

Abstract
Iron is essential for all cells but is toxic in excess, so iron absorption and distribution are tightly regulated. Serum iron is bound to transferrin and enters erythroid cells primarily via receptor-mediated endocytosis of the transferrin receptor (Tfr1). Tfr1 is essential for developing erythrocytes and reduced Tfr1 expression is associated with anemia. The transcription factors STAT5A/B are activated by many cytokines, including erythropoietin. Stat5a/b−/− mice are severely anemic and die perinatally, but no link has been made to iron homeostasis. To study the function of STAT5A/B in vivo, we deleted the floxed Stat5a/b locus in hematopoietic cells with a Tie2-Cre transgene. These mice exhibited microcytic, hypochromic anemia, as did lethally irradiated mice that received a transplant of Stat5a/b−/− fetal liver cells. Flow cytometry and RNA analyses of erythroid cells from mutant mice revealed a 50% reduction in Tfr1 mRNA and protein. We detected STAT5A/B binding sites in the first intron of the Tfr1 gene and found that expression of constitutively active STAT5A in an erythroid cell line increased Tfr1 levels. Chromatin immunoprecipitation experiments confirmed the binding of STAT5A/B to these sites. We conclude that STAT5A/B is an important regulator of iron update in erythroid progenitor cells via its control of Tfr1 transcription.
Introduction
Iron is an essential metal, used primarily by erythrocytes for hemoglobin synthesis. Intracellular iron levels must be controlled because iron can participate in redox reactions leading to the generation of damaging free radicals. Since there is no mechanism for regulated iron excretion, iron intake and distribution are tightly regulated. Serum iron circulates bound to transferrin and is taken up by cells via receptor-mediated endocytosis as iron-bound transferrin binds the transferrin receptor (Tfr1).1 Mice lacking Tfr1 display severe anemia and die before embryonic day 12.5 (E12.5), thus supporting its critical role in erythropoiesis.2 Tfr1+/− mice survive to adulthood but have microcytic, hypochromic erythrocytes, which are most likely due to a reduction of cell surface Tfr1 leading to iron-deficient erythropoiesis.2
Hepcidin, a secreted peptide hormone produced primarily by hepatocytes, is a key regulator of iron homeostasis. Hepcidin binds the iron exporter ferroportin, causing its internalization and degradation. Dietary iron enters circulation by traversing intestinal epithelial cells, exiting through ferroportin. Similarly, tissue macrophages that have phagocytosed senescent red blood cells release iron back into circulation through ferroportin. Therefore, hepcidin binding to ferroportin prevents dietary iron uptake and the release of iron from tissue macrophages. Hepcidin production responds to iron stores and erythroid demand and thereby balances iron release into the serum.3
STAT5A and STAT5B are transcription factors that are activated by numerous cytokines, including erythropoietin (Epo).4–6 Upon binding of Epo to its receptor (EpoR), the receptor-associated tyrosine kinase Jak2 activates STAT5A/B, which in turn regulates erythropoiesis.7–10 However, the molecular understanding of this process remains fragmented. Mice from which the entire Stat5a/b locus has been deleted (Stat5a/b−/− mice) display severe anemia and die perinatally,8 precluding definitive analysis of the role of STAT5A/B in erythropoiesis in vivo. In particular, the perinatal lethality has prevented studies of the role of STAT5A/B in iron homeostasis.
To address the function of STAT5A/B in erythropoiesis in vivo, we deleted the Stat5a/b locus in hematopoietic stem cells (HSCs) through the use of mice that carried the Stat5a/b locus flanked by loxP sites and also a Tie2-Cre (TC) transgene11,12 (Stat5a/bf/f; TC), which is known to be active in HSCs. Stat5a/bf/f; TC neonates and adult mice displayed microcytic, hypochromic anemia and liver iron deposition. Here we analyze the molecular basis of the microcytic, hypochromic anemia and link STAT5A/B to transcriptional regulation of Tfr1.
Methods
Mice and genotype analysis
Stat5a/b−/− mice were generated using Cre-mediated recombination.8 Stat5a/bfl/fl mice8 were bred with mice carrying the Tie2-Cre transgene.11,12 Genotyping was performed by polymerase chain reaction (PCR) as described.8,12 The Stat5a/b floxed allele was amplified with primers 5′-AGCAGCAACCAGAGGACTAC-3′ and 5′-TACCCGCTTCCATTGCTCAG-3′. Primers specific for the recombined Stat5a/b allele were 5′-AGCAGCAACCAGAGGACTAC-3′ and 5′-CCCATTATCACCTTCTTTACAG-3′. Tie2-Cre primers were 5′-CGCATAACCAGTGAAACAGCATT-GC-3′ and 5′-CCCTGTGCTCAGACAGAAATGAGA-3′. Wild-type recipient mice (6- to 8-week-old B6.SJL-CD45.1 mice) were purchased from The Jackson Laboratory (Bar Harbor, ME). Animals were handled and housed in accordance with the guidelines of the National Institutes of Health (NIH, Bethesda, MD) Animal Care and Use Committee.
