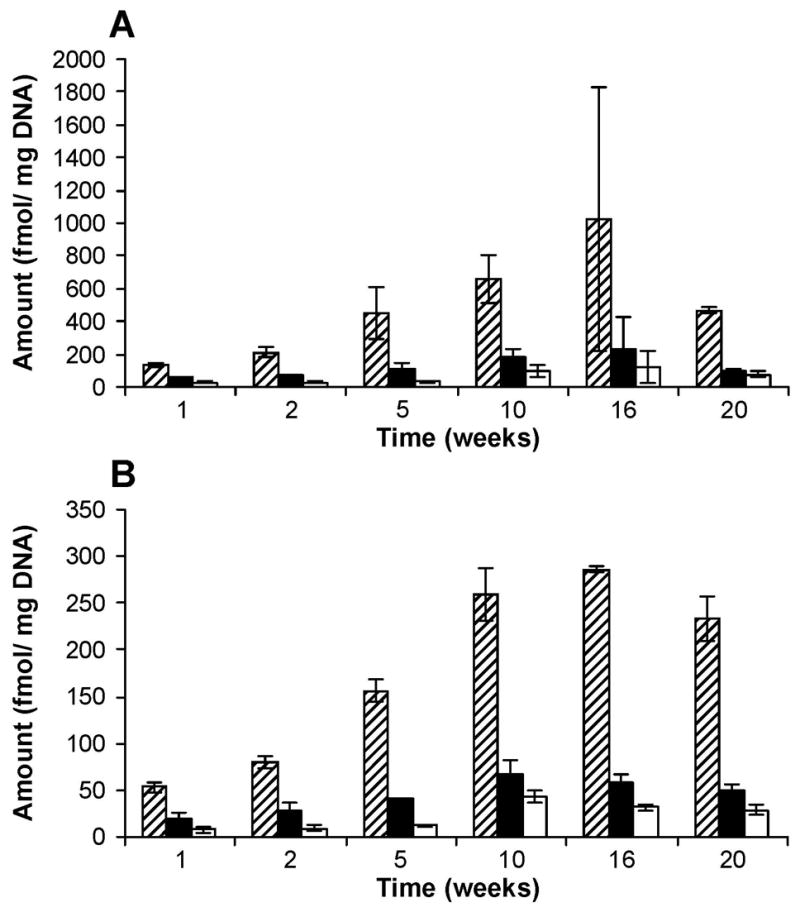
Levels of POB-DNA adducts in lung DNA isolated from rats treated with (A) (R)-NNN and (B) (S)-NNN. Symbol designations are: ▨, O2-POB-dThd; ■, 7-POB-Gua; □, O2-POB-Cyt. Each value is the mean ± S.D. of single analyses of DNA samples from three rats per group.
Comparison of POB-DNA adduct levels in esophageal, liver and lung DNA
Adduct formation was favored from (S)-NNN in esophagus and liver, but from (R)-NNN in lung. Adduct levels in the esophagus from (S)-NNN-treated rats were significantly higher than other tissues and groups at weeks 1, 2 and 5 (P < 0.05). There was no significant difference between adduct levels in the esophagus from (S)-NNN treatment and those in the lung from (R)-NNN-treated rats at weeks 10, 16 and 20, while both were significantly higher than the other tissues and groups (P < 0.05) (Figure 4A–C). Compared to esophagus and lung, the amounts of POB-DNA adducts in liver were quite low with a preference for (S)-NNN. At each time point, adduct levels in the liver from (R)-NNN-treated rats were significantly lower than in the other tissues and groups (P < 0.05). For (S)-NNN, levels of POB-DNA adducts in esophagus were 2–6 times higher than those in liver and 3–13 times higher than those in lung. For (R)-NNN, levels of POB-DNA adducts in esophagus during the first two weeks treatment were the highest, followed by lung and liver. Subsequently, adduct levels from (R)-NNN in lung exceeded those in esophagus and liver. The maximal adduct level in lung from (R)-NNN treatment was 1370 fmol/mg DNA, comparable to the maximal amount in esophagus from (S)-NNN treatment (1380 fmol/mg DNA) (Figure 4A, C). For both (R)-NNN and (S)-NNN, O2-POB-dThd was the predominant adduct found in liver and lung (Figure 6 and 8). However, 7-POB-Gua was the major adduct found in esophagus (Figure 3). The maximal total adduct level for (S)-NNN in esophagus was found after 10 weeks, while the maximal levels were reached after 16 weeks in liver and lung for both (R)-NNN and (S)-NNN (Figure 4A–C).
