

Abstract
Chronic lymphocytic leukemia (CLL) is the most common adult leukemia, and resistance to fludarabine-based therapies is a major challenge in CLL treatment. Because CLL cells are known to have elevated levels of reactive oxygen species (ROS), we aimed to test a novel ROS-mediated strategy to eliminate fludarabine-resistant CLL cells based on this redox alteration. Using primary CLL cells and normal lymphocytes from patients (n = 58) and healthy subjects (n = 12), we showed that both fludarabine-resistant and -sensitive CLL cells were highly sensitive to β-phenylethyl isothiocyanate (PEITC) with mean IC50 values of 5.4 μM and 5.1 μM, respectively. Normal lymphocytes were significantly less sensitive to PEITC (IC50 = 27 μM, P < .001). CLL cells exhibited intrinsically higher ROS level and lower cellular glutathione, which were shown to be the critical determinants of CLL sensitivity to PEITC. Exposure of CLL cells to PEITC induced severe glutathione depletion, ROS accumulation, and oxidation of mitochondrial cardiolipin leading to massive cell death. Such ROS stress also caused deglutathionylation of MCL1, followed by a rapid degradation of this cell survival molecule. Our study demonstrated that the natural compound PEITC is effective in eliminating fludarabine-resistant CLL cells through a redox-mediated mechanism with low toxicity to normal lymphocytes, and warrants further clinical evaluation.
Introduction
Chronic lymphocytic leukemia (CLL) is manifested by progressive accumulation of morphologically mature but immunologically dysfunctional B lymphocytes. CLL is the most common adult leukemia in the western countries. A hallmark of CLL cells is their resistance to apoptosis, leading to prolonged cell survival and development of drug resistance.1 The use of fludarabine-based therapies and, more recently, the availability of therapeutic antibodies have improved treatment outcomes.1 However, CLL remains incurable. Although currently available agents such as fludarabine, cyclophosphamide, and rituximab are effective in inducing complete remission, many patients eventually develop drug resistance leading to disease relapse.2,3 In particular, CLL cells with unfavorable cytogenetic alterations such as deletion of chromosome 17p with loss of p53 are often resistant to fludarabine and cyclophosphamide.4,5 Similarly, CLL cells from patients in advanced disease stages or with a history of prior chemotherapy exhibit elevated oxidative stress,6 and thus may have a greater potential to acquire additional mutations and genetic abnormalities, leading to drug resistance and disease progression. This presents a significant challenge in treatment of CLL. Thus, it is important to identify novel agents capable of killing drug-resistant CLL cells with low toxicity to normal cells.
One logical strategy to develop new anticancer agents with high therapeutic selectivity is to exploit the biologic difference between normal and cancer cells. Previous studies suggest that many cancer cells exhibit increased production of reactive oxygen species (ROS) associated with oncogenic transformation and mitochondrial dysfunction.7–9 Compared with normal lymphocytes, CLL cells were shown to display a substantial increase in ROS associated with oxidative DNA damage and mitochondrial DNA (mtDNA) mutations, especially in patients who had undergone prior therapy with DNA-damaging agents.10–13 Because mitochondrial respiratory chain is the major site of ROS generation due to electron bifurcation from the transport complexes, it is conceivable that dysfunction of mitochondrial respiration would increase electron leakage and lead to elevated ROS generation. Interestingly, the loss of p53 seems to promote mtDNA mutations and enhance ROS production in cultured cell lines.14 Multiple mechanisms, including mitochondrial dysfunction and metabolic stress, likely contribute to ROS stress in CLL cells. From the therapeutic standpoint, one important question is how to use the elevated ROS stress in CLL cells to develop effective new strategies to kill the leukemia cells. Although the increase of ROS in cancer cells is often viewed as an adverse event due to its role in promoting genomic instability and cell proliferation,9,15,16 high levels of ROS can also induce cancer cell death, a desired outcome that chemotherapy attempts to achieve. This may provide a rationale to exploit the intrinsic oxidative stress to develop new strategies that turn the toxic effect of ROS against CLL cells, using proper redox-modulating agents.
Our recent study showed that the natural compound PEITC effectively disabled the glutathione antioxidant system and preferentially killed ovarian cancer cells with increased ROS generation under the influence of Ras oncogenic signal, but exhibited low toxicity to nontumorigenic ovarian epithelial cells with low ROS output.17 Based on these observations, we hypothesized that CLL cells, due to their significant elevation of ROS generation, are likely to be highly dependent on the antioxidant system to keep redox balance, and that these cells would be particularly sensitive to PEITC. On the other hand, normal lymphocytes may better tolerate PEITC treatment, because of their low basal ROS output and normal metabolic regulation. Importantly, because the fludarabine-resistant CLL cells in advanced disease stages, with prior chemotherapy, or without p53 function tend to have higher ROS, these cells are likely to be sensitive to PEITC. The current study was designed to test these possibilities and to evaluate the therapeutic selectivity of PEITC and its potential for use in treatment of refractory CLL.
Methods
Reagents
PEITC, 3-(4,5dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT), N-acetylcysteine (NAC), 9-β-D-arabinofuranosyl-2-fluoro-adenine (F-ara-A, the nucleoside form of fludarabine), cumene hydroperoxide, glutathione peroxidase, NADH (reduced nicotinamide adenine dinucleotide), NADPH (reduced nicotinamide adenine dinucleotide phosphate), glutathione (GSH [reduced glutathione] and GSSG [glutathione disulfide]), 2-vinylpyridine, metaphosphoric acid, and propidium iodide (PI) were purchased from Sigma-Aldrich (St Louis, MO). Ficoll-lite Lympho H was from Atlanta Biological (Lawrenceville, GA). CM-H2DCF-DA, nonyl acridine orange (NAO) and CM-FDA were purchased from Invitrogen (Carlsbad, CA). Z-VAD-fmk, Z-DEVD-fmk, annexin V–fluorescein isothiocyanate (annexin V–FITC), caspase-3 activity assay kit, recombinant active caspase-3 and antibodies for poly (ADP-ribose) polymerase (PARP) and BCL2 were from BD Biosciences (San Jose, CA). Annexin V–phycoerythrin (annexin V–PE) was from CalTag Laboratories (Burlingame, CA). BCA protein assay kit was from Pierce Biotechnology (Rockford, IL). MG-132 was from EMD Biosciences/Calbiochem (San Diego, CA). PEITC was dissolved in dimethyl sulfoxide (DMSO) and freshly diluted in culture media before use. The final DMSO concentration in media was less than 0.1% (vol/vol). Antibody against gamma-glutamyl cysteine synthetase (GSH1) and MCL1 were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti–caspase-3 was from Cell Signaling Technology (Danvers, MA). Antibody to GSH was from Virogen (Watertown, MA).
Isolation of CLL cells and normal lymphocytes
Primary leukemia cells were isolated from the peripheral blood samples of 58 CLL patients diagnosed according to National Cancer Institute criteria.18 Proper informed consents under a research protocol approved by the Institutional Review Board of M. D. Anderson Cancer Center were obtained from all patients in accordance with the Declaration of Helsinki before sample collection. Specimens from CLL patients with or without prior therapy, newly diagnosed or relapsed, were all included. Patient demographic information and relevant clinical parameters are listed in Table S1 (available on the Blood website; see the Supplemental Materials link at the top of the online article). CLL cells were isolated from blood samples by density gradient centrifugation19 and incubated in RPMI 1640 medium supplemented with 10% FBS and penicillin (100 U/mL) + streptomycin (100 μg/mL) overnight before drug incubations were started. Normal lymphocytes were isolated from blood samples of 12 healthy donors using similar procedures.