RNA isolation from erythroid cells and quantitative real-time PCR analysis of Stat5 deletion efficiency and Tfr1 and DMT1 expression
Ter119-positive and -negative cells were isolated from fetal liver or adult bone marrow and spleen using magnetic positive selection with anti-Ter119 microBeads (Miltenyi Biotec, Auburn, CA) according to the manufacturer's instructions. RNA was extracted and purified using an RNeasy Plus mini kit (Qiagen, Valencia, CA). Total RNA (0.5 μg) was reverse transcribed into cDNA using the Superscript III First-strand synthesis Supermix (Invitrogen, Frederick, MD). Taqman real-time quantification of mRNA transcript levels was performed using mouse-specific FAM STAT5a primer Mm00839861_m1, Tfr1 primer Mm00441941_ml, DMT1 primer Mm00435363_ml (Applied Biosystems, Foster City, CA), and VIC β-actin primers for normalization. The assay was run on a 7900 HT fast real-time PCR system (Applied Biosystems) and analyzed with SDS2.3 Software (Applied Biosystems).
Complete blood count
Neonates and adult mice were bled from the mandibular vein into 1.5-mL microcentrifuge tubes through heparinized capillary hematocrit tubes (Drummond Scientific, Broomall, PA). Complete blood count (CBC) was measured by HEMAVET multispecies hematology system-HV950FS (Drew Scientific, Dallas, TX) per the manufacturer's instructions.
Giemsa staining
Blood smears were made using peripheral blood from the mouse mandibular vein. Slides were fixed in 100% methanol for 30 minutes, rinsed in water, and stained with freshly made 10% Giemsa for 30 minutes. The right femur was isolated from E18.5 embryos, fixed in neutral buffered formalin (Fisher Scientific, Pittsburgh, PA) at 4°C overnight, dehydrated, and embedded in paraffin. Tissue blocks were sectioned at 5 μm. Bone sections were deparaffinized and stained with freshly made 10% Giemsa for 30 minutes.
Images were captured on an Olympus (Tokyo, Japan) BX51 light microscope equipped with Plan 10×/0.25, Plan-NEOFLUAR 20×/0.50, and UPlan Fl 60×/1.25 Oil Iris lenses and a Nikon (Tokyo, Japan) digital still DXM 1200 camera using ACT-1 (version 2.6.3.0) software. Captured images were processed by Adobe Photoshop (version 9) and Illustrator (version 10) software (Adobe Systems, San Jose, CA).
Tissue iron analyses
Liver and spleen samples were digested in acid and tissue non–heme iron was determined as described previously.13
Serum iron analyses
Serum was separated from whole blood that had been collected by retro-orbital bleed in microtainer serum separator tubes (Becton Dickinson, Franklin Lakes, NJ) and stored at − 20°C. Serum iron and unsaturated iron binding capacity (UIBC) were determined using the Serum Iron/UIBC kit (ThermoDMA, Louisville, CO) according to the manufacturer's instructions.
Perls Prussian blue iron staining
Liver tissues were fixed in neutral buffered formalin (Fisher Scientific) at 4°C overnight, dehydrated, and embedded in paraffin. Tissue blocks were sectioned at 5 μm. Liver sections were deparaffinized and hydrated in distilled water, and incubated in stock potassium ferrocyanide solution for 5 minutes and in working potassium ferrocyanide–hydrochloric acid solution for 20 minutes. Sections were rinsed in distilled water and counterstained in nuclear fast red solution for 5 minutes. Sections were washed in running water and dehydrated in 95% and 100% alcohol, cleared in xylene, and then mounted with Permount (Fisher Scientific).
RNA isolation and quantitative real-time PCR analysis of hepcidin levels
Total RNA was isolated from liver tissue that had been snap-frozen at − 80°C and stored in RNALater (Qiagen). Livers were homogenized in RNA STAT-60 (Leedo Medical Laboratories, Houston, TX). RNA was extracted according to the manufacturer's instructions. Total RNA was treated with DNase I (Roche, Indianapolis, IN) to remove trace DNA, and cDNA was synthesized using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA), according to the manufacturer's instructions. Real-time quantification of mRNA transcript levels was performed using the iQ SYBR Green Supermix (Bio-Rad), according to the manufacturer's instructions. Hepcidin (Hamp1) mRNA was amplified using primers mHamp1-F 5′-CTGAGCAGCACCACCTATCTC-3′ and mHamp1-R 5′-TGGCTCTAGGCTATGTTTTGC-3′. β-Actin (Actb) mRNA was amplified as an internal control using primers mbact F 5′-ACCCACACTGTGCCCATCTA-3′ and mbact R 5′-CACGCTCGGTCAGGATCTTC-3′.14 Standard curves for hepcidin and β-actin were generated from dilutions of cDNA made from liver in parallel to other experimental samples. Samples were run in triplicate and results were reported as the ratio of the mean values for hepcidin to β-actin.