Discussion
NNN, given in the drinking water, is an esophageal carcinogen in rats (1). A number of in vitro studies have shown that 2′-hydroxylation of NNN is favored in rat esophagus compared to 5′-hydroxylation (13–14,16–17), and POB-DNA adducts derived from NNN 2′-hydroxylation have been detected in rat esophagus cultured with [5-3H]NNN, as measured by HPB released by acid hydrolysis (10). The results of the current study are consistent with these findings and demonstrate for the first time the in vivo formation of POB-DNA adducts in the esophagus of NNN-treated rats. Individual POB-DNA adducts were detected in rat esophageal DNA from both (R)-NNN and (S)-NNN-treated rats, with (S)-NNN treatment producing 3–5 times more adducts than (R)-NNN treatment, a significant difference. These results are consistent with those of a previous study, in which (S)-NNN was metabolized by cultured rat esophagus predominantly by 2′-hydroxylation, while (R)-NNN was preferentially hydroxylated at the 5′-position (19). There is evidence that a high-affinity cytochrome P450 enzyme is present in rat esophagus, and that this enzyme primarily catalyzes NNN 2′-hydroxylation (16). This enzyme also is apparently stereoselective, preferring 2′-hydroxylation of (S)-NNN over (R)-NNN. It has been shown clearly that cytochrome P450 2A3 does not play an important role in NNN 2′-hydroxylation in the rat esophagus (25–26). The identity of the high affinity cytochrome P450 enzyme remains unknown.
All NNN tumorigenicity studies to date were carried out with racemic NNN. 2′-Hydroxylation is believed to be the major bioactivation pathway for NNN in rats based on previous studies in which it was the predominant pathway in both rat esophagus and nasal cavity, target tissues for NNN tumorigenicity. 2′-Hydroxylation of NNN producesPOB-DNA adducts, which were detected in these tissues as HPB-releasing DNA adducts. Since levels of POB-DNA adducts in rat esophagus generated from (S)-NNN treatment were significantly higher than those from (R)-NNN treatment, our data suggest that (S)-NNN is more tumorigenic to rat esophagus than (R)-NNN. A tumorigenicity study using (R)- and (S)-NNN is underway to test this hypothesis. Considering that (S)-NNN is the predominant enantiomer in tobacco products, human cancer risk assessed on the basis of tumorigenicity studies using racemic NNN may be underestimated.
7-POB-Gua was the major adduct detected in esophageal DNA, followed by O2-POB-dThd and O2-POB-Cyt. This order of relative adduct levels is similar to that found in in vitro studies using 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone (NNKOAc, 3) (20, 22), which generates intermediates 4 and 8, except that O6-POB-dGuo, the second most abundant adduct in those studies, was not observed here. 7-POB-Gua, O2-POB-dThd and O2-POB-Cyt persisted in the esophagus throughout the experiment. These results are different from those in liver and lung DNA. In liver and lung DNA of rats treated with enantiomers of NNN as well as in rats treated with NNK and enantiomers of its metabolite NNAL (21), O2-POB-dThd was the predominant adduct and accumulated during treatment, while the percentage of 7-POB-Gua to total POB-DNA adducts decreased over time. These results suggest that there is low repair of 7-POB-dGuo in the rat esophagus, as compared to liver and lung, although we cannot exclude the possibility that the relatively rapid cell turnover in the rat esophagus (10 days) (27) may affect the relative adduct persistence data in esophagus vs. lung and liver.
