

Abstract
N′-Nitrosonornicotine (NNN) is believed to play an important role as a cause of cancer in people who use tobacco products and is considered to be a human carcinogen. NNN requires metabolism to form DNA adducts, which are absolutely critical to its carcinogenic properties. Previous studies have identified cytochrome P450-catalyzed 2′- and 5′-hydroxylation of NNN as potential DNA adduct forming metabolic pathways. 5′-Hydroxylation is the more prevalent of these in monkeys and humans, and is known to generate mutagenic intermediates, but the DNA adducts formed by this pathway have never been characterized. In this study, we used 5′-acetoxyNNN as a stable precursor to 5′-hydroxyNNN, and investigated its esterase-catalyzed reactions with deoxyguanosine (dGuo) and DNA. Adducts resulting from carbocation and oxonium ion intermediates, produced by the spontaneous decomposition of 5′-hydroxyNNN, were identified. The carbocation pathway resulted in the formation of 2-[2-hydroxy-5-(3- pyridyl)pyrrolidin-1-yl]deoxyinosine (12) which was characterized by comparison to an independently synthesized standard. Treatment of 12 with NaBH3CN produced two diastereomers of 2-[2-(3-pyridyl)pyrrolidin-1-yl]deoxyinosine (14), and their absolute configurations at the 2-position were determined by comparison to synthetic standards. The oxonium ion pathway produced diastereomers of N2[5-(3-pyridyl)tetrahydrofuran-2-yl]dGuo (16), identified by comparison to synthetic standards. The absolute configuration at the 5- position was determined by establishing the stereochemistry of the enantiomers of 5-(3-pyridyl)- 2-hydroxytetrahydrofuran at the 5-position, and allowing these to react individually with dGuo. Treatment of 16 with NaBH3CN produced N2[4-hydroxy-4-(3-pyridyl)but-1-yl]dGuo (18) which was also synthesized independently. Using liquid chromatography-electrospray ionization-tandem mass spectrometry with selected reaction monitoring, adducts 12 and 16 were identified as products of the reactions of 5′-acetoxyNNN with dGuo. Similarly, adducts 14 and 18 were identified as products of the reaction of 5′-acetoxyNNN with DNA followed by NaBH3CN treatment and enzymatic hydrolysis. These results provide the first structural characterization of DNA adducts that can be formed by 5′-hydroxylation of NNN.
Introduction
Two critical biological properties of tobacco products- addiction and carcinogenicity- are represented by the structurally related compounds nicotine (1) and N′-nitrosonornicotine (NNN, 2). Nicotine is the principal known addictive agent in tobacco products and is the reason why most people cannot break their tobacco habit in spite of a strong desire to quit (1). Nicotine is not carcinogenic, but replacement of its methyl group by a nitroso- group produces the carcinogen NNN (2–4). NNN, which is present in both unburned tobacco and tobacco smoke, causes tumors of the esophagus and nasal cavity in rats, and the respiratory tract in mice, hamsters, and mink (2–4). NNN and the related nitrosamine 4-(methylnitrosamino)-1-(3- pyridyl)-1-butanone (NNK, 3) elicit oral tumors when co-applied to the rat oral cavity (5). NNN is believed to play a significant role as a cause of cancers of the oral cavity, esophagus, nasal cavity, and perhaps other tissues in people who use tobacco products (2,6). The International Agency for Research on Cancer ranks NNN and NNK in Group 1, carcinogenic to humans (7).
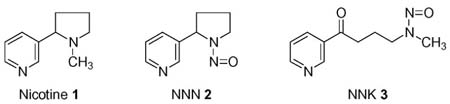
NNN requires metabolism, catalyzed principally by cytochrome P450 enzymes, to produce its mutagenic and, presumably, its carcinogenic effects (4). DNA adduct formation, which results in miscoding and the formation of permanent mutations, is critical in this process (8). Two pathways of NNN metabolism, 2′-hydroxylation and 5′-hydroxylation, illustrated in Scheme 1, can produce electrophiles that react with DNA, forming adducts (4). 2′-Hydroxylation of NNN initially produces 2′-hydroxyNNN (5) which spontaneously ring opens yielding diazohydroxide 7. This intermediate, which is also formed metabolically from NNK, is known to react with DNA, and several adducts have been characterized (9–11). 5′-Hydroxylation of NNN initially gives 5′-hydroxyNNN (6) which ring opens yielding diazohydroxide 8. Although the mutagenicity of 5′-acetoxyNNN (4), a stable precursor to 5′-hydroxyNNN (6), has been known for nearly 30 years (12), and the 5′-hydroxylation pathway is a prevalent one in the metabolism of NNN in monkeys and in human tissues (13,14), there are no reports in the literature on DNA adduct formation by 5′-hydroxylation of NNN. In this paper, we describe the first characterization of deoxyguanosine (dGuo) and DNA adducts resulting from this pathway of NNN metabolism.
Scheme 1.
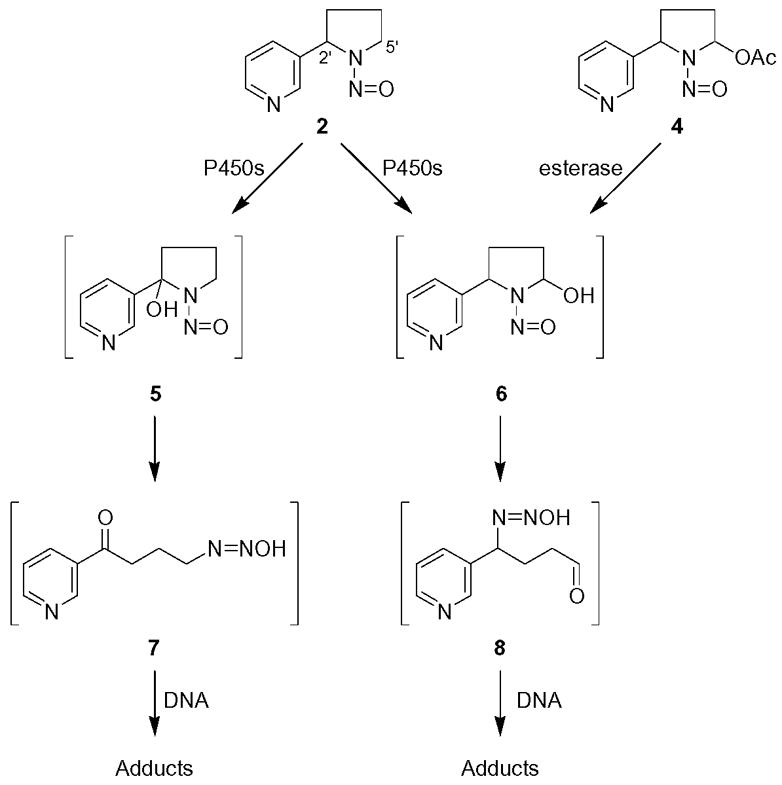
Metabolic activation of NNN (2) by cytochrome P450s and esterase catalyzed hydrolysis of 5′-acetoxyNNN (4)
Materials and Methods
Caution
NNN and 5′-acetoxyNNN are carcinogenic and mutagenic and therefore should be handled with extreme care, using appropriate clothing and ventilation at all times.
All chemicals were obtained from Sigma-Aldrich Chemical Co. (Milwaukee, WI), unless noted otherwise.
Apparatus and Assay Conditions
HPLC was carried out with Waters Associates (Milford, MA) systems equipped with a model 996 photodiode array detector or a model 440 UV-visible detector set to 254 nm, unless noted otherwise. The following solvent elution programs were used, with linear solvent gradients: system1, a 300 mm × 3.9 mm, 10μ C-18 Bondclone column (Phenomenex, Torrance, CA) eluted at 1 mL/min with 0% CH3OH in H2O to 100% CH3OH over the course of 60 min; system 2, a 250 mm × 10 mm, 10μ, C-18 Vydac 201TP column (Separations Group, Hesperia, CA) eluted at 3 mL/min with 0% CH3OH in H2O to 100% CH3OH over the course of 60 min; system 3, a 250 mm × 6 mm, 5μ Luna C18 column (Phenomenex) eluted isocratically at 0.7 μL/min with 70% CH3OH in H2O (20 μL of 20 mM oxalic acid solution was injected immediately before each chromatographic run); system 4, a 0.46 cm × 25 cm Beckman Ultrasphere ODS 5μ C-18 column (Beckman Coulter, Fullerton, CA) eluted with 20 to 60% CH3OH in H2O over the course of 30 min at a flow rate of 1 mL/min, and then held at 60% CH3OH for 10 min.