Cytotoxicity assays
Drug-induced cell death was determined by flow cytometry after double-staining 106 cells with annexinV–FITC and PI as described.11 Incubation of CLL cells in fresh medium for 48 hours resulted in 5% to 30% spontaneous apoptosis, consistent with previous observations.20 Therefore, in all experiments, control samples at each time points were collected and the drug-induced apoptosis was calculated by subtracting “spontaneous” apoptosis from total apoptosis. To determine the drug effect on cell viability, cells were seeded in 96-well plates at 5 × 105 cells/well (200 μL), and cell viability after 72 hours of drug exposure was determined using MTT assay. For comparison between CLL cells and normal lymphocytes, the drug incubation time was reduced to 30 hours to minimize the influence of spontaneous apoptosis, which increased with culture time. The selective cytotoxic effect of PEITC against CLL was observed in both 30-hour and 72-hour incubations.
Detection of ROS
Cellular ROS contents were measured by incubating cells (5 × 105 cells) with 1 μM CM-H2DCF-DA for 60 minutes and analyzed by flow cytometry as described.17 To ensure the consistency of the assay conditions for patient samples at different days, we used a reference cell line (Raji cells) stained under identical conditions as a control for comparable parameter settings.13
Analysis of cellular glutathione and glutathione peroxidase enzyme activity
A glutathione assay kit (Cayman Chemical, Ann Arbor, MI) was used to measure cellular glutathione. After preparing cell extracts by sonication and deproteination, GSH and GSSG were determined as described.17 Cellular GPX enzyme activity was assayed using continuous spectrophotometric rate determination.17,21 Cellular protein extracts were added to a mixture of 2 mM GSH, 0.1 U glutathione reductase and 0.2 mg/mL NADPH, and reaction was initiated by adding cumene hydroperoxide (1 mM). Glutathione peroxidase (GPX) activity was calculated based on the slope of NADPH oxidation (ΔA340 nm/min), the coefficient of NADPH, the protein concentrations, and the dilution factors.
Assays of mitochondrial oxidative damage, cytochrome c release, and caspase-3 activation
Mitochondrial membrane lipid oxidation was detected by measuring the oxidation of cardiolipin, using NAO as a fluorescence dye.22 A cytochrome c release assay kit (EMD Biosciences/Calbiochem) was used to measure the loss of mitochondrial cytochrome c according to the manufacturer's recommended procedures. A caspase-3 activation assay kit (BD Biosciences) was used to determine the levels of activated caspase-3. After cells were incubated with or without PEITC, 106 cells/sample were permeabilized, fixed, and stained with FITC-conjugated antibody specific for activated caspase-3 and analyzed by flow cytometry. Procaspase-3 and activated caspase-3 were also detected by Western blot analysis.
Assays of MCL1 expression, cleavage, and glutathionylation
MCL1 and BCL2 protein levels were determined by Western blotting. An in vitro assay was used to determine the cleavage of MCL1 by caspases. Cell lysates were prepared by sonicating 5 × 107 CLL cells in 200 μL PBS containing 0.1% CHAPS and the samples were centrifuged at 5000 × g for 10 minutes to remove unbroken cells and debris. The lysates were dialyzed in cold PBS using Slide-A-Lyzer Dialysis Cassettes with 3-kDa cutoff (Pierce Biotechnology) to remove endogenous glutathione. To test the effect of glutathionylation on cleavage of MCL1, the dialyzed lysates were diluted in reaction buffer (40 mM HEPES and 20% glycerol) and pre-incubated with 2 mM GSSG, 2 mM GSH, 0.5 mM NADH, or 0.5 mM NADPH at 37°C for 10 minutes. Then, 40 ng of recombinant caspase-3 (prewarmed 5 minutes at 37°C) was added and incubated for 1 hour. The reactions were stopped by addition of 2× reducing Laemni buffer. Cleavage of MCL1 and PARP were detected by Western blot analysis. Cellular glutathionylation of MCL1 was determined by immunoprecipitation with an anti-GSH antibody under nonreducing conditions, followed by Western blot analysis,23 using anti-MCL1 antibody.
Determination of thiol contents in whole cells
To monitor cellular thiols, dual staining with CellTracker Green 5-chloro-methylfluorescein diacetate (CMFDA) for thiols24 and annexin V–PE for cell death were performed and analyzed by flow cytometry. Because CMF fluorescence is stable in the cells for at least 72 hours, we stained cells with CMFDA before annexin-PE. Briefly, after incubation with 20 μM F-ara-A for 72 hours, 106 cells were washed with PBS twice to remove thiols in media before staining with 0.5 μM CMFDA in PBS for 15 minutes. Then, the cells were washed once with PBS, stained with 5 μL of annexin V–PE for 15 minutes, washed, and resuspended in 500 μL binding buffer for dual-color (red/green) flow cytometric analysis. The population of healthy cells (annexin V–negative) was gated, and the thiol levels of the gated population were quantified in the histogram of CMFDA.
Statistical analyses
Bar graphs and plots were generated using Prizm software (GraphPad, San Diego, CA). Data were expressed as means plus or minus 95% confidence interval (CI). Unpaired t test was used to compare the mean values of different experimental groups. Paired t test was used to compare control and drug-treated samples from the same patient. A nonparametric test (Mann-Whitney) was used to test the difference between 2 groups when the data contained one or more zero values. One-way analysis of variance (ANOVA) was used to compare the mean values of the control samples with that of the tested samples with multiple time points. Histograms from flow cytometry were obtained and analyzed using the CellQuestPro software. Correlation between ROS levels and drug sensitivity was analyzed using spearman correlation analysis. P values less than .05 were considered statistically significant.
Results
PEITC effectively eliminates fludarabine-resistant CLL cells with minimal toxicity to normal lymphocytes
We first used MTT assay to compare the drug sensitivity of primary CLL cells isolated from the blood samples of CLL patients. As illustrated in Figure 1, the sensitivity of primary CLL cells to F-ara-A (the active form of fludarabine for in vitro incubation) was highly heterogeneous, with the IC50 values (defined as the concentration that caused 50% loss of cell viability) ranged from 0.6 μM to 106 μM. Among the 40 patient samples tested in this fashion, 22 samples were considered F-ara-A–sensitive (IC50 < 10μM), and 18 samples were F-ara–resistant (IC50 > 10 μM). Figure 1A compared the dose-dependent loss of cell viability of 4 representative F-ara–sensitive CLL samples (in blue) and 4 representative F-ara-A–resistant CLL samples (in red) after treatment with F-ara-A (Figure 1A left panel) or PEITC (Figure 1A right panel). The dose response to PEITC in the F-ara-A–sensitive CLL cells was almost identical to that of F-ara–resistant CLL cells. The quantitative cytotoxic data for each CLL patient sample are compared in Figure 1B. The F-ara-A IC50 values for the F-ara-A–sensitive and –resistant groups were 3.4 (± 0.8) μM and 38.6 (± 13.1) μM, respectively (P < .001). In contrast, both F-ara-A–sensitive and –resistant CLL cells were equally sensitive to PEITC, with the IC50 values of 5.1 (± 0.5) μm and 5.4 (± 0.5) μm, respectively (P = .39).
Figure 1.