Like 7-methyldGuo, 7-POB-dGuo can undergo spontaneous depurination under physiological conditions, thus releasing 7-POB-Gua and leaving an apurinic (AP) site. 7-MethyldGuo has a half-life of less than 6.5 h at neutral pH at 37 °C (28). The depurination rate of 7-POB-dGuo is faster than that of 7-methyldGuo with a half-life of 3 h at neutral pH at 37 °C (6). AP sites are cytotoxic and mutagenic in mammalian cells, causing G: C → T: A transversions (29). The repair mechanism for 7-POB-dGuo is unknown, while 7-methydGuo is removed by the base excision repair pathway (30). In mammalian cells, 3-methyladenine glycosylase (MPG) mediates the repair of 7-methyldGuo by cleavage of 7-methyGua from the DNA backbone, thus leaving AP sites, followed by removal of AP sites by AP endonuclease. The gap is filled by a DNA polymerase and a ligase (30). A similar mechanism is expected for repair of 7-POB-dGuo. Mammalian MPGs are constitutively expressed and widely distributed in tissues (31–32). The highest level of rat MPG is found in colon, followed by heart and other tissues, while there are no data available for MPG levels in rat esophagus (32). In contrast to 7-POB-Gua, O6-POB-dGuo was below the limit of detection in rat esophageal DNA, indicating that it was efficiently repaired by O6-alkylguanine-DNA-alkyltransferase (AGT) in the esophagus. AGT is widely expressed in rat tissues including liver, lung, and esophagus (33). Collectively, these results indicate that the high sensitivity of the rat esophagus to NNN tumorigenicity may result from stereoselectivity and regioselectivity of NNN bioactivation together with low repair of POB-DNA adducts. It is also possible that poor repair of 7-methyldGuo may play a role in the commonly observed rat esophageal carcinogenicity of methylalkyl nitrosamines (34).
This is the first study to detect individual POB-DNA adducts in liver DNA from (R)-NNN and (S)-NNN-treated rats. POB-DNA adducts were previously found in liver DNA from rats treated with a single s.c. injection of [5-3H]NNN by analysis of HPB released by acid hydrolysis (8). Total adduct levels in (S)-NNN-treated rats were 4–12 times higher than those in (R)-NNN-treated rats, a significant difference. However, adduct levels in rat liver, a non-target tissue of NNN tumorigenicity, were significantly lower than those in esophagus of rats treated with either enantiomer. The results are consistent with a previous study, in which 2′-hydroxylation of NNN in cultured rat esophagus was 7 times greater than in cultured rat liver (13) 5′-Hydroxylation was the dominant metabolic pathway in rat liver. In liver microsomes, the ratio of 2′- to 5′-hydroxylation of NNN was 0.13 when 10 mM NNN was used and 0.71–0.23 when the concentrations of NNN were 1–2000 μM (16, 35).
There is no previous study on NNN metabolism and DNA adduct formation in rat lung. This study clearly demonstrates that both (R)-NNN and (S)-NNN undergo 2′-hydroxylation, producing POB-DNA adducts in rat lung. In contrast to esophageal and liver DNA, (fl)-NNN produces 2–4 times more total POB-DNA adducts in lung DNA than does (S)-NNN, a significant difference at all time points except for 16 weeks. Cytochrome P450 2A3, which is expressed in rat lung (36) may be responsible for (R)-NNN bioactivation. It is a good catalyst of both 2′- and 5′-hydroxylation of (R)-NNN, but does not catalyze 2′-hydroxylation of (5)-NNN (25–26) There must be other P450s in rat lung which catalyze 2′-hydroxylation of (S)-NNN. Similar to the results in liver DNA, O2-POB-dThd was the predominant adduct detected in lung and accumulated throughout the experiment. O6-POB-dGuo was not detected due to AGT repair. The maximal adduct levels were found at 16 weeks for both (R)-NNN and (S)-NNN treatment. The patterns of formation and accumulation of POB-DNA adducts in liver and lung from NNN-treated rats are consistent with those in liver and lung from rats chronically treated with NNK and NNAL in the drinking water (21), as would be expected based on common pathways involving intermediate 8 (Scheme 1). These adduct level patterns could result from the effects of chronic NNK treatment on enzymes involved in NNK metabolic activation and detoxification, as discussed in the accompanying paper (21).