LC-ESI-MS was carried out in systems 5–7 as follows. System 5 used an Agilent 1100 Series capillary flow HPLC ion trap MS system (Agilent Technologies, Palo Alto, CA) with a 150 mm × 0.5 mm Zorbax Extend-C-18, 3.5 μm column (Agilent) eluted at 13.0 μL/min with 10% CH3OH in 15 mM NH4OAc (pH 5.8) to 60 % CH3OH over the course of 30 min. N2 was used as drying gas (200 °C, 5 L/min) and as nebulizing gas (15 psi). The system also contained an in-line UV detector. The mass spectrometer was operated in the full scan mode (m/z 100–800), with target-ion abundance of 30,000, maximum accumulation time of 300 ms, and fragmentation amplitude of 0.9 V. MS/MS experiments were performed with a CID gas pressure of 1.5 mTorr and CID energy of 30 V. System 6 used a Thermo Finnigan LCQ Deca instrument (Thermo Finnigan LC-MS Division, San Jose, CA)) interfaced with an Alliance 2690 HPLC multisolvent delivery system (Waters) equipped with an SPD-10A UV-Vis detector (Shimadzu Scientific Instruments, Columbia, MD) and a 300 mm × 3.9 mm, 10μ C-18 Bondclone column eluted at 0.5 mL/min with 5% CH3OH in 40 mM NH4OAc to 35% CH3OH over the course of 60 min, then from 35% to 70% CH3OH in 30 min, then from 70% CH3OH to 100% CH3OH in 5 min. The ESI source was set as follows: voltage, 2.0 kV; capillary temperature 275 °C. MS/MS data were acquired with the following parameters: parent mass, m/z 417, isolation width, 1.5 amu; activation amplitude, 30%; activation Q, 0.25; activation time 30 ms. System 7 used a TSQ Quantum Ultra instrument (Thermo Finnigan LC-MS Division, San Jose, CA) interfaced with an Agilent 1100 Series capillary flow HPLC system and a 150 × 0.5 mm, 5μ Zorbax SB-18 column (Agilent) eluted at 10.0 μL/min with 10% CH3OH in 15 mM NH4OAc (pH 5.8) to 70% CH3OH over the course of 40 min. The ESI source was set in the positive mode as follows: voltage, 4 kV; current, 20 μA; and ion transfer tube, 150 °C. MS/MS experiments used the selected reaction monitoring (SRM) mode, m/z 415 → m/z 299. The collision energy was 13 eV, and the argon collision gas pressure was 0.8 mTorr.
High resolution mass spectra (HRMS) were determined with a Finnigan-MAT 90/95 instrument (Thermo Finnigan MAT, Bremen, Germany).
NMR spectra were recorded in DMSO-d6 or CDCl3 using Varian Unity (Varian, Inc., Palo Alto, CA) spectrometers operated at either 600 or 800 MHz and standard 5 mm tubes or 3 mm Shigemi tubes (Shigemi, Inc., Allison Park, PA).
Reaction of 5′-acetoxyNNN(4) with dGuo
5′-AcetoxyNNN (4, 59.0 mg, 0.25 mmol) (15) was allowed to react with dGuo (13.2 mg, 0.05 mmol) in 5 mL of 0.1M phosphate buffer, pH 7.0, in the presence of porcine liver esterase (90 units) at 37 °C for 1h. Products 12 and 16 were separated using system 6 and characterized by LC-ESI-MS using system 7, and by comparison to synthetic standards.
Reaction of 5′-acetoxyNNN(4) with DNA
5′-AcetoxyNNN (4, 12.0 mg, 0.05 mmol) was allowed to react with calf thymus DNA (20 mg) in 5 mL of 0.1M phosphate buffer, pH 7.0, in the presence of porcine liver esterase (90 units) at 37 °C for 1h. The DNA was precipitated by addition of ethanol, then washed with 70% ethanol and 95% ethanol. The DNA (1.2 mg) was dissolved in 1 mL of 10 mM Tris HCl/5 mM MgCl2 buffer, pH 7.0. NaBH3CN (10 mg) was added and the mixture was incubated at 37 °C for 30 min and the DNA was then hydrolyzed enzymatically (16). The hydrolysate was further purified by solid phase extraction on C18 Seppak Cartridges (Waters) as described (16). Products 14 and 18 were eluted with CH3OH and analyzed by LC-ESI-MS using system 7 (starting from 5% CH3OH in 15 mM NH4OAc, pH 5.8 for 10 min, then to 95% CH3OH over the course of 60 min, collision energy 1.3 mTorr) and by comparison to synthetic standards.
5-(Benzhydrylidenamino)-5-(3-pyridyl)-1-pentene (20). 2-Aza-1,1-diphenyl-3-(3-pyridyl)-1- propene (19) (17) (2.7 g, 9.9 mmol) was dissolved in anhydrous THF (50 mL) under N2 and the mixture was cooled to −78 °C. Lithium diisopropylamide (14 mL, 26 mmol) was added dropwise via syringe over a 15 min period. The reaction mixture turned a deep purple color as the temperature rose to −40 °C. A solution of 4-bromo-1-butene (1.24 mL, 12.2 mmol) in THF (15 mL) was added via syringe over a period of 20 min. The mixture was allowed to stir at 40 °C for 8 h. The pH was adjusted to 6 with 10% HCl and the mixture was extracted with CHCl3 (5 × 30 mL). The organics were combined, dried (Na2SO4), filtered, and condensed. The resulting oil was purified by column chromatography with elution by hexanes/ethyl acetate (1:1) and ethyl acetate giving 20 (1.1 g, 3.4 mmol, 39%) as a yellow oil.
1H-NMR (CDCl3) δ 8.47 (ddd, J=4.8, 1.8 Hz, 1H, pyr-6H), 8.40 (d, J=1.8 Hz, 1H, pyr-2H), 7.73 (dd, J=1.82, 2.4 Hz, 1H, pyr-4H), 7.72 (dd, J=1.82, 2.4 Hz, 1H, pyr-4H), 7.67 (d, J=7 Hz, 2H, PhH), 7.48–7.32 (m, 6H, PhH), 7.23 (m, 1H, pyr-5H), 7.02 (d, J=3.6 Hz, 2H, PhH), 5.71 (m, 1H, 2H), 4.93 (dd, J=1.2, 16.8 Hz, 1H, 1Ha), 4.88 (d, J=10.2 Hz, 1H, 1Hb), 4.39 (dd, J=4.8, 5.4 Hz, 1H, 5H), 2.03 (m, 2H, 4H), 1.88 (m, 2H, 3H); ESI-MS m/z 327.1 (M + H)+.
5-(Benzhydrylidenamino)-5-(3-pyridyl)-1,2-pentanediol (21)
Compound 20 (0.5 g, 1.5 mmol) was dissolved in anhydrous THF (25 mL) and treated with aqueous OsO4 (0.08M, 1mL) and 4- methylmorpholine N-oxide (270 mg, 2.3 mmol). The reaction was allowed to stir at room temperature for 3 h and was quenched with 10% NaHSO3 (15 mL). The volatiles were removed at reduced pressure and the remaining material was extracted with ethyl acetate (2 × 30 mL). The organics were combined, dried (Na2SO4), filtered, and condensed to give 21 as a brown oil (500 mg, 1.39 mmol, 93%).
1H-NMR (CDCl3) δ 8.40 (m, 1H, pyr-6H), 8.32 (m, 1H, pyr-2H), 7.77 (d, J=6.6Hz, 2H, Ph), 7.66 (dd, J=1.8, 2.4 Hz, 1H, pyr-4H), 7.62–7.23 (m, 6H, PhH), 7.18 (m, 1H, pyr-5H), 7.02 (d, J=3.6 Hz, 2H, PhH), 4.38 (dd, J=6.6, 6.6 Hz, 1H, 5H), 3.61 (m, 1H, 2H), 3.52 (m, 1H, 1Ha), 3.34 (m, 1H, 1Hb), 2.31–1.82 (m, 2H, 4H), 1.44–1.18 (m, 2H, 3H); ESI-MS m/z 361.1 (M + H)+.