Flow cytometry
Fetal livers were mechanically dissociated in phosphate-buffered saline with 0.5% bovine serum albumin (PBS/0.5% BSA) through a 40-μm strainer and resuspended in PBS/0.5% BSA. Cells were incubated with a phycoerythrin (PE)–conjugated α-Ter119 antibody, FITC-conjugated α-CD71 (Tfr1) antibody, and 7-AAD (7-amino-actinomycin D) for 15 minutes at room temperature. Blood from fetuses, neonates, or adults was collected and stained with a PE-conjugated α-CD71 antibody for 15 minutes at room temperature. After washing with PBS/0.5% BSA, cells were stained with Reti-COUNT (thiazole orange) reagent (BD Biosciences, San Diego, CA) for 30 minutes at room temperature followed directly by fluorescence-activated cell sorting (FACS) analysis. Constitutively active STAT5A-transfected MEL cells were stained with PE-conjugated α-phospho-STAT5A (Y694) or PE-conjugated α-CD71 antibodies for 15 minutes at room temperature. Cells were washed with PBS/0.5% BSA and FACS analysis was performed. All antibodies were purchased from BD Pharmingen (San Diego, CA) and antibody titers were determined before experiments. FACS analyses were carried out using FACSCalibur (BD Biosciences). Data analyses were performed using FlowJo (TreeStar, Eugene, OR).
Microarray analysis
Isolated Ter119-positive fetal liver cells from 3 to 5 E14.5 embryos of the same genotype were combined, and total RNA was extracted using TRIzol reagent (Life Technologies, Bethesda, MD) with 2 additional ethanol precipitations. RNA quality was verified using an Agilent Bioanalyzer (Agilent Technologies, Palo Alto, CA). Microarray analyses were performed using Affymetrix Mouse Genome 430 2.0 array GeneChips (Affymetrix, Santa Clara, CA). Microarray signals were analyzed using the Affymetrix RMA algorithm. Up- and down-regulated genes were selected based on P values less than .05 and fold changes of more than 1.5 or less than 1.5 as assessed by ANOVA using Partek Pro software (Partek, St Louis, MO). Microarray data have been deposited in Gene Expression Omnibus (GEO) under accession number GSE11777.15
Retrovirus production and transfection with constitutively active STAT5A
Recombinant mouse stem cell virus (MSCV) vectors expressing STAT5A (R20, constitutively active mouse STAT5A), in which STAT5A was mutated at the N-terminus and amino acids 1 to 136 were deleted, and green fluorescent protein (GFP) were gifts from Dr Richard Moriggl (Vienna, Austria). The MEL cell line was maintained in Dulbecco modified Eagle medium (DMEM) with 10% FBS, penicillin, and streptomycin. MEL cells that express constitutively active mouse STAT5A were established by transfecting MEL cells with STAT5A/GFP retroviral vectors. MSCV-GFP–transfected MEL cells were set as control. MEL cells were transfected with the cell line Optimization Nucleofector kit (Amaxa, Gaithersburg, MD). Nucleofector Solution L (Amaxa) in combination with program A-20 was used in transfection. Four to 5 hours after transfection, 1.5% DMSO was added to the medium. The cultures were maintained at 37°C with 5% CO2. After 72 hours, the MSCV-STAT5A-GFP– and MSCV-GFP–transfected MEL cells were collected, and activated STAT5A and Tfr1 expression levels were analyzed by FACS.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assays were performed as previously described16 using the Upstate Biotechnology ChIP kit (Temecula, CA). STAT5A/GFP- or GFP-transfected MEL cells were sorted by BD FACSAria Flow Cytometer (BD Biosciences) and were cross-linked immediately using 1% formaldehyde (Protocol Formalin; Fisher Scientific). Cell lysates were sonicated and immunoprecipitated with an α-Stat5 antibody (R&D Systems, Minneapolis, MN) or rabbit serum as a control (Upstate Biotechnology). Immunoprecipitated DNA was eluted and amplified by real-time PCR using a 7900 HT fast real-time PCR system (Applied Biosystems) and analyzed using SDS2.3 Software (Applied Biosystems). Sequence-specific primers and probes used for amplification of the putative STAT binding sites (GAS sites) within the Tfr1 gene were as follows: GAS 1: 5′-CATGGTAGGATCTCAGTTCATGGC-3′ and 5′-CAGGTTACTGAAGGCTTACGATGG-3′; GAS 2: 5′-CTCCCAAGTGCTAGGATTAAAGGC-3′ and 5′-GACTGGATGCTGAATAGAGGTGGG-3′; GAS 3: 5′-GCTCTAGCGATTGGGTCTGTTTC-3′ and 5′-GCCTCTTGCCTCCCAAGTACTAG-3′. Sequence primers outside the GAS sites were as follows: 5′-AGTGTCACGGACATTTAGAGGGG-3′ and 5′-CAGCGTTCAGACCTATTCTGCC-3′. As a positive control, primers that detect the binding site for STAT5A/B on the IGF-1 gene were designed based on the sequences in rat.17 The sequence primers were 5′-GCATATGTCTCTGAAAGGGGTGA-3′ and 5′-GGCACAAGCTAGCCGATGGTTAG-3′, which detect 2 GAS sites in intron 2. All Ct values were normalized to the values from primers outside the Tfr1 GAS sites (2 kb away from GAS 3 site) (ΔCt), and then ΔCt values were normalized to rabbit serum (ΔΔCt). The results (2̂ ΔΔCt) were expressed as fold enrichment relative to GFP control.