Cytotoxic effect of PEITC in fludarabine-sensitive and resistant CLL cells. (A) Cytotoxicity of PEITC and F-ara-A in primary CLL cells, after 72-hour incubation detected by MTT assay. Results of 8 representative patient samples (4 F-ara-A–sensitive CLL in blue, S-1 to S-4; 4 F-ara-A–resistant CLL in red, R-1 to R-4) are shown. Each data point represents the mean of triplicate determinations. (B) Comparison between the concentrations required to cause a loss in cell viability by 50% (IC50) of F-ara-A and PEITC in F-ara-A–sensitive (n = 22) and –resistant (n = 18) CLL cells. Cells with an IC50 < 10 μM F-ara-A were considered fludarabine-sensitive, whereas those with an IC50 > 10 μM were considered fludarabine-resistant.
The promising cytotoxic activity of PEITC against F-ara-A–resistant CLL cells led us to test whether this compound is toxic to normal lymphocytes, using both annexin V/PI and MTT assays. As shown in Figure 2A,B, normal lymphocytes were significantly less sensitive to PEITC (IC50 = 27 μM, n = 11) compared with primary CLL cells (IC50 = 4.8 μM, n = 13; P < .001). In Figure 2A, 10 μM PEITC, which caused almost complete loss of CLL cell viability in a 30-hour incubation, was minimally toxic to normal lymphocytes (80% viability). Determination of cytotoxicity of PEITC at 72 hours also confirmed that normal lymphocytes are significantly less sensitive to this compound (IC50: 24.5-43.5 μM) compared with CLL cells during the same incubation (72-hour IC50: approximately 4.5 μM, Figure S1). Similarly, annexin-V/PI staining showed that incubation with 5 μM PEITC for 24 hours causes more than 70% cell death (n = 18), whereas significantly less cell death (18%) was observed in normal lymphocytes (n = 7, P < .001, Figure 2C,D). These suggest that PEITC preferentially killed CLL cells. Based on our previous observation that PEITC causes glutathione depletion in ovarian cancer cells,17 we speculated that PEITC might disturb redox homeostasis and induce ROS accumulation in CLL cells. Indeed, incubation with 5 μM PEITC for 2 hours resulted in a considerable accumulation of ROS in CLL cells but only a moderate ROS increase in normal lymphocytes (Figure 2E), indicating a substantial difference between the 2 cell types in their redox regulation and responses to oxidative stress.
Figure 2.

Selective killing of primary CLL cells by PEITC. (A) Cytotoxicity of PEITC in primary CLL cells (n = 6, black solid symbols) and normal lymphocytes (n = 4, open symbols), after 30-hour incubation detected by MTT assay. Each data point represents the mean of duplicate measurements. (B) Comparison of the mean IC50 of PEITC in CLL cells (n = 13) and normal lymphocytes (n = 11). Each bar represents the mean and 95% CI. (C) Cell death induced by 5 μM PEITC (24 hours) in primary CLL cells and normal lymphocytes detected by flow cytometric analysis (annexin V/PI double staining). Representative dot plots are shown. (D) Quantitative comparison of cell death induced by PEITC (5 μM, 24 hours) as in C. Percentage of drug-induced cell death was calculated by subtracting the spontaneous death in the control from the overall cell death in the PEITC-treated samples for each time point. The black and white bar represents the mean and 95% CI of 18 CLL patient samples and 7 normal blood samples, respectively. (E) Induction of ROS increase in primary CLL cells and normal lymphocytes by PEITC (5 μM, 2 hours), detected by flow cytometry using 1 μM DCF-DA. Representative histograms for CLL cells and normal lymphocytes are shown.
High ROS and low glutathione are intrinsic properties of CLL cells and key determinants of sensitivity to PEITC
The preferential ROS accumulation in CLL cells treated with PEITC led us to speculate that CLL cells may be intrinsically under oxidative stress and therefore more vulnerable to further stress by PEITC. To test this possibility, we compared the basal ROS levels in fresh CLL cells isolated from 33 CLL patients with that of normal lymphocytes from 12 healthy donors. As shown in Figure 3A,B, primary CLL cells exhibited a significant increase in ROS content compared with normal lymphocytes (P < .001). Further analyses revealed that the elevated ROS in CLL cells was associated with attenuation of antioxidant capacity, since there was a significant decrease in cellular glutathione (both GSH and GSSG), whereas glutathione peroxidase (GPX) enzyme activity in CLL cells was comparable with that of normal lymphocytes (Figure 3C,D). Because GSH can be oxidized to GSSG for exportation outside the cells and glutathione reductase activity is similar in CLL and normal lymphocytes,25 the GSSG/GSH ratios were similar in both cell types (Figure 3C). Western blot analysis showed an elevation of gamma-glutamyl-cysteine synthetase (GSH1) protein expression in CLL cells (Figure 3E), suggesting a possible compensatory mechanism of CLL cells in response to ROS stress and increased consumption of glutathione. Because compared with normal cells, CLL cells are under intrinsic ROS stress with lower glutathione, it is conceivable that further abrogation of the glutathione antioxidant system by PEITC would severely impact CLL cells and induce cell death. In fact, the sensitivity to PEITC in CLL cells was strongly correlated with basal ROS level (Figure 3F; P < .001). In contrast, there appeared no obvious correlation between F-ara-A sensitivity and basal ROS levels (Figure 3G; P > .100).
Figure 3.

Alterations of redox states in primary CLL cells. (A) Increase of basal ROS in primary CLL cells, detected by flow cytometry using 1 μM DCF-DA. Representative histograms for CLL cells and normal lymphocytes are shown. (B) Quantitative comparison of the basal ROS levels between normal lymphocytes and primary CLL cells from 12 healthy donors and 33 CLL patients. Each bar represents the mean and 95% CI. (C) Decrease of basal reduced and oxidized glutathione (GSH and GSSG) in primary CLL cells (n = 9), compared with that of normal lymphocytes (n = 5). Each bar represents the mean and 95% CI. (D) GPX enzyme activities of normal lymphocytes (n = 4) and primary CLL cells (n = 8). Each bar represents the mean and 95% CI. (E) Basal expression levels of glutathione synthesis enzyme GCS (GSH1) in lymphocytes from 3 healthy donors and 3 CLL patient samples. (F) Correlation between the IC50 values of PEITC (MTT assay) and the basal ROS levels in primary CLL cells (n = 18). Spearman correlation coefficient r = −0.872, P < .001. (G) Lack of correlation between the IC50 values of F-ara-A (MTT assay) and basal ROS levels in primary CLL cells (n = 21). Spearman correlation coefficient r = −0.368, P = .100.
PEITC kills CLL cells through depletion of glutathione and ROS-mediated damage to mitochondria
Based on our previous observation that PEITC induced cell death by abrogating the glutathione system,17 we tested the relationship between cell death and glutathione change in CLL cells. As shown in Figure 4A, PEITC (5 μM) caused substantial amount of cell death, which was detectable at 6 hours and reached more than 70% cell death by 24 hours (n = 23). Analysis of glutathione revealed that this compound induced a rapid glutathione depletion, detectable at 1 hour and reached more than 70% depletion in 3 hours (Figure 4B, n = 9). Because during the first few hours of PEITC incubation there was no significant drug-induced cell death, it is likely that the depletion of glutathione was a primary event that triggered ROS elevation and subsequent cell death. Consistently, supplementation of the glutathione precursor NAC to the cell culture medium largely prevented the PEITC-induced glutathione depletion (Figure 4C), and significantly suppressed ROS increase (Figure 4D) and drug-induced cell death (Figure 4E, n = 7). These data together suggest that PEITC killed CLL cells through depletion of glutathione, leading to ROS-mediated cell death.
Figure 4.