5-Amino-5-(3-pyridyl)-1,2-pentanediol (22)
Compound 21 (0.5 g, 1.4 mmol) was dissolved in 12 mL ethanol containing 0.5M NH2OH. HCl and the mixture was allowed to stir at room temperature overnight, condensed under reduced pressure, and the residue suspended in 4 mL H2O. Ion-exchange chromatography (Dowex 1X8-200, strongly basic) with elution by H2O gave 22 (250 mg, 1.28 mmol, 91%) as a clear purplish oil. 1H-NMR (D2O) δ 8.34 (s, 1H, pyr-6H), 8.29 (dd, J=1.2, 3.6 Hz, 1H, pyr-2H), 7.71 (dd, J=1.8, 4.2 Hz, 1H, pyr-4H), 7.32 (dd, J=3.6, 4.2 Hz, 1H, pyr-5H), 3.82 (dd, J=7.2, 6.6 Hz, 1H, 5H), 3.52 (m, 1H, 2H), 3.38 (m, 1H, 1Ha), 3.26 (m, 1H, 1Hb), 1.80–1.58 (m, 2H, 4H), 1.38–1.04 (m, 2H, 3H); ESI-MS m/z 197.1 (M + H)+ HRMS Calc’d for C10H16N2O2 (M + H)+ 197.1285, found 197.1278
O6[(Trimethylsilyl)ethyl]-N2[1,2-dihydroxy-5-(3-pyridyl)pent-5-yl]dGuo (24)
Compound 22 (7.5 mg, 0.038 mmol) was added to a mixture of O6[(trimethylsilyl)ethyl]2-fluoroinosine (23, 9.5 mg, 0.25 mmol, ChemGene Corp, Wilmington, MA) and N,N-diisopropylethylamine (50 μL) in 200 μL DMSO. The mixture was stirred at 65 °C for 7 h, cooled, and the solvents were removed under vacuum. The residue was purified with HPLC system 2, retention time 42 min, giving 24 (4.5 mg 0.008 mmol, 22%). 1H-NMR (DMSO-d6) δ 8.58 (m, 1H, pyr-2H), 8.39 (m, 1H, pyr- 6H), 8.04 (s, 1H, dGuo-C8H), 7.77 (m, 1H, pyr-4H), 7.46 (bs, 1H, dGuo-N2H), 7.28 (m, 1H, pyr- 5H), 6.20 (dd, J=7.2, 7.2 Hz, 1H, 1′H), 5.27 (bs, 1H, 3′OH), 4.93 (m, 2H, 5H and 1OH), 4.42–4.33 (m, 5H, 5′OH, 2OH, OCH2CH2Si and 3′H), 3.80 (m, 1H, 4′H), 3.53 (m, 1H, 5′Ha), 3.43–3.2 (m, 4H, 1H, 2H and 5′Hb), 2.56 (m, 1H, 2′Hb), 2.00–1.60 (m, 2H, 4H), 1.44–1.15 (m, 2H, 3H), 1.08 (m, 2H, OCH2CH2Si), 0.065 (s, 9H, Si(CH3)3); ESI-MS m/z 547.1 (M + H)+; HRMS Calc’d for C25H38N6O6Si (M + Na)+ 569.2514, found 569.2506
N2[1,2-dihydroxy-5-(3-pyridyl)pent-5-yl]dGuo (25)
Compound 24 (4.5 mg, 0.008 mmol) was dissolved in 5% acetic acid (1 mL) and the mixture was stirred at room temp for 3 h, concentrated at reduced pressure and the residue purified using HPLC system 2, giving 25, retention time 21 min (2 mg, 0.0045 mmol, 56%). 1H-NMR (DMSO-d6) δ 8.59 (s, 0.5H, pyr- 2H), 8.57 (s, 0.5 H, pyr-2H), 8.44 (m, 1H, pyr-6H), 7.88 (s, 0.5H, dGuo-C8H), 7.84 (s, 0.5H, dGuo-C8H), 7.76 (d, J=8Hz, 0.5H, pyr-4H), 7.74 (d, J=8Hz, 0.5H, pyr-4H), 7.36 (m, 1H, pyr- 5H), 7.10 (bs, 1H, dGuo-N2H), 6.08 (dd, J=6.4, 5.6Hz, 0.5H, 1′H), 6.04 (dd, J=6.4, 5.6 Hz, 0.5H, 1′H), 4.94–4.85 (m, 2H, 5H and 1OH), 4.31 (m, 1H, 3′H), 3.78 (m, 1H, 4′H), 3.59 (m, 1H, 5′Ha), 3.51–3.27 (m, 4H, 5′Ha, 5′Hb, 1Ha and 2H), 3.20 (m, 1H, 1Hb), 2.44 (m, 1H, 2′H), 2.13 (m, 0.5H, 2′H), 2.06 (m, 0.5H, 2′H), 2.02–1.77 (m, 2H, 4H), 1.57–1.16 (m, 2H, 3H); ESI-MS m/z 447.1 (M + H)+. MS/MS of m/z 447; 331[(M + H) − deoxyribose]+; HRMS Calc’d for C20H26N6O6 (M + Na)+ 469.1806, found 469.1822
2[2-Hydroxy-5-(3-pyridyl)pyrrolidin-1-yl]deoxyinosine (12)
Sodium periodate (200 μL, 80 mM in 0.01 M phosphate buffer, pH 7) was added to a solution of 25 (2 mg, 0.0045 mmol) in phosphate buffer (200 μL, 0.01M, pH 7). The mixture was stirred at room temperature for 25 min; HPLC showed complete disappearance of starting material. HPLC purification using system 2, retention time 20–24 min, gave a mixture of diastereomers of 12 (1 mg, 0.0024 mmol, 54%). In HPLC system 1 (instead of H2O, 15 mM NH4OAc pH 5.0 was used), retention times were 27.5, 28.3, 28.9, and 29.6 min. UV (λmax) of the 4 diastereomers: 27.5 min, 209.8, 258.0, 285.0 (sh); 28.3 min 211.0, 260.3, 285.0 (sh); 28.9 min 212.2, 260.3, 285.0 (sh); 29.6 min 209.8, 258.0, 285.0 (sh).
1H-NMR (DMSO-d6) δ 8.63 (d, J=2.4 Hz, 0.5H, pyr-2H), 8.59 (d, J=2.4 Hz, 0.5H, pyr-2H), 8.41 (m, 1H, pyr-6H), 7.89 (s, 0.5H, dGuo-C8H), 7.82 (s, 0.5H, dGuo-C8H), 7.78 (m, 0.5H, pyr-4H), 7.73 (m, 0.5H, pyr-4H), 7.37–7.28 (m, 1H, pyr-5H), 6.57 (s, 1H, 2-OH), 6.02–5.93 (m, 1H, 1′H), 5.69 (m, 1H, 2H), 5.13 (dd, J=7.2, 8.0 Hz, 0.5H, 5H), 5.10 (dd, J=7.2, 8.0 Hz, 0.5H, 5H), 4.26–4.21 (m, 1H, 3′H), 3.80–3.71 (m, 1H, 4′H), 3.57 (m, 0.5H, 5′Ha), 3.49–3.38 (m, 1.5H, 5′Ha and 5′Hb), 2.67 (m, 2′H), 2.42 (m, 1H, 4Ha), 2.34 (m, 2′H), 2.21 (m, 2′H), 2.09 (m, 2′H and 3Ha), 2.07–1.86 (m, 2′H, 3Hb and 4Hb), 1.81 (m, 2′H), 1.71(m, 2′H). LC-ESI-MS and HPLC-UV analysis were carried out using system 5, retention time 17–21 min; MS (positive ion) m/z 415 (M + H)+; MS/MS of m/z 415; m/z 299; SRM analysis (m/z 415→299) was carried out using system 7, retention time 23.3–25.6 min; HRMS calculated for C19H22NaN6O5, (M + Na)+; 437.1544 found, 437.1536.
2-(R)- and 2-(S)-2[2-(3-Pyridyl)pyrrolidin-1-yl]deoxyinosine (14). (R)- or (S)-Nornicotine (18) (1.8 mg, 0.012 mmol), O6[(trimethylsilyl)ethyl]2-fluoroinosine (23, 3.0 mg, 0.008 mmol), and Et3N (60 μL) were dissolved in DMSO (400 μL) and heated at 60 °C for 36 h. The mixture was condensed under reduced pressure, and purified with HPLC system 2 (eluted at 3 mL/min with 10% CH3OH in H2O to 100% CH3OH in 60 min), giving 2-(R)-14, retention time 19.7 min (800 μg, 0.002 mmol, 25%) and 2-(S)-14, retention time 21.2 min (800 μg, 0.002 mmol, 25%). 1H-NMR (DMSO-d6) 2-(R)-14 δ 8.44 (d, J=2.4Hz, 1H, pyr-2H), 8.42 (dd, J=1.2, 4.8Hz, 1H, pyr- 6H), 7.80 (s, 1H, dGuo-C8H), 7.56 (d, J=7.8Hz, 1H, pyr-4H), 7.30 (dd, J=4.8, 7.8 Hz, 1H, pyr- 5H), 5.94 (m, J=6.0 Hz, 1H, 1′H), 5.24 (d, J=4.8Hz, 1H, 2H), 5.17 (d, J=4.2 Hz, 1H, 3′-OH), 4.83 (t, J=5.4 Hz, 1H, 5′-OH), 4.19 (br, 1H, 3′H), 3.90 (m, 1H, 5Ha), 3.74 (m, 1H, 4′H), 3.61 (m, 1H, 5Hb), 3.54 (m, 1H, 5′Ha), 3.47 (m, 1H, 5′Hb), 2.39 (m, 2H, 3H), 1.97 (m, 2H, 2′H), 1.81–1.94 (m, 2H, 4H). LC-ESI-MS and HPLC-UV analysis were carried out using system 5, 2-(R)-14 retention time 19.1 min; MS (positive ion) m/z 399 (M + H)+; MS/MS of m/z 399; m/z 283; HRMS calculated for C19H22NaN6O4, 421.1595 (M + Na)+; found, 421.1616.