Apoptosis assay
Spleens were taken from 8- to 10-week-old Stat5a/bf/f; TC mice or littermate control mice and were resuspended in PBS/0.5% BSA. Ter119-positive cells were separated using magnetic positive selection with anti-Ter119 microBeads (Miltenyi Biotec) according to the manufacturer's instructions. An aliquot of the Ter119-positive cells were stained with annexin V–PE apoptosis detection kit I (BD Pharmingen) following the manufacturer's instructions and then incubated with an APC-conjugated α-Ter119 antibody (BD Pharmingen) for 15 minutes at room temperature. FACS analysis was performed as described in “Flow cytometry.” The splenic Ter119-positive cells were cultured for 24 hours in IMDM with 15% FBS, 1% BSA, 200 μg/mL holo transferrin, 10 μg/mL recombinant human insulin, 2 mM l-glutamine, 10−4 M β-mercaptoethanol, and 5 U erythropoietin and then subjected to FACS analysis for apoptosis at the indicated time points.
Transplantation
Embryonic day-14.5 (E14) fetal liver cells were harvested from Stat5a/b−/− fetuses or control littermates and dispersed with a 21-gauge needle. The cells were resuspended in phosphate-buffered saline containing 2% fetal bovine serum. Cells (1 × 106) were injected via the lateral tail vein into recipient mice that were lethally irradiated with 11Gγ 4 to 5 hours before injection. FACS analysis was used to determine the reconstitution of donor cells. PerCP-Cy5.5–conjugated CD45.2 was used as donor cell marker; and PE-conjugated CD45.1 was used as recipient cell marker. Mice were killed 5 months after transplantation and analyzed.
Results
Deletion of the Stat5a/b locus in the germ line and hematopoietic cells
Stat5a/b+/− mice8 were generated through the deletion of the Stat5a/b locus flanked by loxP sites by an MMTV-Cre transgene, which is active in the female germ line.18 The Stat5a/b locus was selectively deleted in hematopoietic cells in mice carrying 2 floxed Stat5a/b alleles and a Tie2-Cre transgene11 (Stat5a/bf/f; TC mice). Quantitative real-time PCR confirmed the deletion of the Stat5a/b locus in Stat5a/bf/f; TC mice, and Stat5a/b mRNA levels in fetal Ter119-positive and -negative cells were reduced by more than 90% (data not shown).
Stat5a/b+/− males and females were mated, and from the more than 2000 mice weaned only 9 were Stat5a/b−/−. Prior to parturition, Stat5a/b−/− fetuses were present in a normal Mendelian ratio, suggesting that Stat5a/b−/− mice died perinatally. Newborn Stat5a/b−/− mice were anemic with hematocrits of approximately 1.6 (16%; Table 1) and died of unknown cause within hours after delivery. In contrast, Stat5a/bf/f; TC mice were born at the expected Mendelian ratio. Stat5a/bf/f; TC neonates had hematocrits of approximately 2.5 (25%) compared with 4.7 (47%) in controls, and hematocrits in adult mutant mice remained low at 2.9 (29%; Table 1). Both red cell size (mean corpuscular volume) and hemoglobin content (MCH) were significantly reduced in Stat5a/b−/− and Stat5f/f; TC neonates as well as in adult Stat5a/bf/f; TC mice (Table 1), demonstrating that loss of STAT5A/B results in microcytic, hypochromic anemia. Increased red cell distribution width (RDW) in Stat5a/bf/f; TC neonates and adult mice was suggestive of iron deficiency. Stat5a/b−/− neonates were severely anemic, although their RDWs were not elevated, most likely due to a complete inability to produce normal cells. Consistent with the measured parameters, peripheral blood smears of Stat5a/bf/f; TC embryos and neonates subjected to Giemsa staining (Figure 1) showed microcytosis, anisocytosis, poikilocytosis, and hypochromia. The number of mature red blood cells was greatly reduced in Stat5a/bf/f; TC fetuses and neonates compared with wild-type controls, and a higher ratio of nucleated erythroid cells was observed (Figure 1). The morphology of bone marrow from Stat5a/bf/f; TC mice established the presence of red cell hypoplasia (Figure S1, available on the Blood website; see the Supplemental Materials link at the top of the online article). These findings all are consistent with a defect in delivery of iron to developing erythroblasts.
Table 1.
Hematologic indices of neonate and adult mice
| RBC, 109/L | Hb, g/L | HCT | MCV, fl | MCH, pg | RDW, % | |
|---|---|---|---|---|---|---|
| Adults | ||||||
| Control | 9600 ± 300 | 141 ± 3 | 4.95 ± 0.07 | 51.9 ± 1.2 | 15.2 ± 0.3 | 19.1 ± 0.2 |
| Stat5a/bf/f; TC | 6600 ± 500* | 84 ± 5* | 2.86 ± 0.19* | 47.4 ± 0.9* | 13.9 ± 0.3* | 23.2 ± 1.4* |
| Neonates | ||||||
| Control | 4500 ± 400 | 133 ± 14 | 4.73 ± 0.35 | 112.4 ± 2.6 | 30.1 ± 0.8 | 20.7 ± 0.3 |
| Stat5a/bf/f; TC | 2600 ± 200* | 67 ± 5* | 2.55 ± 0.21* | 100.1 ± 3.8* | 26.6 ± 1.1* | 28.8 ± 1.1* |
| Stat5a/b−/− | 1900 ± 600* | 36 ± 10* | 1.6 ± 0.03* | 84.8 ± 1.6* | 19.2 ± 0.3* | 19.6 ± 0.8 |
RBC indicates red blood cell count; Hb, hemoglobin; HCT, hematocrit; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; and RDW, red blood cell distribution width.