After 10 weeks of the experiment, adduct levels in lung DNA of (R)-NNN-treated rats were not significantly different from those in esophagus of (S)-NNN-treated rats. However, rat lung is not a known target tissue for NNN tumorigenicity. If POB-DNA adduct formation is an important mechanism for NNN tumorigenicity in the rat esophagus, why doesn’t NNN also induce lung tumors given comparable levels of POB-DNA adducts in rat lung? First, the effects of DNA adduct formation on tumor induction may be different between lung and esophagus. Lung may not be as sensitive as esophagus. At the same 10 ppm doses of NNK and NNN, the lung carcinogen NNK produces significantly higher levels of POB-DNA adducts than does NNN in lung (21). POB-DNA adduct levels in lung from NNK-treated rats are 7–12 times higher than those from (R)-NNN-treated rats and 23–30 times higher than those from (S)-NNN-treated rats (21). NNK treatment also produces methyl adducts and these, in combination with POB-DNA adducts, may be important in rat lung tumorigenesis (1). Second, diminished survival caused by (S)-NNN-induced esophageal tumors could have precluded the development of lung tumors in rats by (R)-NNN. Since moderately high levels of POB-DNA adducts are produced in lung of (R)-NNN-treated rats, (R)-NNN could be a lung carcinogen if administrated for sufficiently long periods.
5′-Hydroxylation of NNN may also be an important bioactivation pathway. 5′-Hydroxylation is favored in cultured mouse lung and hamster trachea, which are target tissues for NNN tumorigenicity in these species (13, 37). DNA adducts are formed from 5′-hydroxylation of NNN and have been recently characterized in our laboratory (12). A quantitative HPLC-ESI-MS/MS method for analysis of DNA adducts derived from 5′-hydroxylation of NNN is under development. Quantitation of DNA adducts generated from both 2′- and 5′-hydroxylation of NNN would provide useful information for understanding the mechanism of NNN tumorigenesis.
In summary, individual FOB-DNA adducts were identified and quantified for the first time in esophageal, liver and lung DNA isolated from (R)-NNN and (S)-NNN-treated rats. In rat esophagus, (S)-NNN treatment generated significantly higher levels of POB-DNA adducts than (R)-NNN treatment, which confirms the preferential metabolism of (S)-NNN by 2′-hydroxylation in the rat esophagus. The results suggest that (S)-NNN is more tumorigenic to the rat esophagus than (R)-NNN. For both (R)-NNN and (S)-NNN, total adduct levels in esophagus were higher than those in liver, supporting the concept that 2′-hydroxylation of NNN is the major bioactivation pathway for esophageal tumorigenesis. In contrast to esophagus and liver, (R)-NNN treatment produced more POB-DNA adducts than (S)-NNN treatment in rat lung, which suggests an important role for cytochrome P450 2A3 in NNN metabolism in the rat lung.
Supplementary Material
Supporting Information Available: Levels of POB-DNA adducts in esophageal, liver and lung DNA of (R)- and (S) NNN-treated rats, expressed as fmol per mg DNA; levels of POB-DNA adducts in rats treated with (R)- and (S)-NNN after 20 weeks of experiment, expressed as fmol per μmol appropriate nucleoside; statistical comparison of levels of each individual POB-DNA adduct in esophageal, liver and lung DNA. This material is available free of charge via the Internet at http://pubs.acs.org.
Scheme 1.

Overview of NNN metabolism and DNA adduct formation. NTH: neutral thermal hydrolysis.
Acknowledgments
The authors acknowledge Bruce Lindgren of the University of Minnesota Cancer Center Biostatistics Core Facility for statistical analysis. Mass spectrometry was carried out in the Analytical Biochemistry Core Facility. The core facilities are supported in part by grant CA-77598 from the National Cancer Institute. We thank Dr. Edward J. McIntee for synthesis of (R)- and (S)-NNN, and Adam R. Benoit and Dr. David Lahti for their help in animal experiments and DNA isolation, Bob Carlson for manuscript preparation and submission. Stephen S. Hecht is an American Cancer Society Research Professor, supported in part by ACS grant RP-00–138. This work was supported by Grant CA-81301 from the National Cancer Institute.