PEITC killed CLL cells mediated by glutathione depletion of ROS stress. (A) Time course of CLL cell killing by 5 μM PEITC, analyzed by annexin-PI assay. Representative dot plots (left panel) and quantitation of cell death (right panel) are shown. Percentage of drug-induced cell death was calculated by subtracting the spontaneous cell death from the overall cell death in the PEITC-treated sample. Each bar represents the mean and 95% CI (n = 23). (B) Depletion of cellular glutathione in CLL cells after exposure to 5 μM PEITC for indicated times. Each bar represents the mean and 95% CI (n = 9 CLL samples for each time point). (C) Effect of NAC on PEITC-induced glutathione depletion. CLL cells were preincubated with 1 mM NAC for 1 hour before exposure to 5 μM PEITC for 5 hours. Each bar represents the mean and 95% CI (n = 4). (D) Effect of NAC on PEITC-induced ROS accumulation is shown. CLL cells were preincubated with 1 mM NAC for 1 hour before exposure to 5 μM PEITC for 2 hours. Each bar represents the mean and 95% CI from assays of 3 different CLL samples. (E) Effect of NAC on PEITC-induced cell death. CLL cells were pre-incubated with 1 mM NAC for 1 hour before exposure to 5 μM PEITC for 24 hours. Cell death was detected by annexin/PI assay. Each bar represents the mean and 95% CI from assays of 7 CLL samples.
Because mitochondria are the major source of ROS production, and rely on the import of cellular glutathione as a major antioxidant,26 we further tested the possibility that the loss of cellular glutathione might cause a rapid redox imbalance and ROS-mediated mitochondrial damage. As shown in Figure 5A, incubation of CLL cells with 5 μM PEITC led to a rapid oxidation of cardiolipin detectable by loss of interaction with fluorescent dye NAO. NAO fluorescent signal losses were 15% and 75% after PEITC incubation at 2 hours and 4 hours, respectively, consistent with the time-course of glutathione depletion. Previous studies showed that cardiolipin can stabilize cytochrome c in mitochondria, and loss of cardiolipin can lead to cytochrome c release and caspase activation.27 As shown in Figure 5B,C, exposure to 5 μM PEITC induced mitochondrial cytochrome c release and activation of caspase-3, with the time courses slightly behind the cardiolipin oxidation. To test if cell death induced by PEITC was caspase-mediated, the pan-caspase inhibitor Z-VAD-fmk was added to CLL cells before they were exposed to PEITC. As shown in Figure 5D, the caspase inhibitor partially reduced cell death from 59% (PEITC alone) to 37% (PEITC + Z-VAD-fmk; P < .05). Furthermore, the PEITC-induced activation of caspase-3 was significantly prevented by NAC (Figure 5E), whereas Z-VAD-fmk failed to prevent the depletion of glutathione (Figure 5F). These results suggest that glutathione depletion was upstream of caspase activation and cell death.
Figure 5.

PEITC induces CLL cell death through oxidative damage to mitochondria. (A) Induction of oxidative damage to cardiolipin in CLL cells by PEITC (5 μM). Cardiolipin oxidation was measured by flow cytometry using NAO staining.17 M1 indicates the gating of the subpopulation of CLL cells that lost cardiolipin signal due to oxidation. Representative histograms of the time course experiments in a CLL patient sample are shown. Similar results were obtained using another sample. (B) Loss of mitochondrial cytochrome c induced by 5 μM PEITC in CLL cells. The overlays of the control (gray shade) and PEITC-treated (black line) samples show the distribution of mitochondrial cytochrome c fluorescent intensity of each cell population, with the mean value of the relative intensity indicated. Representative histograms of a CLL patient sample are shown. Similar results were obtained using another sample. (C) Caspase-3 activation in CLL cells treated with 5 μM PEITC, measured by flow cytometry using FITC-conjugated antibody specific for active caspase-3. M1 indicates the gating of subpopulation of cells with positive caspase-3 activation. Representative histograms of a CLL patient sample are shown. Similar results were obtained using 2 other different patient samples. (D) Partial suppression of PEITC-induced cell death by Z-VAD-fmk. CLL cells were preincubated with 20 μM Z-VAD-fmk for 30 minutes before incubation with 5 μM PEITC for 24 hours. Cell death was detected by annexin V/PI assay. Each bar represents the mean and 95% CI (n = 5 CLL samples). (E) Suppression of PEITC-induced caspase-3 activation by NAC. CLL cells were preincubated with 1 mM NAC for 1 hour before incubation with 5 μM PEITC for 5 hours. Procaspase-3 was detected by Western blot and quantified by densitometry and normalized with β-actin. Each bar represents the mean and 95% CI of 3 different CLL samples. (F) No effect of Z-VAD-fmk on PEITC-induced glutathione depletion was found. CLL cells were preincubated with 20 μM Z-VAD-fmk for 30 minutes before incubation with 5 μM PEITC for 5 hours. Each bar represents the mean and 95% CI of 3 different CLL samples.
Induction of rapid degradation of the antiapoptotic protein MCL1 by PEITC through deglutathionylation
Because antiapoptotic proteins such as BCL2 and MCL1 are known to play key roles in cancer cell survival, and MCL1 is particularly important in conferring drug resistance in CLL cells,28 we tested the effect of PEITC on BCL2 and MCL1 in CLL cells. Interestingly, PEITC caused a rapid decrease of MCL1 but not BCL2 in both F-ara-A–sensitive and –resistant CLL cells (Figure 6A,B). Because a high level of MCL1 was shown to mediate resistance to fludarabine in vivo and in vitro,28,29 abrogation of MCL1 by PEITC likely facilitated the killing of F-ara-A–refractory CLL cells. As MCL1 may be a better substrate for caspase than BCL2,30 we used the pan-caspase inhibitor Z-VAD-fmk to test if it could prevent PEITC-induced MCL1 degradation, and showed that the 20 μM Z-VAD-fmk largely suppressed MCL1 degradation in PEITC-treated cells (Figure 6C). Similar effect was observed by using the specific inhibitor to caspase-3, Z-DEVD (data not shown), suggesting that caspase-3 might be a major protease that cleaved MCL1.
Figure 6.

Effect of PEITC on MCL1 stability and its glutathionylation state in CLL cells. (A) Time-dependent effect of 5 μM PEITC on MCL1 and BCL2 protein levels in F-ara-A–sensitive and –resistant CLL cells, detected by Western blot analysis. (B) Quantitation of MCL1 protein after exposure to PEITC as described in panel A. Each bar represents the mean and 95% CI from 7 F-ara-A–sensitive or 6 of F-ara-A–resistant CLL patient samples. (C) Suppression of PEITC-induced MCL1 degradation by caspase inhibitor Z-VAD-fmk. Cells were pretreated with 20 μM Z-VAD-fmk for 30 minutes before exposure to 5 μM PEITC. A representative Western blot from experiments with a CLL sample is shown. Similar results were obtained using another sample. (D) Suppression of PEITC-induced MCL1 degradation by NAC. CLL cells were preincubated with 1 mM NAC for 1 hour before exposure to 5 μM PEITC for 5 hours. Each bar represents the mean and 95% CI of 4 different CLL samples. (E) Glutathionylation prevented caspase-3–mediated cleavage of MCL1 in vitro. Dialyzed CLL lysates were incubated with 0.5 mM NADH, 0.5 mM NADPH, 2 mM GSH, or 2 mM GSSG for 10 minutes, and then exposed to recombinant caspase-3 for 60 minutes. Cleavage of MCL or PARP was detected by Western blot analysis. Representative results are shown. (F) PEITC treatment reduced the level of glutathionylation of MCL1 in CLL cells. CLL cells were exposed to 5 μM PEITC for 2.5 hours. Glutathionylated protein was immunoprecipitated (IP) using anti-GSH, and the levels of glutathionylated MCL-1 were analyzed using immunoblotting (IB) with anti-MCL1 antibody. Because PEITC induces rapid degradation of MCL1 protein, 3-fold higher amount of protein from the PEITC-treated sample was used for IP (input) to allow the detection of MCL1 signal. (G) Depletion of total glutathione and ratio of GSH/GSSG after 5 μM PEITC treatment as in panel F. Addition of 1 mM NAC restored cellular glutathione. Each stacked bar (GSH/GSSG) represents the average of duplicate measurements.