P < .05 compared with control. n = 10 adults and n = 20 neonates for each group.
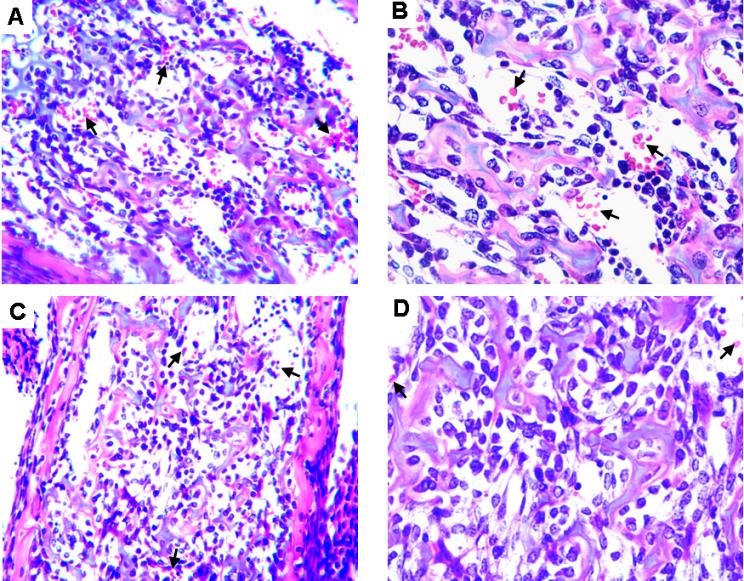
Figure 1.

Peripheral blood smears from control, Stat5a/bf/f; TC, and Stat5a/b−/− E14.5 embryos and neonates, subjected to Giemsa staining. Peripheral blood smears revealed microcytosis, anisocytosis, and hypochromia in Stat5a/b mutant mice, suggestive of iron deficiency. An increase in nucleated erythroblasts (long →) and reticulocytes (short →) further indicates stressed erythropoiesis in mutant mice.  indicates mature red blood cells. Original magnifications: top and middle panels, ×200; bottom panel, ×600.
indicates mature red blood cells. Original magnifications: top and middle panels, ×200; bottom panel, ×600.
To determine whether phenotypic differences observed in Stat5a/b−/− and Stat5a/bf/f; TC mice compared with controls are autonomous to the hematopoietic lineage or due to changes in other tissues potentially affected by the loss of STAT5A/B, we transplanted Stat5a/b−/− fetal liver cells (CD45.2) into lethally irradiated hosts (CD45.1). Five months after transplantation, mice that received a transplant of control fetal liver cells had hematocrits of 3.8 (38%), whereas mice that received a transplant of Stat5a/b−/− cells had hematocrits of 2.6 (26%; Table 2). Both MCV and MCH were reduced in mice that received a transplant of Stat5a/b−/− cells. These data support our conclusion that loss of STAT5A/B in hematopoietic cells alone is sufficient to cause microcytic, hypochromic anemia. In lysed peripheral blood from mice that received a transplant of control cells, 95% of cells were CD45.2 (donor derived). In contrast, only 63% of these cells from mice that received a transplant of Stat5a/b−/− cells were C45.2, suggesting that surviving control recipient cells had a competitive advantage over Stat5a/b−/− cells (data not shown).
Table 2.
Hematologic indices of mice that received a transplant of Stat5a/b−/− or control fetal liver cells
| RBC, 109/L | Hb, g/L | HCT | MCV, fl | MCH, pg | RDW, % | |
|---|---|---|---|---|---|---|
| Control | 8800 ± 200 | 120 ± 3 | 3.83 ± 0.08 | 43.6 ± 0.2 | 13.7 ± 0.1 | 17.1 ± 0.2 |
| Stat5a/b−/− | 6100 ± 300* | 82 ± 5* | 2.63 ± 0.13* | 41.9 ± 0.6* | 12.9 ± 0.3* | 17.3 ± 0.5 |
The mice were analyzed 5 months after transplantation.
RBC indicates red blood cell count; Hb, hemoglobin; HCT, hematocrit; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; and RDW, red blood cell distribution width.
P < .05 compared with control. n = 5 in each group.