Abbreviations
- AGT
O6-alkylguanine-DNA-alkyltransferase
- ANOVA
analysis of variance
- AP
apurinic
- HPB
4-hydroxy-1-(3-pyridyl)-1-butanone
- HPLC-ESI-MS/MS
high performance liquid chromatography-electrospray ionization-tandem mass spectrometry
- MPG
mammalian 3-methyladenine glycosylase
- NNK
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
- NNKOAc
4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone
- NNAL
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol
- NNN
N′-nitrosonornicotine
- O2-POB-dCyd
O2-[4-(3-pyridyl)-4-oxobut-1-yl]-2′-deoxycytidine
- O2-POB-Cyt
O2-[4-(3-pyridyl)-4-oxobut-1-yl]cytosine
- O2-POB-dThd
O2-[4-(3-pyridyl)-4-oxobut-1-yl]thymidine
- O6-POB-dGuo
O6-[4-(3-pyridyl)-4-oxobut-1-yl]-2′-deoxyguanosine
- 7-POB-dGuo
7-[4-(3-pyridyl)-4-oxobut-1-yl]-2′-deoxyguanosine
- 7-POB-Guo
7-[4-(3-pyridyl)-4-oxobut-1-yl]guanine
- POB
4-(3-pyridyl)-4-oxobut-1-yl
- REML
restricted maximum likelihood
- SRM
selected reaction monitoring
References
- 1.Hecht SS. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem Res Toxicol. 1998;11:560–603. doi: 10.1021/tx980005y. [DOI] [PubMed] [Google Scholar]
- 2.Hecht SS. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nat Rev Cancer. 2003;3:733–744. doi: 10.1038/nrc1190. [DOI] [PubMed] [Google Scholar]
- 3.International Agency on Research for Cancer. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Vol. 89. IARC; Lyon, France: 2006. Smokeless tobacco and tobacco-specific nitrosamines. in press. [Google Scholar]
- 4.Hecht SS, Rivenson A, Braley J, DiBello J, Adams JD, Hoffmann D. Induction of oral cavity tumors in F344 rats by tobacco-specific nitrosamines and snuff. Cancer Res. 1986;46:4162–4166. [PubMed] [Google Scholar]
- 5.Wang L, Spratt TE, Liu XK, Hecht SS, Pegg AE, Peterson LA. Pyridyloxobutyl adduct O6-[4-oxo-4-(3-pyridyl)butyl]guanine is present in 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone-treated DNA and is a substrate for O6-alkylhuanine-DNA alkyltransferase. Chem Res Toxicol. 1997;10:562–567. doi: 10.1021/tx9602067. [DOI] [PubMed] [Google Scholar]
- 6.Wang M, Cheng G, Sturla SJ, Shi Y, McIntee EJ, Villalta PW, Upadhyaya P, Hecht SS. Identification of adducts formed by pyridyloxobutylation of deoxyguanosine and DNA by 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone, a chemically activated form of tobacco specific carcinogens. Chem Res Toxicol. 2003;16:616–626. doi: 10.1021/tx034003b. [DOI] [PubMed] [Google Scholar]
- 7.Hecht SS, Villalta PW, Sturla SJ, Cheng G, Yu N, Upadhyaya P, Wang M. Identification of O2-substituted pyrimidine adducts formed in reactions of 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone and 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanol with DNA. Chem Res Toxicol. 2004;17:588–597. doi: 10.1021/tx034263t. [DOI] [PubMed] [Google Scholar]
- 8.Hecht SS, Spratt T, Trushin N. Evidence for 4-(3-pyridyl)-4-oxobutylation of DNA in F344 rats treated with tobacco-specific nitrosmaines 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and N′-nitrosonornicotine. Carcinogenesis. 1988;9:161–165. doi: 10.1093/carcin/9.1.161. [DOI] [PubMed] [Google Scholar]
- 9.Spratt TE, Trushin N, Lin D, Hecht SS. Analysis for N2-(pyridyloxobutyl)deoxyguanosine adducts in DNA of tissues exposed to tritium-labeled 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and N′-nitrosonornicotine. Chem Res Toxicol. 1989;2:169–173. doi: 10.1021/tx00009a008. [DOI] [PubMed] [Google Scholar]