1H-NMR (DMSO-d6) 2-(S)-14 δ 8.45 (d, J=1.8 Hz, 1H, pyr-2H), 8.42 (dd, J=1.8, 4.8Hz, 1H, pyr- 6H), 7.85 (s, 1H, dGuo-C8H), 7.58 (d, J=8.4 Hz, 1H, pyr-4H), 7.31 (dd, J=4.8, 7.8 Hz, 1H, pyr- 5H), 5.97 (m, J=6.0 Hz, 1H, 1′H), 5.31 (d, J=6.6 Hz, 1H, 2H), 5.21 (d, J=3.6 Hz, 1H, 3′OH), 4.79 (t, J=6.0 Hz, 1H, 5′OH), 4.22 (br, 1H, 3′H), 3.90 (m, 1H, 5Ha), 3.72 (dd, J=4.8, 7.8 Hz, 1H, 4′H), 3.58 (m, 1H, 4″Hb), 3.41 (m, 1H, 5′Ha), 3.31 (m, 1H, 5′Hb), 2.37 (m, 2H, 3H), 1.99 (m, 2H, 2′H), 1.84–1.94 (m, 2H, 4H).
LC-ESI-MS and HPLC-UV analysis were carried out using system 5, 2-(S)-14 retention time 20.7 min. MS (positive ion) m/z 399 (M + H)+; MS/MS of m/z 399; m/z 283; HRMS calculated for C19H22NaN6O4, 421.1595 (M + Na)+; found, 421.1616.
N2[5-(3-pyridyl)tetrahydrofuran-2-yl]dGuo (16)
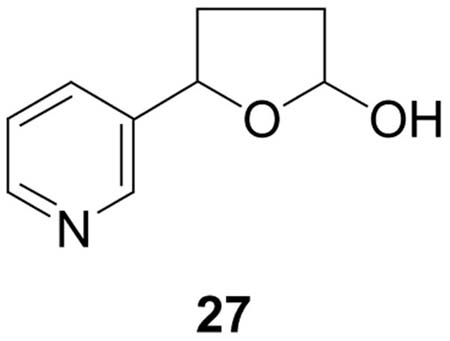
5-(3-Pyridyl)-2-hydroxytetrahydrofuran (27) (19)(5.8 mg, 0.035 mmol) was dissolved in 0.5 mL dry DMF under N2. Trifluoroacetic acid (1μL) and dGuo (10 mg, 0.035 mmol) were added and the mixture was allowed to stir at room temperature for 4 days. Reaction progress was monitored by HPLC system 1. Retention times were: dGuo (16.2 min); 27 (23.7 min); diastereomers of 16 (peak 1, 29.2; peak 2, 30.8; peak 3, 32.2; peak 4, 32.8 min). Material eluting from 27 to 34 min was collected and the solvent was removed to give 16 (3.5 mg, 0.008 mmol, 22%). 1H NMR (DMSO-d6) δ 10.5 (bs, 1H, dGuo-N1- H), 8.54 (d, J=6.3 Hz, 1H, pyr-2H), 8.46 (m, 1H, pyr-6H), 7.95 (s, 1H, dGuo-C8H), 7.76 (dd, J=1.6, 6.4 Hz, 1H, pyr-4H), 7.35 (m, 2H, pyr-5H and dGuo-N2H), 6.16 (dd, J=8.3, 8.3 Hz, 1H, 1′H), 6.05 (d, J=6.4 Hz, 0.5H, 2H), 5.91 (d, J=6.4 Hz, 0.5H, 2H), 5.28 (d, J=4.4 Hz, 1H, 3′OH), 5.04 (dd, J=7.1, 7.1 Hz, 0.5H, 5H), 4.96 (dd, J=7.1, 7.1 Hz, 0.5H, 5H), 4.87 (dd, J=5.4, 5.4 Hz, 1H, 5′OH), 4.35 (m, 2H, 3′H), 3.79 (m, 1H, 4′H), 3.53 (m, 1H, 5′Ha), 3.47 (m, 1H, 5′Hb), 2.57 (m, 2′Ha), 2.41 (m, 1H, 3Ha), 2.35 (m, 1H, 4Ha), 2.18 (m, 1H, 2′Hb), 1.92–1.77 (m, 2H, 3Hb and 4Hb); UV (CH3OH/15mM NH4OAc) λmax peaks 1–4 207.7, 255.8. LC-ESI-MS and HPLC-UV analysis were carried out using system 5 (see Figure 5), retention times: peak 1, 19.0; peak 2, 19.5, peak 3, 20.9, peak 4, 22.1 min; MS (positive ion) m/z 415 (M + H)+; MS/MS of m/z 415; m/z 299; SRM analysis (m/z 415→m/z 299) was carried out using system 7, retention time 26–28 min; HRMS calculated for C19H22NaN6O5, 437.1544 (M + Na)+; found, 437.1549.
Figure 5.
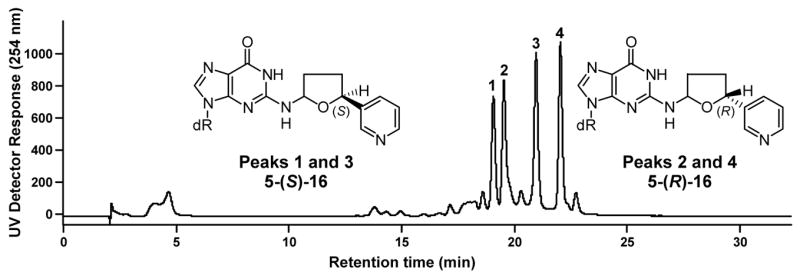
Chromatogram obtained upon HPLC analysis (using the in-line UV detector of LC-ESI-MS system 5) of adduct 16.
[1-(1,3-Dioxan-2-yl)-4-(3-pyridyl)but-4-yl]α-methoxy-α-(trifluoromethyl)phenyl acetate (32)
A mixture of 1-(1,3-dioxan-2-yl)-4-oxo-4-(3-pyridyl)butane (30) (19) (8.0 mg, 0.036 mmol) and NaBH4 (100 mg) in CH3OH (5 mL) was stirred for 6 h at room temperature. HPLC analysis (system 2) indicated complete disappearance of starting material. The pH of the reaction mixture was adjusted to 7.0 with 1 N HCl, and 31 was purified using HPLC system 2, retention time 27 min (7.0 mg, 0.031 mmol, 86%). Compound 31 (6.6 mg, 0.030 mmol) was allowed to react with (R)-(−)-α-methoxy-α-(trifluoromethyl)phenylacetic acid chloride (22.3 mg, 0.088 mmol) in CH2Cl2 as described (20). The mixed diastereomers of 32 were collected in HPLC system 2 (6.45 mg, 0.015 mmol, 50%), and separated with HPLC system 3; retention times 23.1 and 24.6 min. 1H NMR (23.1 min peak, carbinol (S)-32) δ 8.60 (s, 1H, pyr-2H), 8.55 (d, J=3.5 Hz, 1H, pyr-6H), 7.64 (d, J=6.0, 1H, pyr-4H), 7.38–7.30 (m, 5H, aromatic), 7.26 (dd, J=5.0, 8.0 Hz, 1H, pyr-5H), 5.99 (dd, J=6.0, 8.0 Hz, 1H, butyl-4H), 4.47 (t, J=5.0 Hz, 1H, butyl-1H), 4.04 (m, 2H, OCH2,), 3.69 (m, 2H, OCH2), 3.42 (d, J=1.0 Hz, 3H, OCH3), 2.08–1.92 (m, 2H, butyl-3H), 1.67 (m, 1H, OCH2CH), 1.56–1.47 (m, 2H, butyl-2H), 1.31–1.28 (dd, J=1.0, 14Hz, 1H, OCH2CH); MS (positive ion ESI) m/z 440 (M + 1), MS/MS of m/z 440; m/z 206 (loss of O(CO)C(CF3)(Ph)(OCH3).
1H-NMR (24.6 min peak, carbinol (R)-32) δ 8.53 (s, 1H, pyr-2H or pyr-6H), 8.49 (s, 1H, pyr-2H or pyr-6H), 7.46 (d, J=8.0 Hz, 1H, pyr-4H), 7.38–7.30 (m, 5H, PhH), 7.20 (m, 1H, pyr-5H), 5.94 (dd, J=6.0, 8.0 Hz, 1H, butyl-4H), 4.52 (t, J=5.0 Hz, 1H, butyl-1H), 4.07 (dd, J=4.0Hz, 2H, OCH2), 3.71 (m, 2H, OCH2), 3.53 (d, J=1.0Hz, 3H, OCH3), 2.14–1.92 (m, 2H, butyl-3H), 1.7–1.56 (m, 3H, OCH2CH + butyl-2H), 1.31 (m, 1H, OCH2CH); MS (positive ion ESI) m/z 440, MS/MS of m/z 440; m/z 206.
5-(R)-(+) and 5-(S)-(−) 5-(3-Pyridyl)-2-hydroxytetrahydrofuran (27)
Each diastereomer of 32 (4.4 mg, 0.010 mmol) was treated with 0.1N NaOH, 37 °C, 12 h and the reaction mixture was adjusted to pH 7.0 with 1 N HCl, to give (R)-(+)- and (S)-(−)-31 (1.6 mg, 0.010 mmol, 98%), purified by HPLC system 2. Each of (R)-(+)- and (S)-(−)-31 was then treated with 10% aq H2SO4 as described (19) to give 5-(R)-(+)- and 5-(S)-(−)- 27. Products were collected using HPLC system 2: 5-(R)-(+)-27, retention time 23 min (1.0 mg, 0.006 mmol, 61 %, ) and 5-(S)-(−)-27, retention time 23 min (1.0 mg, 0.006 mmol, 61 %, ).