Analysis of tissue and serum iron
Microcytic, hypochromic anemia is commonly caused by insufficient iron available to developing erythrocytes. To determine whether Stat5a/b mutant mice had a defect in iron metabolism, tissue and serum non–heme iron levels were analyzed. We measured non–heme iron content in liver tissue from Stat5a/bf/f; TC and wild-type adult mice and Stat5a/bf/f; TC and wild-type neonates and found that the livers of Stat5a/bf/f; TC mice were iron loaded compared with wild-type controls (Figure 2A). In addition, Prussian blue iron staining of liver sections revealed periportal hepatocyte iron staining in Stat5a/bf/f; TC livers but not in controls (Figure 2E). There was no significant difference in spleen non–heme iron content by tissue iron assay or Prussian blue staining (data not shown). Serum iron levels (Figure 2B) and transferrin saturation (Figure 2C) were elevated in Stat5a/bf/f; TC mice compared with controls. These data demonstrate that Stat5a/bf/f; TC mice have sufficient or excess total body iron, suggesting that the anemia is not caused by overall iron insufficiency. Therefore, the anemia is likely caused by an inability of the erythrocytes to assimilate iron. To confirm cell autonomy, we analyzed iron parameters in lethally irradiated control mice that had received a transplant of control or Stat5a/b−/− fetal liver cells. Liver iron levels from mice that received a transplant of Stat5a/b−/− cells were increased more than 2.5-fold compared with controls (Figure 2F), and serum iron levels and transferrin saturation were also significantly increased (Figure 2G,H).
Figure 2.

Iron overload in the absence of STAT5A/B. Iron content and hepcidin expression in control and Stat5a/bf/f; TC mice (A-E) and in mice that received a transplant of control or Stat5a/b−/− fetal liver cells (F-I). (A) Non–heme liver iron in adult and neonate female control and Stat5a/bf/f; TC mice. n = 5 neonates; n = 6 adults. (B,C) Serum iron (B) and transferrin saturation (C) in adult female control and Stat5a/bf/f; TC mice. n = 12 in each group. (D) Hepcidin mRNA expression in neonate and adult control and Stat5a/bf/f; TC mice (males and females). Hepcidin expression in control neonate and adult mice is set to 1. n = 9 in each group. (E) Perls Prussian blue iron staining of liver paraffin sections from adult female control and Stat5a/bf/f; TC mice. Original magnification, ×100 in top panel and ×200 in bottom panel. Non–heme iron is stained blue. (F) Non–heme liver iron. (G) Serum iron. (H) Serum transferrin saturation. (I) Hepcidin mRNA expression. Hepcidin expression in mice that received a transplant of control cells is set to 1. n = 5 in each group for all the measurements. *P < .05 compared with control samples. P values were determined by 2-tailed unpaired t test. Error bars represent SEM.
Hepcidin mRNA levels were analyzed in liver tissue from Stat5a/bf/f; TC and control adult and neonate mice by quantitative real-time PCR. Hepcidin mRNA levels were approximately 2.5-fold higher in mutant neonates than in wild-type controls, and approximately 1.9-fold higher in mutant adults than in wild-type controls (Figure 2D). Similarly, hepcidin levels in mice that received a transplant of Stat5a/b−/− fetal liver cells were elevated 2-fold over controls (Figure 2I).
Transferrin receptor expression in erythroid cells
Iron uptake into erythrocytes occurs by receptor-mediated endocytosis of iron transferrin bound to Tfr1. We assessed cell surface Tfr1 levels on similar populations of reticulocytes from control and Stat5a/bf/f; TC mice by FACS, using reticulocyte RNA content as a measure of cell maturity. We determined that Tfr1 levels were decreased by approximately 50% in adult mutant mice (Figure 3A,B). Tfr1 levels also were significantly reduced in fetal liver Ter119-positive cells from Stat5a/b−/− mice (Figure 3B). As determined from FACS analysis forward scatter, neonate and adult Stat5a/bf/f; TC Ter119-positive cells were 6% to 7% smaller than controls, and Stat5a/b−/− Ter119-positive cells were 23% smaller (data not shown). Gated Ter119-positive cells from lethally irradiated recipient mice that had received a transplant of Stat5a/b−/− cells also expressed significantly less Tfr1 (Figure 3C).
Figure 3.

Decreased Tfr expression in the absence of STAT5A/B. Transferrin receptor expression in control and Stat5a/b mutant Ter119-positive cells and (A-C) transferrin receptor mRNA expression in Stat5a/bf/f; TC and control adult mice (D). (A,B) Gated Ter119-positive cells were analyzed for fluorescence of an α-CD71 (Tfr1) antibody in fetal liver and adult bone marrow and spleen cells. Peripheral blood data were generated by double staining with the α-CD71 antibody and thiazole orange dye, which stains RNA and can be used to isolate reticulocytes of a narrow age range as the amount of RNA changes significantly during maturation. (A) Representative data from 8 adult peripheral blood analyses. Top panels show the gate for thiazole orange–positive cells (right) in the whole red blood cell population (left panel). (B) Quantification of Tfr1 protein levels, expressed as the median measurements of α-CD71 fluorescence. E14 data were generated from Stat5a/b−/− embryos. n = 10 in each group. Adult data were generated from Stat5a/bf/f; TC mice. n = 6 for each group. (C) Quantification of Tfr1 protein levels in mice that received a transplant of control or Stat5a/b−/− fetal liver cells, expressed as the median measurements of α-CD71 fluorescence. n = 5 for each group. FL indicates fetal liver; PB, peripheral blood; and BM, bone marrow. *P < .05 compared with control mice. Error bars represent SEM. (D) Total RNA was extracted from Ter119-positive cells isolated from bone marrow and spleen. Tfr1 expression levels in control mice are set to 1. n = 3 in control group; n = 4 in Stat5a/bf/f; TC group. *P < .05 compared with control mice. Error bars represent SEM.