Interestingly, supplementation of NAC to replenish glutathione significantly protected MCL1 degradation induced by PEITC (Figure 6D). This suggests that the depletion of glutathione might play a role in MCL1 cleavage. Because glutathionylation of certain proteins may affect their functions and stability,23,31 we speculated that MCL1 may be a target of glutathionylation, and its glutathionylation status might affect susceptibility to caspase-cleavage. To test the possibility, we first examined whether in vitro glutathionylation of MCL1 could protect it from cleavage by caspase-3. Because glutathionylation can occur spontaneously by thiol-disulfide exchange with GSSG,31 we preincubated cell-free protein lysates with GSSG before subjected to digestion by caspase-3. Western blot analysis showed that MCL1 could be cleaved by recombinant caspase-3 in vitro and addition of GSSG can significantly protect MCL1 from cleavage (Figure 6E top panel). Apparently, the effect was not due to the reducing power of glutathione since there were no protective effects by GSH, NADH, or NADPH. Whereas the protection of MCL1 by GSSG was obvious, the protective effect on PARP cleavage was rather limited (Figure 6E bottom panel), suggesting the direct protection of MCL1 by glutathionylation rather than an inhibitory effect on caspase enzyme activity. Furthermore, using anti-GSH antibody to pull down glutathionylated protein, followed by Western blot analysis of MCL1, we found a significant glutathionylation of MCL1 in the control CLL cells (Figure 6F). Interestingly, exposure to PEITC resulted in a substantial decrease of glutathionylated MCL1 (Figure 6F top panel), despite more MCL-1 protein from the PEITC-treated cells used as the input (Figure 6F bottom panel). In a parallel experiment using the CLL cells from the same patient, cellular GSH and GSSG were found consistently depleted by PEITC (Figure 6G). Interestingly, PEITC only caused a decrease in glutathionylation of certain proteins such as MCL1, but the majority of glutathionylated proteins remained unchanged (data not shown). Together, these data suggest a novel mechanism of MCL1 stabilization through a glutathionylation-dependent mechanism, and that removal of glutathionation from MCL1 through depletion of glutathione by PEITC rendering it more susceptible to cleavage by caspase-3.
PEITC effectively eliminated subpopulation of F-ara-A–resistant CLL cells
Because minimal residual disease (MRD) after therapy is an important cause of CLL disease relapse in clinic,32 we further tested the ability of PEITC to remove the CLL cells that were resistant to F-ara-A. First, we compared the percentage of the residual viable cells after F-ara-A treatment with that of PEITC incubation. As shown in Figure 7A, even in the F-ara-A–sensitive CLL cells, there was a substantial percentage of residual viable cells (20%) after a 72-hour incubation with 30 μM F-ara-A, but incubation with 10 μM PEITC almost completely eliminated all CLL cells. Strikingly, in a F-ara-A–resistant CLL sample, incubation with 100 μM F-ara-A for 72 hours still resulted in 45% viable cells, but almost all CLL cells were eliminated by 10 μM PEITC (Figure 7B). The quantitative comparison of the percentage of residual viable CLL cells after in vitro treatment with 20 μM F-ara-A or with 10 μM PEITC in multiple CLL patient samples is shown in Figure 7C. Among the 22 CLL patient samples that were sensitive to F-ara-A, the residual viable cells after treatment with 20 μM F-ara-A or 10 μM PEITC was 22.1% and 6.1%, respectively (P < .001). In the 18 CLL samples that were F-ara-A–resistant, the percentages of residual viable cells after treatment with 20 μM F-ara-A or 10 μM PEITC were 53.4% and 7.5%, respectively (P < .001). Interestingly, the subpopulation of viable CLL cells resistant to F-ara-A (20 μM) seemed to have a higher level of cellular thiols as detected by dual staining with CMFDA and annexin V–PE (Figure 7D gated annexin-negative subpopulation R1).
Figure 7.

Comparison of residual viable cells after treatment with F-ara-A or PEITC. (A) Representative cell viability curves of F-ara-A–sensitive CLL cells treated with various concentrations of F-ara-A or PEITC for 72 hours and cell viability was measured by MTT assay. (B) Representative cell viability curves of F-ara-A–resistant CLL cells treated with various concentrations of F-ara-A or PEITC for 72 hours. (C) Quantitative comparison of percentage of viable cells after treatment with 20 μM F-ara-A or 10 μM PEITC (72 hours, MTT assay) in F-ara-A–sensitive and –resistant CLL cells. Each data point represents the mean of triplicate measurements for each patient sample. (D) Comparison of cellular thiols in control (untreated) CLL cells and in the residual viable CLL cells after treatment with 20 μM F-ara-A for 72 hours, using CMFDA and annexin V–PE dual staining flow cytometric analysis. Viable cells were defined as annexin V–negative subpopulation, which was gated as R1 (D left and middle panels). Cellular thiol levels in the R1 cell population of the control sample and the F-ara-A–treated sample were shown on the right panel. Representative plots of 3 different patients are shown.
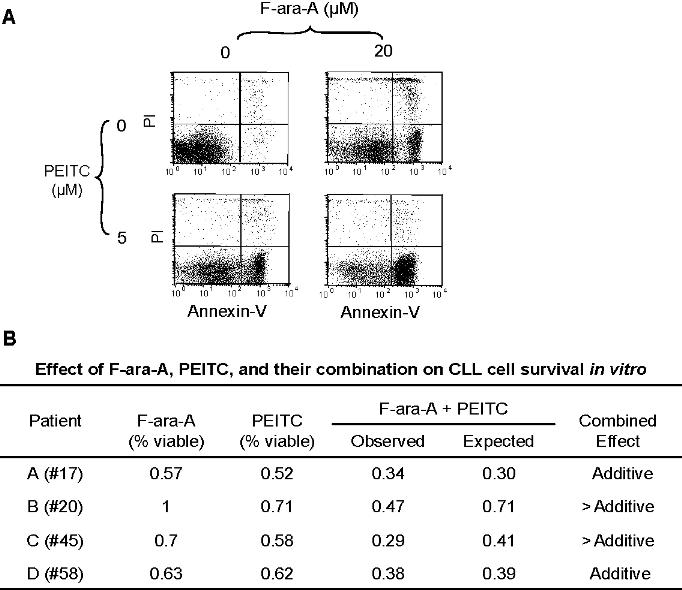
The promising cytotoxicity of PEITC in fludarabine-resistant CLL cells prompted us to test whether combination of PEITC and fludarabine could increase cytotoxic effect against CLL cells. After CLL cells were incubated with 20 μM F-ara-A for 44 hours, PEITC (5 μM) was added for an additional 4 hours. Analysis of cell death using annexin V–PI assay in 4 different CLL patient samples showed that the combination of 2 compounds has an additive or more than additive effect under these experimental conditions (Figure S2).