Determination of absolute configuration of 5-(R)-(+) 5-(3-pyridyl)-2-hydroxytetrahydrofuran (27) 5-(R)-(+)-27 (3.2 mg, 0.019 mmol) was heated under reflux in xylene (5 mL) in the presence of Ag2CO3 on Celite (5.9 mg, 0.1 mmol) for 2 h at 160 °C as described (19). Lactone 33 (2.0 mg, 0.012 mmol, 63%) was purified using HPLC system 2. It was then heated under reflux with 6N HCl as described (21) for 1 h, cooled and the mixture neutralized to pH 7.0 with 6N NaOH. This gave hydroxy acid 34 which was purified with HPLC system 4 (1.0 mg, 0.006 mmol, 50%). Hydroxy acid 34 was allowed to react with acidic methanol followed by (S)- (−)-α-methylbenzyl isocyanate to give methyl 4-[α-methylbenzylcarbamoyl]-4-(3- pyridyl)butanoate (MMPB, 35) as described (22,23). The mixture was purified with HPLC system 4, and LC-ESI-MS and HPLC-UV analysis were carried out using system 5, except that there was a hold at 60% CH3OH for an additional 10 min. MS (positive ion ESI) m/z 343 (M + H)+; MS/MS of m/z 343; m/z 178. The single diastereomer co-eluted with standard methyl 4-(R)- [(S)- α-methylbenzylcarbamoyl]-4-(3-pyridyl)butanoate ((R,S)-MMPB, 35).
N2[5-(R)-(3-Pyridyl)tetrahydrofuran-2-yl]dGuo and N2[5-(S)-(3-pyridyl)tetrahydrofuran-2- yl]dGuo (16) 5-(R)-(+)- or 5-(S)-(−)-27 (1.45 mg, 0.009 mmol) was each allowed to react with dGuo (2.5 mg, 0.009 mmol) in DMF (0.5 mL) and trifluoroacetic acid (1 μL) for 4 days at room temperature, as described above. The mixture was purified using HPLC system 1. LC-ESI-MS and HPLC-UV analysis were carried out using system 5: retention time of the 5-(R)-isomers 19.7 min (peak 2) and 22.7 min (peak 4); MS (positive ion ESI) m/z 415 (M + H)+, MS/MS of m/z 415, m/z 299 [(M − deoxyribose) + H]+; retention time of the 5-(S)-isomers 19.0min (peak 1) and 21.4 min (peak 3); MS (positive ion ESI) m/z 415, MS/MS of m/z 415, m/z 299.
N2[4-Hydroxy-4-(3-pyridyl)but-1-yl]dGuo (18)
Racemic adduct 16 (1.2 mg, 0.003 mmol) and NaBH3CN (10 mg) in methanol:H2O (1:1) (5 mL) were stirred overnight at room temperature. The pH of the mixture was adjusted to 7.0 with 0.1N HCl, and it was extracted with CHCl3. The extracts were concentrated and 18 was purified using HPLC system 1 (retention time 29.8 min). Standard 18 was prepared by treating adduct 29 (24) (1.0 mg, 0.0024 mmol) with NaBH4 (10 mg) in CH3OH (5 mL). The HPLC retention time, UV, MS (system 7), and 1H-NMR spectra of 18 prepared from 16 matched those of 18 prepared from 29.
Results
Scheme 2 shows that diazohydroxide 8, formed from 5′-hydroxylation of NNN as illustrated in Scheme 1, could decompose to produce a series of electrophiles including diazonium ion 9, carbocation 10, and oxonium ion 15. Based on previous work on DNA adduct formation from α-hydroxylation of N-nitrosopyrrolidine (16), we hypothesized that two dGuo adducts resulting from 5′-hydroxylation of NNN would be 12 and 16. We investigated the reactions of 5′-acetoxyNNN (4, Scheme 1) with dGuo and DNA to test this hypothesis. The data described below demonstrate that stereoisomers of 12 and 16 were indeed formed in these reactions.
Scheme 2.

Adduct formation from diazohydroxide 8 and derived intermediates
Liquid chromatography-electrospray ionization-tandem mass spectrometry with selected reaction monitoring (LC-ESI-MS/MS-SRM) analysis of m/z 415→299, corresponding to the transition [M + H]+→[(M − deoxyribose) + H]+, of the products of reaction of 5′-acetoxyNNN with dGuo produced the chromatogram illustrated in Figure 1. We proposed that the peaks eluting at 23.6 min corresponded to adduct 12 (Scheme 2). Standard adduct 12 was synthesized as summarized in Scheme 3. Aminodiol 22 was prepared by alkylation of 19 with 4-bromo-1- butene followed by OsO4 oxidation and deprotection. Reaction of 22 with protected 2- fluoroinosine 23 gave 24, which was deprotected and treated with NaIO4 to yield the desired adduct 12, as a mixture of 4 diastereomers, characterized by 1H-NMR, UV, and LC-ESI-MS. In our hands, this material was somewhat unstable and prone to decomposition, possibly because of equilibrium with Schiff base 13 and open-chain amino aldehyde 11 (Scheme 2). However, its retention time in LC-ESI-MS/MS System 7 was the same as that of the 23.6 min peak in Figure 1. Treatment of synthetic adduct 12 with NaBH3CN produced 2 diastereomers of 14 (Scheme 2), as indicated in Figure 2. Two major peaks were observed by HPLC with UV detection and LCESI- MS/MS. The same 2 peaks were cleanly produced upon NaBH3CN treatment of the reaction mixture of 5′-acetoxyNNN and dGuo. Their structures were confirmed by reaction of nornicotine (26) with protected 2-fluoroinosine 23 (shown for (R)-26 in Scheme 4), producing 2 diastereomers of 14, which were characterized by UV, 1H-NMR, and MS. The 2 diastereomers of 14 were identical to those formed in the NaBH3CN reduction of synthetic 12 or in the reduction of 12 produced in the reaction of 5′-acetoxyNNN with dGuo. The absolute configuration of carbon 2 of the pyrrolidine ring of 14 (bearing the pyridine ring) was assigned by separate reactions of 2-(R)- and 2-(S)-nornicotine with protected 2-fluoroinosine 23 (shown for (R)-26 in Scheme 4). Thus, peak 1 in Figure 2 was assigned as the 2-(R)-isomer while peak 2 was assigned as the 2-(S)-isomer. Collectively, these results established the structure of the 23.6 min peak of Figure 1 as diastereomers of adduct 12.
Figure 1.
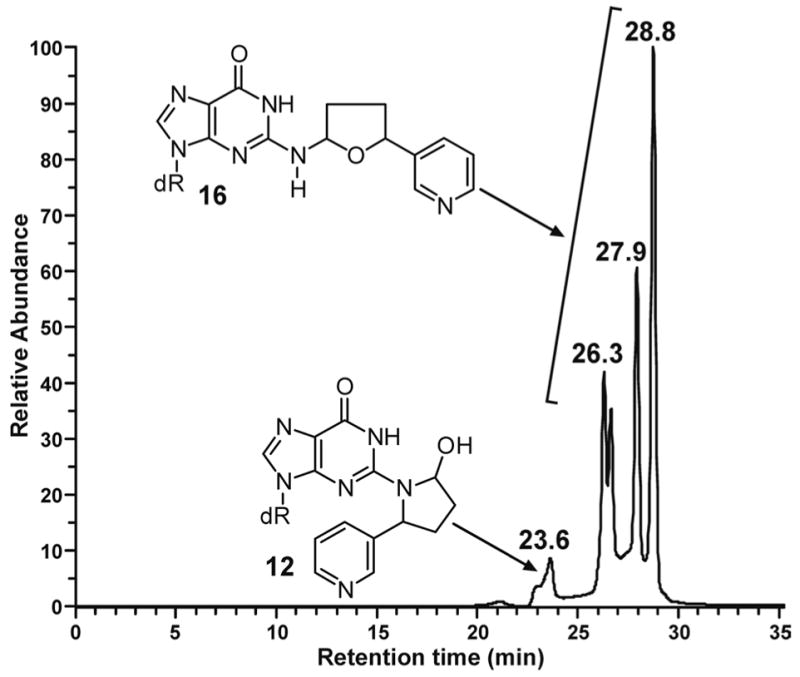
Chromatogram obtained upon LC-ESI-MS/MS-SRM analysis, at m/z 415→299 (system 7), of the products of reaction of 5′-acetoxyNNN (4) and dGuo
Scheme 3.

Synthesis of adduct 12
Figure 2.