To investigate whether the reduction in cell surface Tfr1 was due to a change in transcriptional regulation, we isolated total RNA from Stat5a/b−/− and control fetal liver Ter119-positive cells and subjected it to microarray analyses (Document S1). This approach also allowed us to monitor the expression of other genes involved in iron metabolism and erythropoiesis. Tfr1 mRNA levels were decreased by approximately 30% in Stat5a/b−/− erythroid cells, suggesting that the decrease in Tfr1 protein is due to reduced Tfr1 mRNA. Real-time reverse-transcription (RT)–PCR showed Tfr1 mRNA levels reduced by 60% in bone marrow and 80% in splenic cells from adult Stat5a/bf/f; TC mice (Figure 3D). As erythrocytes have a high demand for iron and obtain it only via transferrin and Tfr1,2,19 this reduction in Tfr1 levels could cause iron-restricted erythropoiesis in the Stat5a/b mutant mice. Microarray analyses also confirmed reduced expression of bona fide STAT5A/B target genes, such as Socs3 and Pim1 (Table 3;0).
Table 3.
Microarray analysis of fetal liver Ter119-positive cells from Stat5a/b−/− or control E18.5 embryos
| Gene symbol | Mean, KO | Mean, WT | Mean ratio, KO/WT | P |
|---|---|---|---|---|
| Socs3 | 67.50 | 601.83 | 0.11 | .00 |
| Stat5a | 29.80 | 221.33 | 0.13 | .01 |
| Stat5b | 110.03 | 571.53 | 0.19 | .01 |
| Pim1 | 4657.77 | 12268.10 | 0.38 | .01 |
| Cish | 540.93 | 1209.23 | 0.45 | .02 |
| Hbb-bh1 | 10666.70 | 22056.97 | 0.48 | .04 |
| Osmr | 111.93 | 176.20 | 0.64 | .25 |
| Bcl2l | 11875.93 | 18033.90 | 0.66 | .14 |
| Osm | 257.67 | 369.60 | 0.70 | .04 |
| Trf | 12056.83 | 22319.70 | 0.54 | .04 |
| Trfr | 2666.63 | 3803.30 | 0.70 | .03 |
| Bcl2l | 2093.47 | 2570.73 | 0.81 | .18 |
| Hbb-y | 57118.80 | 65485.93 | 0.87 | .32 |
| Hba-a1 | 60582.80 | 68668.43 | 0.88 | .31 |
| Ccnd1 | 1399.73 | 1561.93 | 0.90 | .58 |
| Actb | 53338.57 | 58914.40 | 0.91 | .33 |
| Eraf | 58985.63 | 64714.10 | 0.91 | .36 |
| Hbb-b2 | 43897.70 | 45628.30 | 0.96 | .74 |
| Ccnd3 | 12132.47 | 12562.27 | 0.97 | .79 |
| Pim2 | 646.43 | 659.43 | 0.98 | .86 |
| Pim3 | 749.00 | 739.33 | 1.01 | .94 |
| Hba-x | 44065.57 | 42822.47 | 1.03 | .93 |
| Stat3 | 1292.90 | 1196.63 | 1.08 | .13 |
| Gapd | 21993.63 | 20195.33 | 1.09 | .39 |
| Gbif-pending | 269.13 | 236.60 | 1.14 | .59 |
| Stat1 | 766.13 | 660.93 | 1.16 | .50 |
| Epor | 8875.87 | 7350.00 | 1.21 | .14 |
| Jak2 | 4147.47 | 3208.57 | 1.29 | .13 |
| Ireb2 | 911.00 | 917.77 | 0.99 | .94 |
Biologic and technical triplicates were analyzed. Highlighted genes' expression levels differ significantly between mutant and control. Listed genes include those considered erythropoiesis-related genes. The entire data set is available in Table S1.
Induction of Tfr1 expression by constitutively active STAT5A
The decreased amount of Tfr1 mRNA and protein in Stat5a/bf/f: TC erythrocytes suggested that STAT5A/B regulates expression of the Tfr1 gene. To investigate whether STAT5A/B activity leads to the induction of Tfr1 gene transcription and protein expression, we transfected the murine erythroleukemia cell line MEL with a viral vector expressing a constitutively active STAT5A.20 FACS analysis of cells 72 hours after transfection revealed activated STAT5A (Figure 4A,B) and a 2-fold increase in cell surface Tfr1 compared with GFP-transfected controls (Figure 4C,D). These analyses demonstrate that STAT5A induces Tfr1 levels in MEL cells.
Figure 4.

Transferrin receptor expression in murine erythroleukemia (MEL) cells transfected with a plasmid encoding constitutively active STAT5A. MEL cells were transfected with a plasmid encoding constitutively active STAT5A and GFP or GFP alone. (A,C) Histogram from FACS analyses of gated GFP-positive cells. (A) Phospho-STAT5A levels in MEL cells. (C) Transferrin receptor (CD71) expression in MEL cells. Quantification of phospho-STAT5A (B) and Tfr1 levels (D) in GFP- and STAT5A/GFP-transfected cells. Representative data are from 1 of 3 individual experiments. Error bars represent SEM. *P < .05 compared with GFP-transfected cells.