Discussion
Intrinsic oxidative stress in cancer has long been recognized to play important roles in maintaining malignant phenotype and promoting disease progression.9 However, its therapeutic implications have yet to be determined. Here, we show that it is possible to exploit the redox alterations in primary CLL cells as a basis to preferentially kill the malignant cells including those resistant to standard therapeutic agent fludarabine, using ROS-modulating agents such as PEITC. This conclusion is based on several lines of evidence. (1) The primary CLL cells isolated from patients exhibited 2 major redox alterations: a significant increase in basal ROS levels and a decrease in cellular glutathione. (2) The low glutathione and high ROS stress rendered CLL cells highly vulnerable to further disruption of the glutathione antioxidant system by PEITC, leading to severe further ROS accumulation, oxidative mitochondrial membrane damage, and rapid cell death. (3) CLL cells either sensitive or resistant to fludarabine were all under intrinsic stress and thus sensitive to PEITC-induced ROS stress and cell death. (4) The antiapoptotic protein MCL1, which plays an important role in fludarabine-resistance, was stabilized by glutathionylation. PEITC caused a decrease of glutathionylation, rendering MCL1 more susceptible to cleavage by caspase-3. The redox-mediated abrogation of MCL1 survival signal likely accelerated cell death and facilitated the removal of the fludarabine-refractory CLL cells.
Our study suggests that the intrinsic oxidative stress in CLL cells and the increased dependency on glutathione antioxidant system may provide a biologic basis for selective elimination of the leukemia cells. This could be accomplished using a proper redox-modulating agent PEITC. A pharmacokinetic study showed that an oral dose of 40 mg PEITC resulted in a plasma concentration in the micromolar range within 3 to 8 hours.33 Thus, the effective concentrations of PEITC seem achievable in humans. Furthermore, since PEITC at the concentrations of 5 to 10 μM causes a rapid killing of CLL cells within 5 to 10 hours, it is feasible to maintain such drug concentrations in plasma for the duration necessary to eliminate CLL cells. As this compound is present in commonly consumed vegetables and has low toxicity toward normal cells, it merits further preclinical and clinical evaluation. A recent study showed that it is possible to generate a CLL xenograft model by injection of primary CLL cells from patients into NOD/SCID mice, which developed leukemia that mimics characteristics of CLL.34 It would be interesting to test the in vivo activity of PEITC in this disease model.
The in vitro therapeutic selectivity of PEITC was observed in experiments directly comparing primary CLL cells isolated from patients and normal lymphocytes from healthy donors. Most CLL cells are B lymphocytes, whereas the normal lymphocytes from healthy donors contain more T cells than B cells. This raises a possibility that PEITC might preferentially kill B cells. In a separate study, we purified normal B cells from 4 normal blood samples, and showed that the normal B lymphocytes were significantly less sensitive to PEITC than CLL cells, suggesting a major difference between normal B cells and CLL cells. Since purification of B cells from T cells requires technical procedures that may disturb metabolism and redox status, further studies are required to overcome this technical challenge and to develop suitable methods to compare ROS states and drug sensitivity of T cells with that of B cells.
Although there was a consistent correlation between the levels of ROS accumulation and the degrees of PEITC-induced cell death, we noticed that the glutathione precursor NAC completely suppressed the PEITC-induced glutathione loss (Figure 4C) and prevented caspase-3 cleavage (Figure 5E), but only partially prevented ROS accumulation and cytotoxicity (Figure 4D,E). These suggest that the cytotoxic action of PEITC in CLL cells may involve not only depletion of glutathione but also other mechanisms. Recent studies suggest that protein modification and activation of ERK and JNK pathways may contribute to PEITC cytotoxicity.35,36
Glutathione is a key regulator of cell death and survival of cancer cells, including leukemia cells.37,38 PEITC is known to conjugate with GSH, leading to its exportation and depletion of cellular glutathione. This is thought to be a major mechanism of PEITC-induced ROS stress in cancer cells.17 Since GSH-PEITC conjugation is a reversible reaction catalyzed by glutathione-s-transferase (GST), which seemed up-regulated after PEITC treatment,39 it would be interesting to test whether CLL cells might express higher levels of GST than normal lymphocytes and thus contribute to their high sensitivity to PEITC.
Besides its direct ROS-scavenging effect, glutathione helps to maintain protein redox status.38 However, the role of thiol redox status in regulating cell survival remains poorly understood. Under mild oxidative stress, protein glutathionylation was shown to regulate the functions of multiple proteins.23,31 Here, we showed that the antiapoptotic protein MCL1 is a novel substrate of glutathionylation in CLL cells, and that such glutathionylation regulates its stability. Removal of glutathionylation by the glutathione-depleting agent PEITC rendered MCL1 susceptible to rapid proteolytic cleavage. Interestingly, a previous study revealed a highly positive-charged domain of MCL1, which adapts a relatively open structure.40 This may render MCL1 more attractive to the negatively charged thiol moiety of glutathione, as seen in other glutathionylation targets such as thioredoxin.31,41 Previous work showed that hydrogen peroxide induces phosphorylation of MCL1 through activation of c-Jun N-terminal kinase.42 Here, we provide evidence that MCL1 may be a redox-sensitive protein and its glutathionylation status could affect its stability. Because MCL1 has a critical role in prolonging the CLL cell survival, especially in a tumor-stroma context,43,44 and is strongly associated with resistance to fludarabine,28,29 rituximab,45 chlorambucil, and prednisone,46 the thiol-mediated abrogation of MCL1 by PEITC suggests a novel mechanism that may be exploited to overcome drug resistance by combining PEITC with these anti-CLL agents.
Our study suggests that glutathionylation of MCL1 may affect its stability. It is unclear if glutathionylation of MCL1 might affect the functions of this protein. The detectable level of glutathionylated MCL1 in untreated CLL cells (Figure 6F) suggested that such modification occurs constitutively in the leukemia cells, which are under intrinsic ROS stress. However, it remains to be determined if MCL1 could exist as glutathionated protein in unstressed cells and whether glutathionylation could modulate its functional state a similar fashion as protein phosphorylation. In human MCL1 protein, there are 2 cysteine residues (Cys16, Cys286) that potentially can be glutathionylated. Even though the functional importance of these cysteine residues in MCL1 has not been well studied, recent report showed that the first 79 amino acids of MCL1 (including Cys16) regulates its localization and antiapoptotic activity.47 Furthermore, a structural study revealed a disulfide linkage between Cys286 of MCL1 and Cys75 of NOXA (a MCL1 regulator).48 Further studies are required to elucidate the functional significance of glutathionylation on these cysteine residues in MCL1.
Clinical studies showed that CLL cells with loss of p53 due to cytogenetic alterations such as 17p deletion or point mutations are often resistant to fludarabine therapy and have poor clinical outcomes.2–5 Therefore, new agents capable of effectively killing p53-null CLL cells would have significant clinical impact. Among 30 CLL samples tested for genetic abnormalities, 7 samples had deletion of 17p. Interestingly, 5 of the 7 samples exhibited resistance to F-ara-A (IC50 > 10 μM), consistent with the crucial effect of p53 on sensitivity to fludarabine. Importantly, these p53-null CLL cells remain sensitive to PEITC. Since the loss of p53 is known to promote genetic instability and mitochondrial dysfunction,49 which not only confers drug resistance but may also promote ROS production,14 it is conceivable that the p53-null CLL cells may have elevated ROS generation and would be highly sensitive to PEITC. Since the loss of p53 is prevalent in cancer and associated with resistance to many standard therapeutic agents,50 the novel ROS-mediated strategy using agents such as PEITC may have potentially broad clinical implications.