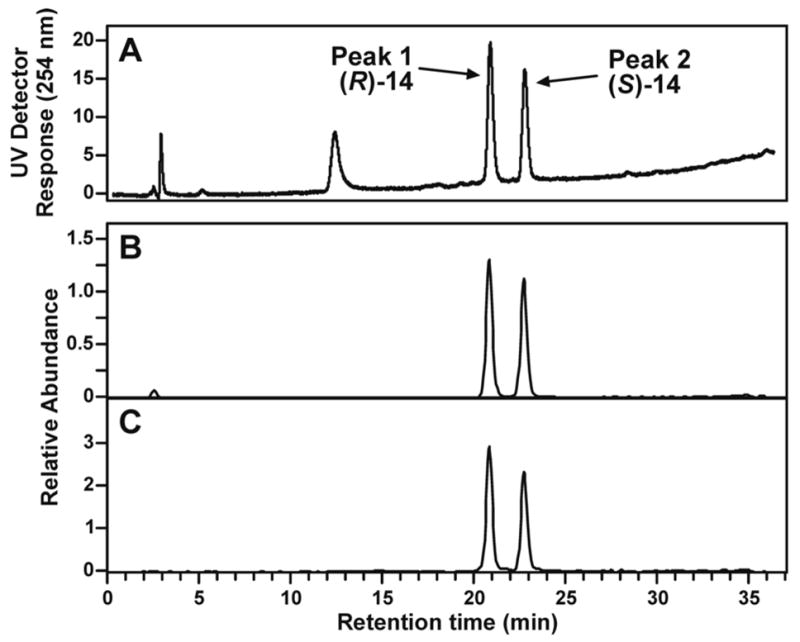
LC-ESI-MS/MS chromatograms (system 5) obtained upon analysis of the reaction of adduct 12 with NaBH3CN: A, detection by UV at 254 nm; B, extracted ion chromatogram of m/z 399 (M + H)+; C, extracted ion chromatogram of m/z 399→283 [(M-deoxyribose) + H]+
Scheme 4.
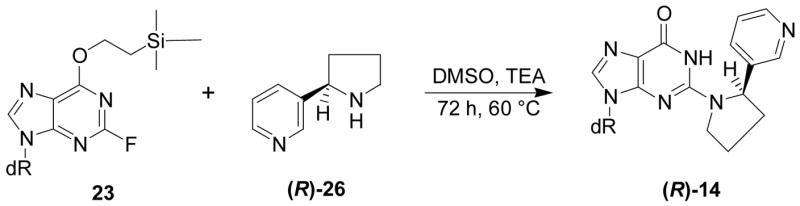
Synthesis of (R)-14
We then turned our attention to the identity of the four peaks eluting from 26.3–28.8 min in Figure 1, proposed to correspond to adduct 16 (Scheme 2). The UV spectra of these peaks are shown in Figure 3A-D. All were essentially identical and characteristic of N2-substituted dGuo derivatives. Standard adduct 16 was prepared by reaction of 5-(3-pyridyl)-2- hydroxytetrahydrofuran (27) with dGuo. LC-ESI-MS analysis of the product produced
Figure 3.
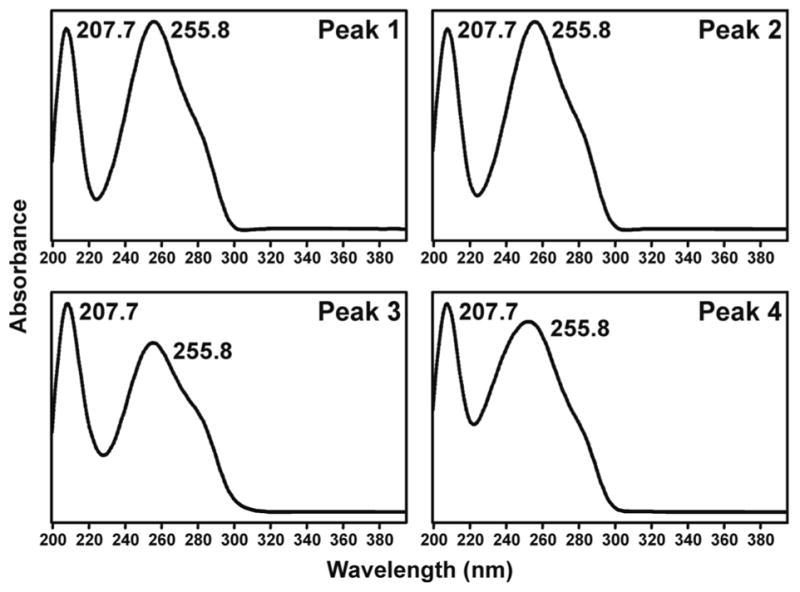
UV spectra of 4 diastereomers of adduct 16, corresponding to the 4 peaks eluting from 26.3–28.8 min in Figure 1
4 main peaks similar to those eluting from 26.3–28.8 min in Figure 1. The UV spectra of these peaks were the same as those shown in Figure 3A-D, from the reaction of 5′-acetoxyNNN with dGuo. The 1H-NMR spectrum of the four peaks was completely consistent with the overall structural assignment as 16.
Further evidence for the structural assignment of 16 was obtained by NaBH3CN treatment of this adduct, which was expected to be in equilibrium with Schiff base 17 (Scheme 2). This reaction produced 18, which was identical to a standard prepared by the reaction of keto aldehyde 28 with dGuo in the presence of NaBH3CN to give 29 (24), followed by reduction (Scheme 5). The identity of the products was established by LC-ESI-MS-selected ion monitoring (SIM) at m/z 417 (M + H)+, and by SRM of the transition m/z 417→ m/z 301. These results conclusively established the overall structure of adduct 16.
Scheme 5.
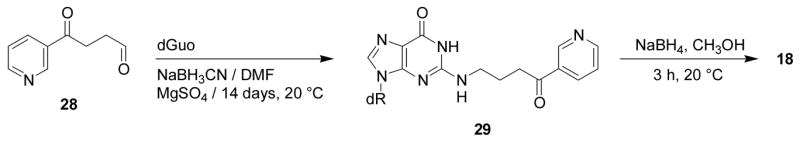
Synthesis of adduct 18
The stereochemistry of 16 was then investigated by synthesizing chiral lactols 27, as illustrated in Scheme 6. Acetal 30 was reduced with NaBH4 providing 31, which was allowed to react with (R)-(−)-α-methoxy-α-(trifluoromethyl)phenylacetic acid chloride. The resulting diastereomeric Mosher esters 32 were separated by HPLC. They were subjected to stepwise hydrolysis, first by base to remove the ester group and then by acid to remove the acetal, yielding hydroxy aldehydes which spontaneously cyclized to the enantiomeric lactols 27.
Scheme 6.

Synthesis and identification of diastereomers of 16
The absolute configurations of the enantiomers of lactol 27 were determined (Scheme 6). One enantiomer was oxidized with Ag2CO3 to give chiral lactone 33 which was hydrolyzed with acid yielding hydroxy acid 34. This was esterified and derivatized with (S)-α-methylbenzyl isocyanate, yielding a single diastereomer of methyl[α-methylbenzylcarbamoyl]-4-(3- pyridyl)butanoate (MMPB, 35). The HPLC retention time, UV, and MS of this diastereomer were compared to those of the previously characterized (R,S)-MMPB and (S,S)-MMPB (35) (22,23). As shown in Figure 4, standard (R,S)-MMPB elutes before (S,S)-MMPB. The diastereomer formed from this enantiomer of lactol 27 co-eluted with the first eluting standard, and was therefore assigned as (R,S)-MMPB, formed from 5-(R)-lactol 27.
Figure 4.

Chromatogram obtained upon HPLC analysis (using the in-line UV detector of LC-ESI-MS system 5) of standard (R,S)- and (S,S)-MMPB (35). (Note: the assignments of (R,S)-MMPB and (S,S)-MMPB in references (22,23) are reversed because they are based on the incorrect assignment of the absolute configuration of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (20,35))
Each chiral lactol 27, of known absolute configuration, was allowed to react with dGuo as shown in Scheme 6, giving adducts 16 with known absolute configuration at the 5-position of the tetrahydrofuran ring. Using these standards and optimized conditions for separation, peaks 1 and 3 of Figure 5 corresponded to the 5-(S)-diastereomers while peaks 2 and 4 corresponded to the 5-(R)-diastereomers. Peaks 1 and 3 and peaks 2 and 4 were interconvertible. Thus, collection of peak 1 from HPLC system 1 and re-injection within 2 h at room temperature produced a mixture of peaks 1 and 3. Similarly, collected peak 2, when re-injected, gave a mixture of peaks 2 and 4. The corresponding peaks in Figure 1 behaved similarly. These results are consistent with the equilibrium involving Schiff base 17, as shown in Scheme 2, and impedes determination of the absolute configuration at the 2-position of adduct 16.
The formation of adducts 12 and 16 in DNA reacted with 5′-acetoxyNNN was then investigated. The DNA was treated with NaBH3CN and enzymatically hydrolyzed. The hydrolysates were analyzed by LC-ESI-MS/MS-SRM. As shown in Figure 6, clear peaks were observed in the chromatograms corresponding to 14 and 18, the reduced forms of adducts 12 and 16, respectively.
Figure 6.