- 10.Murphy SE, Heilblum R, Trushin N. Comparative metabolism of N′-nitrosonornicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone by cultured F344 rat oral tissue and esophagus. Cancer Res. 1990;50:4685–4691. [PubMed] [Google Scholar]
- 11.Trushin N, Rivenson A, Hecht SS. Evidence supporting the role of DNA pyridyloxobutylation in rat nasal carcinogenesis by tobacco-specific nitrosamines. Cancer Res. 1994;54:1205–1211. [PubMed] [Google Scholar]
- 12.Upadhyaya P, McIntee EJ, Villalta PW, Hecht SS. Identification of adducts formed in the reaction of 5′-acetoxy- N′-nitrosonornicotine with deoxyguanosine and DNA. Chem Res Toxicol. 2006;79:426–435. doi: 10.1021/tx050323e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hecht SS, Reiss B, Lin D, Williams GM. Metabolism of N′-nitrosonornicotine by cultured rat esophagus. Carcinogenesis. 1982;3:453–456. doi: 10.1093/carcin/3.4.453. [DOI] [PubMed] [Google Scholar]
- 14.Hecht SS, Young R. Regiospecificity in the metabolism of the homologous cyclic nitrosamines, N′-nitrosonornicotine and N′-nitrosoanabasine. Carcino genesis. 1982;3:1195–1199. doi: 10.1093/carcin/3.10.1195. [DOI] [PubMed] [Google Scholar]
- 15.Brittebo EB, Castonguay A, Furuya K, Hecht SS. Metabolism of tobacco-specific nitrosamines by cultured rat nasal mucosa. Cancer Res. 1983;43:4343–4348. [PubMed] [Google Scholar]
- 16.Murphy SE, Spina DA. Evidence for a high-affinity enzyme in rat esophageal microsomes which α-hydroxylates N′-nitrosonornicotine. Carcinogenesis. 1994;15:2709–2713. doi: 10.1093/carcin/15.12.2709. [DOI] [PubMed] [Google Scholar]
- 17.Stoner GD, Adams C, Kresty LA, Amin SG, Desai D, Hecht SS, Murphy SE, Morse MA. Inhibition of N′-nitrosonornicotine-induced esophageal tumorigenesis by 3-phenethyl isothiocyanate. Carcinogenesis. 1998;19:2139–2143. doi: 10.1093/carcin/19.12.2139. [DOI] [PubMed] [Google Scholar]
- 18.Carmella SG, Mclntee EJ, Chen M, Hecht SS. Enantiomeric composition of N′-nitrosonornicotine and N′-nitrosoanatabine in tobacco. Carcinogenesis. 2000;21:839–843. doi: 10.1093/carcin/21.4.839. [DOI] [PubMed] [Google Scholar]
- 19.McIntee EJ, Hecht SS. Metabolism of N′-nitrosonornicotine enantiomers by cultured rat esophagus and in vivo in rats. Chem Res Toxicol. 2000;13:192–199. doi: 10.1021/tx990171l. [DOI] [PubMed] [Google Scholar]
- 20.Lao Y, Villalta PW, Sturla SJ, Wang M, Hecht SS. Quantitation of pyridyloxobutyl DNA adducts of tobacco-specific nitrosamines in rat tissue DNA by high performance liquid chromatography-electrospray ionization-tandem mass spectrometry. Chem Res Toxicol. 2006;19:674–682. doi: 10.1021/tx050351x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lao Y, Yu N, Kassie F, Villalta PW, Hecht SS. Formation and accumulation of pyridyloxobutyl DNA adducts in F344 rats chronically treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem Res Toxicol. 2006 doi: 10.1021/tx060207r. ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sturla SJ, Scott J, Lao Y, Hecht SS, Villalta PW. Mass spectrometric analysis of relative levels of pyridyloxobutylation adducts formed in the reaction of DNA with a chemically activated form of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Chem Res Toxicol. 2005;18:1048–1055. doi: 10.1021/tx050028u. [DOI] [PubMed] [Google Scholar]
- 23.Wang M, Yu N, Chen L, Villalta PW, Hochalter JB, Hecht SS. Identification of an acetaldehyde adduct in human liver DNA and quantitation as N2-ethyldeoxyguanosine. Chem Res Toxicol. 2006;19:319–324. doi: 10.1021/tx0502948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gupta RC. Enhanced sensitivity of 32P-postlabeling analysis of aromatic carcinogen: DNA adducts. Cancer Res. 1985;45:5656–5662. [PubMed] [Google Scholar]