Supplementary Material
Acknowledgments
The authors thank C. Garcia-Prieto, M.A. Ogasawara, R. LaPushin, Y. Zhou, and W. Lu for technical assistance and helpful discussion.
This work was supported in part by grants CA085563, CA100428, CA109041, and CA16672 from the National Institutes of Health (Bethesda, MD) and by grant CLLGRF-2007PH from CLL Global Research Foundation (Houston, TX). D.T. is a recipient of a scholarship from Anandamahidol Foundation (Bangkok, Thailand) under the royal patronage of His Majesty the King of Thailand, and a recipient of the Lummis Family Fellowship in Biomedical Sciences (Houston, TX).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contributions: D.T. designed and performed research, analyzed data, and drafted the manuscript; H.Z., W.Z., L.F., MD., Y.Z., and Z.C. performed research and analyzed data; H.P. and W.P. provided input into experimental design and data interpretation; W.G.W. and M.J.K. identified clinical specimens, designed research, and interpreted data; and P.H. directed the study design, data analysis, and interpretation, and drafted the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Peng Huang, Department of Molecular Pathology, University of Texas M. D. Anderson Cancer Center, Box 0951, 1515 Holcombe Boulevard, Houston, TX 77030; e-mail: phuang@mdanderson.org.
References
- 1.Keating MJ, Chiorazzi N, Messmer B, et al. Biology and treatment of chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ Program. 2003;1:153–175. doi: 10.1182/asheducation-2003.1.153. [DOI] [PubMed] [Google Scholar]
- 2.Elter T, Hallek M, Engert A. Fludarabine in chronic lymphocytic leukaemia. Expert Opin Pharmacother. 2006;7:1641–1651. doi: 10.1517/14656566.7.12.1641. [DOI] [PubMed] [Google Scholar]
- 3.Byrd JC, Lin TS, Grever MR. Treatment of relapsed chronic lymphocytic leukemia: old and new therapies. Semin Oncol. 2006;33:210–219. doi: 10.1053/j.seminoncol.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 4.Turgut B, Vural O, Pala FS, et al. 17p Deletion is associated with resistance of B-cell chronic lymphocytic leukemia cells to in vitro fludarabine-induced apoptosis. Leuk Lymphoma. 2007;48:311–220. doi: 10.1080/10428190601059829. [DOI] [PubMed] [Google Scholar]
- 5.Grever MR, Lucas DM, Dewald GW, et al. Comprehensive assessment of genetic and molecular features predicting outcome in patients with chronic lymphocytic leukemia: results from the US Intergroup Phase III Trial E2997. J Clin Oncol. 2007;25:799–804. doi: 10.1200/JCO.2006.08.3089. [DOI] [PubMed] [Google Scholar]
- 6.Zhou Y, Hileman EO, Plunkett W, Keating MJ, Huang P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood. 2003;101:4098–4104. doi: 10.1182/blood-2002-08-2512. [DOI] [PubMed] [Google Scholar]
- 7.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–798. [PubMed] [Google Scholar]
- 8.Devi GS, Prasad MH, Saraswathi I, Raghu D, Rao DN, Reddy PP. Free radicals antioxidant enzymes and lipid peroxidation in different types of leukemias. Clin Chim Acta. 2000;293:53–62. doi: 10.1016/s0009-8981(99)00222-3. [DOI] [PubMed] [Google Scholar]
- 9.Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat. 2004;7:97–110. doi: 10.1016/j.drup.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 10.Carew JS, Nawrocki ST, Xu RH, et al. Increased mitochondrial biogenesis in primary leukemia cells: the role of endogenous nitric oxide and impact on sensitivity to fludarabine. Leukemia. 2004;18:1934–1940. doi: 10.1038/sj.leu.2403545. [DOI] [PubMed] [Google Scholar]
- 11.Pelicano H, Feng L, Zhou Y, et al. Inhibition of mitochondrial respiration: a novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J Biol Chem. 2003;278:37832–37839. doi: 10.1074/jbc.M301546200. [DOI] [PubMed] [Google Scholar]
- 12.Oltra AM, Carbonell F, Tormos C, Iradi A, Sáez GT. Antioxidant enzyme activities and the production of MDA and 8-oxo-dG in chronic lymphocytic leukemia. Free Radic Biol Med. 2001;30:1286–1292. doi: 10.1016/s0891-5849(01)00521-4. [DOI] [PubMed] [Google Scholar]
- 13.Hileman EO, Liu J, Albitar M, Keating MJ, Huang P. Intrinsic oxidative stress in cancer cells: a biochemical basis for therapeutic selectivity. Cancer Chemother Pharmacol. 2004;53:209–219. doi: 10.1007/s00280-003-0726-5. [DOI] [PubMed] [Google Scholar]
- 14.Achanta G, Sasaki R, Feng L, et al. Novel role of p53 in maintaining mitochondrial genetic stability through interaction with DNA Pol gamma. EMBO J. 2005;24:3482–3492. doi: 10.1038/sj.emboj.7600819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu C, Miloslavskaya I, Demontis S, Maestro R, Galaktionov K. Regulation of cellular response to oncogenic and oxidative stress by Seladin-1. Nature. 2004;432:640–645. doi: 10.1038/nature03173. [DOI] [PubMed] [Google Scholar]
- 16.Radisky DC, Levy DD, Littlepage LE, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trachootham D, Zhou Y, Zhang H, et al. Selective killing of oncogenic transformed cells through ROS-mediated mechanism by β-phenylethyl isothiocyanates. Cancer Cell. 2006;10:241–252. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 18.Cheson BD, Bennett JM, Rai KR, et al. Guidelines for clinical protocols for chronic lymphocytic leukemia: recommendations of the National Cancer Institute-sponsored working group. Am J Hematol. 1988;29:152–163. doi: 10.1002/ajh.2830290307. [DOI] [PubMed] [Google Scholar]
- 19.Huang P, Sandoval A, Van Den NE, Keating MJ, Plunkett W. Inhibition of RNA transcription: a biochemical mechanism of action against chronic lymphocytic leukemia cells by fludarabine. Leukemia. 2000;14:1405–1413. doi: 10.1038/sj.leu.2401845. [DOI] [PubMed] [Google Scholar]
- 20.Robertson LE, Chubb S, Meyn RE, et al. Induction of apoptotic cell death in chronic lymphocytic leukemia by 2-chloro-2′-deoxyadenosine and 9-beta-D-arabinosyl-2-fluoroadenine. Blood. 1993;81:143–150. [PubMed] [Google Scholar]
- 21.Wen JJ, Vyatkina G, Garg N. Oxidative damage during chagasic cardiomyopathy development: role of mitochondrial oxidant release and inefficient antioxidant defense. Free Radic Biol Med. 2004;37:1821–1833. doi: 10.1016/j.freeradbiomed.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 22.Nomura K, Imai H, Koumura T, Kobayashi T, Nakagawa Y. Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochem J. 2000;351:183–193. doi: 10.1042/0264-6021:3510183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Velu CS, Niture SK, Doneanu CE, Pattabiraman N, Srivenugopal KS. Human p53 is inhibited by glutathionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry. 2007;46:7765–7780. doi: 10.1021/bi700425y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chikahisa L, Oyama Y, Okazaki E, Noda K. Fluorescent estimation of H2O2-induced changes in cell viability and cellular nonprotein thiol level of dissociated rat thymocytes. Jpn J Pharmacol. 1996;71:299–305. doi: 10.1254/jjp.71.299. [DOI] [PubMed] [Google Scholar]