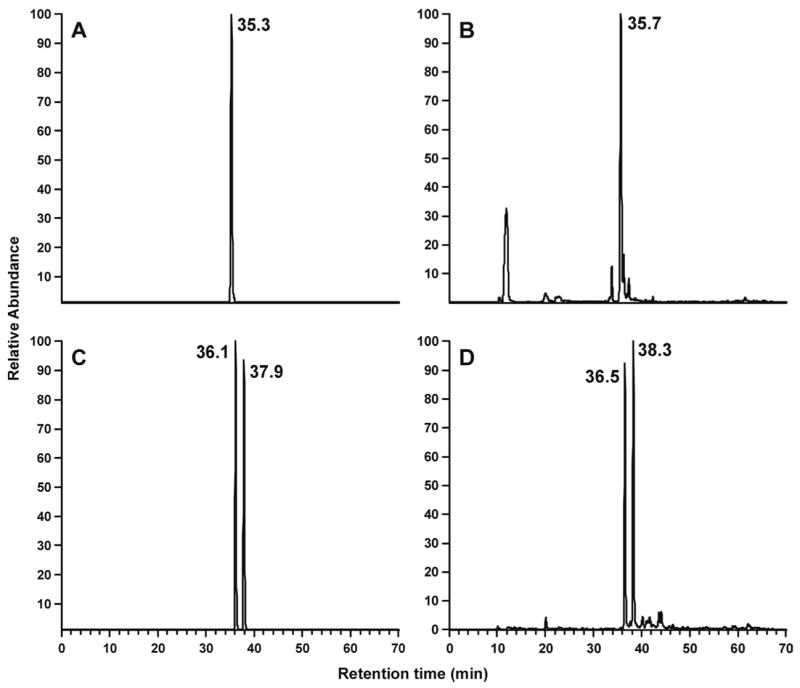
Chromatograms obtained upon LC-ESI-MS/MS-SRM analysis (system 7) of A, standard adduct 18, m/z 417→301; C, standard adduct 14, m/z 399→283; enzymatic hydrolysates of DNA that had been reacted with 5′-acetoxyNNN and treated with NaBH3CN, analyzed for B, m/z 417→301 (adduct 18) and D, m/z 399→283 (adduct 14)
Discussion
This paper reports the first structural characterization of dGuo and DNA adducts formed from NNN 5′-hydroxylation, a known major metabolic pathway of this tobacco-specific carcinogen in primates and humans. Adducts 12 and 16 are formed by reactions of intermediates derived from NNN 5′-hydroxylation- oxonium ions, carbocations, or related species- with the exocyclic amino group of dGuo. Since adducts 12 and 16 introduce a relatively bulky residue into the base pairing region of DNA, it appears likely that they will lead to miscoding or related negative consequences during DNA replication. Such changes are the critical initial steps in the cascade of events leading to tumor formation.
The absolute configurations of the pyridyl-substituted carbon atom in adduct 16 and in 14, the reduced form of adduct 12, were determined in this study. In the case of 16, this was accomplished by establishing the absolute configuration of the pyridyl-substituted carbon in lactol 27, by conversion to MMPB (35) of known configuration, followed by reaction of 27 with dGuo. For 14, the absolute configuration of the pyridyl- substituted carbon was determined by reaction of chiral nornicotine with protected 2- fluoroinosine 23. While few previous studies of nitrosamine-DNA adducts have elaborated absolute stereochemistry (16,25,26), this has been an area of extensive research in the study of DNA adducts of polycyclic aromatic hydrocarbons (27–29). The results of those studies clearly demonstrate that the absolute stereochemistry of DNA adducts is critical in determining their mutagenic and carcinogenic effects. NNN itself is a chiral molecule, and it is known that (S)-NNN is more prevalent in tobacco products than (R)-NNN (30). Further, there are clear differences in cytochrome P450-catalyzed 5′-hydroxylation of the NNN enantiomers (31) as well as in their metabolism in vitro and in vivo in rats (32). Thus, the stereochemical assignments determined here will be important in elucidating the relationship between exposure to NNN enantiomers, their metabolism, and consequent DNA adduct formation.
The structures of adducts 12 and 16 are consistent with observations in our previous studies of N-nitrosopyrrolidine, in which the corresponding adducts, lacking the pyridine ring, were identified in reactions with dGuo and DNA of α-acetoxy-N-nitrosopyrrolidine (16). Adduct 12 and the corresponding adduct of α-acetoxy-N-nitrosopyrrolidine are structurally interesting because they represent the formal replacement of the N-nitroso- group of the α-hydroxy metabolite with deoxyinosine (although the ring nitrogen in the adduct is provided by dGuo). While adducts such as these which are in equilibrium with Schiff bases can be unstable at the nucleoside level, some studies show that Schiff base adducts are quite stable in DNA (25,33). The chemical stability in DNA and persistence of the adducts isolated here will be evaluated in future studies.
The relative importance of 2′-hydroxylation versus 5′-hydroxylation of NNN has been discussed previously (4). Studies in rats favor 2′-hydroxylation as the major metabolic activation pathway of NNN. Thus, 2′-hydroxylation exceeds 5′-hydroxylation in target tissues of NNN in the rat- esophagus and nasal mucosa- while in liver, a non-target tissue, 5′-hydroxylation exceeds 2′-hydroxylation. Hemoglobin and DNA adducts resulting from 2′-hydroxylation have been detected in NNN-treated rats. However, in mouse lung and hamster trachea, target tissues of NNN carcinogenicity, 5′-hydroxylation exceeds or is comparable to 2′-hydroxylation. 5′-Hydroxylation of NNN exceeds 2′-hydroxylation in human liver and in several other human tissues. The relative importance of these two pathways can be more definitively established by determining the levels, persistence, and miscoding potential of specific NNN-DNA adducts. The results presented here are an important step in that direction.
LC-ESI-MS/MS was critical for the isolation and identification of adducts 12 and 16. Reactions of 5′-acetoxyNNN with dGuo and DNA proceed in low yield and it is difficult to identify the products using LC with UV detection because they are overwhelmed by even minor solvolysis products of 5′-acetoxyNNN, all of which have UV absorption of similar intensities to those of the dGuo adducts. LC-ESI-MS/MS on the other hand provides both the required sensitivity and specificity. As currently available instruments routinely have detection limits for DNA adducts in the amol range, it seems likely that we will be able to investigate the presence of adducts 12 and 16 in DNA from tissues of people who use tobacco products and are exposed to several micrograms of NNN daily (7,34). We envision development of a comprehensive MS method for analysis of DNA adducts of both NNN and NNK. Adducts 12 and 16 will be important in this analysis, as they are the only adducts known to date which specifically arise from NNN. All other known adducts can be formed by both methyl hydroxylation of NNK and 2′-hydroxylation of NNN (9,11).
In summary, DNA adducts resulting from the major pathway of NNN metabolism observed in human tissues- 5′-hydroxylation- were characterized for the first time, and the absolute configurations of the pyridyl-substituted carbons were determined. Adducts 12 and 16 are formed by reaction of intermediates derived from NNN 5′-hydroxylation- oxonium ions, carbocations, or related species- with the exocyclic amino group of dGuo. These DNA adducts, which are unique to the tobacco-specific carcinogen NNN, will be important probes for understanding mechanisms of cancer induction by NNN and as potential biomarkers for metabolic activation of NNN in people who use tobacco products.
Acknowledgments
This study was supported by grant CA-81301 from the National Cancer Institute. Stephen S. Hecht is an American Cancer Society Research Professor, supported by grant no RP-00-138. Mass spectrometry was carried out in the Analytical Biochemistry Facility of the Cancer Center, supported in part by Cancer Center Support Grant CA-77598. We thank Yanbin Lao for assistance in obtaining NMR spectra.