- 25.Murphy SE, Isaac IS, Ding X, McIntee EJ. Specificity of cytochrome P450 2 A3-catalyzed α-hydroxylation of N′-nitrosonornicotine enantiomers. Drug Metab Dispos. 2000;28:1263–1266. [PubMed] [Google Scholar]
- 26.Wong HL, Murphy SE, Hecht SS. Cytochrome P450 2A-catalyzed metabolic activation of structurally similar carcinogenic nitrosamines: N′-nitrosonornicotine enantiomers, N-nitrosopiperidine and N-nitrosopyrrolidine. Chem Res Toxicol. 2005;18:61–69. doi: 10.1021/tx0497696. [DOI] [PubMed] [Google Scholar]
- 27.Poorkhalkali N, Helander HF. Cell kinetics of the oesophageal epithelium in the rat: effects of hypergastrinaemia. Scan J Gastroenterol. 2003;38:14–19. doi: 10.1080/00365520310000384. [DOI] [PubMed] [Google Scholar]
- 28.Gates KS, Nooner T, Dutta S. Biologically relevant chemical reactions of N7-alkylguanine residues in DNA. Chem Res Toxicol. 2004;17:839–856. doi: 10.1021/tx049965c. [DOI] [PubMed] [Google Scholar]
- 29.Loeb LA, Preston BD. Mutagenesis by apurinic/apyrimidinic sites. Annu Rev Genet. 1986;20:201–230. doi: 10.1146/annurev.ge.20.120186.001221. [DOI] [PubMed] [Google Scholar]
- 30.Krokan HE, Nilsen H, Skorpen F, Otterlei M, Slupphaug G. Base excision repair of DNA in mammalian cells. FEBS J. 2000;476:73–77. doi: 10.1016/s0014-5793(00)01674-4. [DOI] [PubMed] [Google Scholar]
- 31.Krokan HS, Standal R, Slupphaug G. DNA glycosylases in the base excision repair of DNA. Biochem J. 1997;325:1–16. doi: 10.1042/bj3250001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Department of Genetics; Stanford University, Stanford, CA: http://source.stanford.edu. [Google Scholar]
- 33.Grafstrom RC, Pegg AE, Trump BR, Harris CC. O6-alkylguanine-DNA alkyltransferase activity in normal human tissues and cells. Cancer Res. 1984;44:2855–2857. [PubMed] [Google Scholar]
- 34.Lijinsky W. Cambridge Monographs on Cancer Research. Cambridge University Press; London, UK: 1992. Chemistry and biology of N-nitroso compounds; pp. 282–301. [Google Scholar]
- 35.McCoy GD, Chen CB, Hecht SS. Influence of mixed-function oxidase inducers on the in vitro metabolism of N′-nitrosonornicotine by rat and hamster liver microsomes. Drug Metab Dispos. 1981;9:168–169. [PubMed] [Google Scholar]
- 36.Gopalakrishnan R, Morse MA, Lu J, Weghorst CM, Sabourin C, Stoner LKGD, Murphy SE. Expression of cytochrome P450 2A3 in rat esophagus: relevance to N-nitrosobenzylmethylamine. Carcinogenesis. 1999;20:885–891. doi: 10.1093/carcin/20.5.885. [DOI] [PubMed] [Google Scholar]
- 37.Castonguay A, Lin D, Stoner GD, Radok P, Furuya K, Hecht SS, Schut HAJ, Klaunig JE. Comparative carcinogenicity in A/J mice and metabolism by cultured mouse peripheral lung of N′-nitrosonornicotine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, and their analogues. Cancer Res. 1983;43:1223–1229. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: Levels of POB-DNA adducts in esophageal, liver and lung DNA of (R)- and (S) NNN-treated rats, expressed as fmol per mg DNA; levels of POB-DNA adducts in rats treated with (R)- and (S)-NNN after 20 weeks of experiment, expressed as fmol per μmol appropriate nucleoside; statistical comparison of levels of each individual POB-DNA adduct in esophageal, liver and lung DNA. This material is available free of charge via the Internet at http://pubs.acs.org.