- 25.Farber CM, Kanganis DN, Liebes LF, Silber R. Antioxidant enzymes in lymphocytes from normal subjects and patients with chronic lymphocytic leukaemia: increased glutathione peroxidase activity in CLL B lymphocytes. Br J Haematol. 1989;72:32–35. doi: 10.1111/j.1365-2141.1989.tb07647.x. [DOI] [PubMed] [Google Scholar]
- 26.Fernandez-Checa JC, Kaplowitz N, Garcia-Ruiz C, Colell A. Mitochondrial glutathione: importance and transport. Semin Liver Dis. 1998;18:389–401. doi: 10.1055/s-2007-1007172. [DOI] [PubMed] [Google Scholar]
- 27.Iverson SL, Orrenius S. The cardiolipin–cytochrome c interaction and the mitochondrial regulation of apoptosis, Arch Biochem Biophys. 2004;423:37–46. doi: 10.1016/j.abb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 28.Kitada S, Andersen J, Akar S, et al. Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: correlations with In vitro and In vivo chemoresponses. Blood. 1998;91:3379–3389. [PubMed] [Google Scholar]
- 29.Faria JR, Yamamoto M, Faria RM, EBay J, Oliveira JS. Fludarabine induces apoptosis in chronic lymphocytic leukemia-the role of P53, Bcl-2, Bax, Mcl-1, and Bag-1 proteins. Braz J Med Biol Res. 2006;39:327–333. doi: 10.1590/s0100-879x2006000300003. [DOI] [PubMed] [Google Scholar]
- 30.Michels J, O'Neill JW, Dallman CL, et al. Mcl-1 is required for Akata6 B-lymphoma cell survival and is converted to a cell death molecule by efficient caspase-mediated cleavage. Oncogene. 2004;23:4818–4827. doi: 10.1038/sj.onc.1207648. [DOI] [PubMed] [Google Scholar]
- 31.Ghezzi P. Regulation of protein function by glutathionylation. Free Radic Res. 2005;39:573–580. doi: 10.1080/10715760500072172. [DOI] [PubMed] [Google Scholar]
- 32.Sayala HA, Rawstron AC, Hillmen P. Minimal residual disease assessment in chronic lymphocytic leukaemia. Best Pract Res Clin Haematol. 2007;20:499–512. doi: 10.1016/j.beha.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 33.Liebes L, Conaway CC, Hochster H, et al. High-performance liquid chromatography-based determination of total isothiocyanate levels in human plasma: application to studies with 2-phenethyl isothiocyanate. Anal Biochem. 2001;291:279–289. doi: 10.1006/abio.2001.5030. [DOI] [PubMed] [Google Scholar]
- 34.Dürig J, Ebeling P, Grabellus F, et al. A novel nonobese diabetic/severe combined immunodeficient xenograft model for chronic lymphocytic leukemia reflects important clinical characteristics of the disease. Cancer Res. 2007;67:8653–8661. doi: 10.1158/0008-5472.CAN-07-1198. [DOI] [PubMed] [Google Scholar]
- 35.Mi L, Wang X, Govind S, et al. The role of protein binding in induction of apoptosis by phenethyl isothiocyanate and sulforaphane in human non-small lung cancer cells. Cancer Res. 2007;67:6409–6416. doi: 10.1158/0008-5472.CAN-07-0340. [DOI] [PubMed] [Google Scholar]
- 36.Xu C, Shen G, Yuan X, et al. ERK and JNK signaling pathways are involved in the regulation of activator protein 1 and cell death elicited by three isothiocyanates in human prostate cancer PC-3 cells. Carcinogenesis. 2006;27:437–445. doi: 10.1093/carcin/bgi251. [DOI] [PubMed] [Google Scholar]
- 37.Friesen C, Kiess Y, Debatin KM. A critical role of glutathione in determining apoptosis sensitivity and resistance in leukemia cells. Cell Death Differ. 2004;11(suppl 1):S73–S85. doi: 10.1038/sj.cdd.4401431. [DOI] [PubMed] [Google Scholar]
- 38.Estrela JM, Ortega A, Obrador E. Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci. 2006;43:143–181. doi: 10.1080/10408360500523878. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, Kolm RH, Mannervik B, Talalay P. Reversible conjugation of isothiocyanates with GSH catalyzed by glutathione transferases. Biochem Biophys Res Commun. 1995;206:748–755. doi: 10.1006/bbrc.1995.1106. [DOI] [PubMed] [Google Scholar]
- 40.Day CL, Chen L, Richardson SJ, Harrison PJ, Huang DC, Hinds MG. Solution structure of prosurvival Mcl-1 and characterization of its binding by proapoptotic BH3-only ligands. J Biol Chem. 2005;280:4738–4744. doi: 10.1074/jbc.M411434200. [DOI] [PubMed] [Google Scholar]
- 41.Hansen RE, Ostergaard H, Winther JR. Increasing the reactivity of an artificial dithiol-disulfide pair through modification of the electrostatic milieu. Biochemistry. 2005;44:5899–906. doi: 10.1021/bi0500372. [DOI] [PubMed] [Google Scholar]
- 42.Inoshita S, Takeda K, Hatai T, et al. Phosphorylation and inactivation of myeloid cell leukemia 1 by JNK in response to oxidative stress. J Biol Chem. 2002;277:43730–43734. doi: 10.1074/jbc.M207951200. [DOI] [PubMed] [Google Scholar]
- 43.Pedersen IM, Kitada S, Leoni LM, et al. Protection of CLL B cells by a follicular dendritic cell line is dependent on induction of Mcl-1. Blood. 2002;100:1795–1801. [PubMed] [Google Scholar]
- 44.Petlickovski A, Laurenti L, Li X, et al. Sustained signaling through the B-cell receptor induces Mcl-1 and promotes survival of chronic lymphocytic leukemia B cells. Blood. 2005;105:4820–4827. doi: 10.1182/blood-2004-07-2669. [DOI] [PubMed] [Google Scholar]
- 45.Hussain SR, Cheney CM, Johnson AJ, et al. Mcl-1 is a relevant therapeutic target in acute and chronic lymphoid malignancies: down-regulation enhances rituximab-mediated apoptosis and complement-dependent cytotoxicity. Clin Cancer Res. 2007;13:2144–2150. doi: 10.1158/1078-0432.CCR-06-2294. [DOI] [PubMed] [Google Scholar]
- 46.Saxena A, Viswanathan S, Moshynska O, Tandon P, Sankaran K, Sheridan DP. Mcl-1 and Bcl-2/Bax ratio are associated with treatment response but not with Rai stage in B-cell chronic lymphocytic leukemia. Am J Hematol. 2004;75:22–33. doi: 10.1002/ajh.10453. [DOI] [PubMed] [Google Scholar]
- 47.Germain M, Duronio V. The N terminus of the anti-apoptotic BCL-2 homologue MCL-1 regulates its localization and function. J Biol Chem. 2007;282:32233–32242. doi: 10.1074/jbc.M706408200. [DOI] [PubMed] [Google Scholar]
- 48.Czabotar PE, Lee EF, van Delft MF, et al. Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proc Natl Acad Sci U S A. 2007;104:6217–6222. doi: 10.1073/pnas.0701297104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matoba S, Kang JG, Patino WD, et al. p53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 50.Gasco M, Crook T. p53 family members and chemoresistance in cancer: what we know and what we need to know. Drug Resist Updat. 2003;6:323–328. doi: 10.1016/j.drup.2003.11.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}