References
- 1.Henningfield JE, Benowitz NL. Pharmacology of nicotine addiction. In: Boyle P, Gray J, Henningfield J, Seffrin J, Zatonski W, editors. Tobacco: Science, Policy and Public Health. Oxford University Press; 2004. pp. 129–147. [Google Scholar]
- 2.Hecht SS. Tobacco carcinogens, their biomarkers, and tobacco-induced cancer. Nature Rev Cancer. 2003;3:733–744. doi: 10.1038/nrc1190. [DOI] [PubMed] [Google Scholar]
- 3.Hecht SS, Hoffmann D. Tobacco-specific nitrosamines, an important group of carcinogens in tobacco and tobacco smoke. Carcinogenesis. 1988;9:875–884. doi: 10.1093/carcin/9.6.875. [DOI] [PubMed] [Google Scholar]
- 4.Hecht SS. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem Res Toxicol. 1998;11:559–603. doi: 10.1021/tx980005y. [DOI] [PubMed] [Google Scholar]
- 5.Hecht SS, Rivenson A, Braley J, DiBello J, Adams JD, Hoffmann D. Induction of oral cavity tumors in F344 rats by tobacco-specific nitrosamines and snuff. Cancer Res. 1986;46:4162–4166. [PubMed] [Google Scholar]
- 6.Hecht SS, Hoffmann D. The relevance of tobacco-specific nitrosamines to human cancer. Cancer Surv. 1989;8:273–294. [PubMed] [Google Scholar]
- 7.International Agency for Research on Cancer. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Vol. 89. IARC; Lyon, FR: 2005. Smokeless tobacco and tobacco-specific nitrosamines. in press. [PMC free article] [PubMed] [Google Scholar]
- 8.Preussmann R, Stewart BW. N-Nitroso Carcinogens. In: Searle CE, editor. Chemical Carcinogens. 2. Vol. 2. American Chemical Society; Washington, DC: 1984. pp. 643–828. ACS Monograph 182. [Google Scholar]
- 9.Hecht SS, Villalta PW, Sturla SJ, Cheng G, Yu N, Upadhyaya P, Wang M. Identification of O2-substituted pyrimidine adducts formed in reactions of 4- (acetoxymethylnitrosamino)-1-(3-pyridyl)-1-1butanone and 4-(acetoxymethylnitrosamino)- 1-(3-pyridyl)-1-butanol with DNA. Chem Res Toxicol. 2004;17:588–597. doi: 10.1021/tx034263t. [DOI] [PubMed] [Google Scholar]
- 10.Upadhyaya P, Sturla S, Tretyakova N, Ziegel R, Villalta PW, Wang M, Hecht SS. Identification of adducts produced by the reaction of 4- (acetoxymethynitrosamino)-1-(3-pyridyl)-1-butanol with deoxyguanosine and DNA. Chem Res Toxicol. 2003;16:180–190. doi: 10.1021/tx0256376. [DOI] [PubMed] [Google Scholar]
- 11.Wang M, Cheng G, Sturla SJ, Shi Y, McIntee EJ, Villalta PW, Upadhyaya P, Hecht SS. Identification of adducts formed by pyridyloxobutylation of deoxyguanosine and DNA by 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone, a chemically activated form of tobacco-specific carcinogens. Chem Res Toxicol. 2003;16:616–626. doi: 10.1021/tx034003b. [DOI] [PubMed] [Google Scholar]
- 12.Chen CB, Hecht SS, Hoffmann D. Metabolic α-hydroxylation of the tobacco specific carcinogen, N′-nitrosonornicotine. Cancer Res. 1978;38:3639–3645. [PubMed] [Google Scholar]
- 13.Staretz ME, Murphy SE, Nunes MG, Koehl W, Amin S, Koenig L, Guengerich FP, Hecht SS. Comparative metabolism of the tobacco smoke carcinogens benzo[a]pyrene, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, 4-(methylnitrosamino)- 1-(3-pyridyl)-1-butanol, and N′-nitrosonornicotine in human hepatic microsomes. Drug Metab Dispos. 1997;25:154–162. [PubMed] [Google Scholar]
- 14.Upadhyaya P, Zimmerman CL, Hecht SS. Metabolism and pharmacokinetics of N′-nitrosonornicotine in the patas monkey. Drug Metab Dispos. 2002;30:1115–1122. doi: 10.1124/dmd.30.10.1115. [DOI] [PubMed] [Google Scholar]
- 15.Hecht SS, Chen CB. Hydrolysis of model compounds for α-hydroxylation of the carcinogens, N-nitrosopyrrolidine and N′-nitrosonornicotine. J Organ Chem. 1979;44:1563–1566. [Google Scholar]
- 16.Wang M, McIntee EJ, Shi Y, Cheng G, Upadhyaya P, Villalta PW, Hecht SS. Reactions of α-acetoxy-N-nitrosopyrrolidine with deoxyguanosine and DNA. Chem Res Toxicol. 2001;14:1435–1445. doi: 10.1021/tx010097i. [DOI] [PubMed] [Google Scholar]
- 17.Deo NM, Crooks PA. Regioselective alkylation of N-(diphenylmethyidine)- 3-(aminomethyl)pyridine: a simple route to minor tobacco alkaloids and related compounds. Tetrahedron Lett. 1996;37:1137–1140. [Google Scholar]
- 18.Seeman JI, Chaudarian CG, Secor HV. Synthesis of the enantiomers of nornicotine. J Organ Chem. 1985;50:5419–5421. [Google Scholar]
- 19.Loozen HJJ, Godefroi EF, Besters JSMM. A novel and efficient route to 5-arylated gamma-lactones. J Organ Chem. 1975;40:892–894. [Google Scholar]
- 20.Hecht SS, Spratt TE, Trushin N. Absolute configuration of 4- (methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) formed metabolically from 4- (methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) Carcinogenesis. 1997;18:1851–1854. doi: 10.1093/carcin/18.9.1851. [DOI] [PubMed] [Google Scholar]
- 21.Moore JA, Schwab JM. Unprecedented observation of lactone hydrolysis by the AA12 mechanism. Tetrahedron Lett. 1991;32:2331–2334. [Google Scholar]
- 22.Trushin N, Hecht SS. Stereoselective metabolism of nicotine and tobacco-specific N-nitrosamines to 4-hydroxy-4-(3-pyridyl)butanoic acid in rats. Chem Res Toxicol. 1999;12:164–171. doi: 10.1021/tx980213q. [DOI] [PubMed] [Google Scholar]
- 23.Hecht SS, Hatsukami DK, Bonilla LE, Hochalter JB. Quantitation of 4-oxo-4-(3-pyridyl)butanoic acid and enantiomers of 4-hydroxy-4-(3-pyridyl)butanoic acid in human urine: a substantial pathway of nicotine metabolism. Chem Res Toxicol. 1999;12:172–179. doi: 10.1021/tx980214i. [DOI] [PubMed] [Google Scholar]
- 24.Spratt TE, Trushin N, Lin D, Hecht SS. Analysis for N2- (pyridyloxobutyl)deoxyguanosine adducts in DNA of tissues exposed to tritium-labeled 4- (methylnitrosamino)-1-(3-pyridyl)-1-butanone and N′-nitrosonornicotine. Chem Res Toxicol. 1989;2:169–173. doi: 10.1021/tx00009a008. [DOI] [PubMed] [Google Scholar]
- 25.Wang M, McIntee EJ, Cheng G, Shi Y, Villalta PW, Hecht SS. A Schiff base is a major DNA adduct of crotonaldehyde. Chem Res Toxicol. 2001;14:423–430. doi: 10.1021/tx000234w. [DOI] [PubMed] [Google Scholar]
- 26.Nechev LV, Kozekov I, Harris CM, Harris TM. Stereospecific synthesis of oligonucleotides containing crotonaldehyde adducts of deoxyguanosine. Chem Res Toxicol. 2001;14:1506–1512. doi: 10.1021/tx0100690. [DOI] [PubMed] [Google Scholar]
- 27.Geacintov NE, Cosman M, Hingerty BE, Amin S, Broyde S, Patel DJ. NMR solution structures of stereoisomeric covalent polycyclic aromatic carcinogen - DNA adducts: principles, patterns, and diversity. Chem Res Toxicol. 1997;10:112–146. doi: 10.1021/tx9601418. [DOI] [PubMed] [Google Scholar]
- 28.Szeliga J, Dipple A. DNA adduct formation by polycyclic aromatic hydrocarbon dihydrodiol epoxides. Chem Res Toxicol. 1998;11:1–11. doi: 10.1021/tx970142f. [DOI] [PubMed] [Google Scholar]
- 29.Tan J, Geacintov NE, Broyde S. Principles governing conformations in stereoisomeric adducts of bay region benzo[a]pyrene diol epoxides to adenine in DNA: Steric and hydrophobic effects are dominant. J Am Chem Soc. 2000;122:3021–3032. [Google Scholar]
- 30.Carmella SG, McIntee EJ, Chen M, Hecht SS. Enantiomeric composition of N′-nitrosonornicotine and N′-nitrosoanatabine in tobacco. Carcinogenesis. 2000;21:839–843. doi: 10.1093/carcin/21.4.839. [DOI] [PubMed] [Google Scholar]
- 31.Wong HL, Murphy SE, Hecht SS. Cytochrome P450 2A-catalyzed metabolic activation of structurally similar carcinogenic nitrosamines: N′-nitrosonornicotine enantiomers, N-nitrosopiperidine, and N-nitrosopyrrolidine. Chem Res Toxicol. 2004;18:61–69. doi: 10.1021/tx0497696. [DOI] [PubMed] [Google Scholar]
- 32.McIntee EJ, Hecht SS. Metabolism of N′-nitrosonornicotine enantiomers by cultured rat esophagus and in vivo in rats. Chem Res Toxicol. 2000;13:192–199. doi: 10.1021/tx990171l. [DOI] [PubMed] [Google Scholar]
- 33.Wang M, McIntee EJ, Cheng G, Shi Y, Villalta PW, Hecht SS. Reactions of 2,6-dimethyl-1,3-dioxane-4-ol (aldoxane) with deoxyguanosine and DNA. Chem Res Toxicol. 2001;14:1025–1032. doi: 10.1021/tx0100385. [DOI] [PubMed] [Google Scholar]
- 34.International Agency for Research on Cancer. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Vol. 83. IARC; Lyon, FR: 2004. Tobacco Smoke and Involuntary Smoking; pp. 59–94. [PMC free article] [PubMed] [Google Scholar]
- 35.Hecht SS, Spratt TE, Trushin N. Absolute configuration of 4- (methylnitrosamino)-1-(3-pyridyl)-1-butanol formed metabolically from 4- (methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis. 2000;21:850. [PubMed] [Google Scholar]