Stat5 binds to GAS sites in the Tfr1 gene
To investigate whether STAT5A/B directly induces Tfr1 transcription, we searched for putative STAT5A/B binding sites (GAS sites) surrounding the Tfr1 gene (http://www.ensembl.org).21 We identified 3 GAS consensus sequences in the first intron of Tfr1 (Figure 5A). STAT5A/B previously has been shown to induce transcription via intronic GAS sites in other genes.17 Therefore, we hypothesized that STAT5A/B directly induces Tfr1 transcription by binding to one or more of these putative GAS sites.
Figure 5.

STAT5A/B binding sites in a putative Tfr1 enhancer. (A) Putative STAT5A/B binding (GAS) sites within intron 1 of the Tfr1 gene (underlined). Three conserved GAS sites were identified, with GAS 1 having the highest interspecies conservation. Sites are shown aligned with sequences from other species.21 (B) Chromatin immunoprecipitation (ChIP) analysis of binding of active STAT5A to the putative GAS sites within intron 1 of Tfr1. Biologic and technical triplicates were performed. Error bars represent SEM. *P < .05 compared with GFP-transfected cells.
We used chromatin immunoprecipitation (ChIP) to determine whether STAT5A binds to any of the putative GAS sites. MEL cells were transfected with plasmids encoding constitutively active STAT5A or GFP as a control and STAT5A binding to GAS sites was analyzed. STAT5A binding was detected on all 3 GAS sites, and the most conserved GAS site (site 1) had a 16-fold enrichment compared with the GFP control (Figure 5B). Taken together, these data demonstrate that STAT5A binds to GAS sites in a putative Tfr1 regulatory region and that STAT5A activity leads to increased Tfr1 expression.
Increased apoptosis in the absence of STAT5A/B
Earlier studies have shown that erythroid cells expressing hypomorphic STAT5A/B undergo unscheduled apoptosis due to reduced levels of Bcl-x.9,10 To further establish to what extent apoptosis contributed to the anemia observed in Stat5a/bf/f; TC mice, we analyzed the survival response of splenic Ter119-positive cells to Epo. Ter119-positive cells from mutant and control mice were cultured in the presence of Epo and cell death was determined using annexin V staining (Figure 6). At day 0, 10% of freshly isolated Ter119-positive cells from control mice and 42% from Stat5a/bf/f; TC mice were positive for annexin V. Twenty-four hours after initiating the culture, 53% of control cells and 78% of mutant cells were annexin V positive. The elevated cell death in the absence of STAT5A/B strongly supports the notion that STAT5A/B also controls Epo-mediated cell survival. Bcl-x levels in STAT5A/B-null Ter119-positive cells were only slightly reduced and the significance of this remains to be determined.
Figure 6.

Apoptosis in Stat5a/bf/f; TC and control adult splenic erythroid cells. (A) Freshly isolated splenic Ter119-positive cells were analyzed by FACS using an apoptosis detection kit and Ter119 staining. (B) After 24 hours in culture with Epo, cells were subjected to the same analysis. The left panels are from control mice; the right panels are from Stat5a/bf/f; TC mice. Populations shown were gated as Ter119 positive and 7AAD negative. Representative profile from 1 of 3 individual experiments.
Discussion
Deletion of the Stat5a/b genes in HSCs and in the germ line results in microcytic, hypochromic anemia, and mutant mice display increased serum iron levels and liver iron deposition. In the absence of STAT5A/B, Tfr1 levels on erythroid precursors are reduced by 50%. Furthermore, STAT5A/B binds to GAS sites within the first intron of the Tfr1 gene and induces Tfr1 gene expression. Based on these data, we suggest that STAT5A/B controls erythropoiesis in part by regulating Tfr1 expression, which in turn allows unimpeded iron acquisition by erythroid cells.
Stat5a/b−/− neonates display a hematocrit of 1.6 (16%) compared with 4.7 (47%) in controls. They die within hours after birth, which has precluded detailed studies of the in vivo function of STAT5A/B in adults.8 To further understand the molecular basis of the erythropoietic defects, we deleted the Stat5a/b locus specifically in HSCs and endothelial cells using the Tie2-Cre transgene. Complete blood count analyses revealed that mutant mice were anemic (decreased red blood cell count, hemoglobin, and hematocrit), and had decreased mean corpuscular volume and mean corpuscular hemoglobin, indicating a microcytic, hypochromic anemia. The microcytic anemia was more profound in Stat5a/b−/− neonates than in Stat5a/bf/f; TC mice, suggesting that there might be additional defects from the whole body absence of STAT5A/B. Stat5a/b−/− neonates were smaller than control littermates or Stat5a/bf/f; TC mice. Possible explanations include more severe anemia, placental insufficiency, or a defect in IGF signaling.
Microcytic, hypochromic anemia is most commonly caused by insufficient iron acquisition by developing erythroid precursors. Stat5a/b mutant mice had increased serum iron levels and increased hepatocyte iron deposition, suggesting that the microcytic, hypochromic anemia was the result of insufficient erythroid iron utilization. The presence of abundant periportal hepatocyte iron further supported the hypothesis that excess unused iron had been removed from serum. Although some cells can use non–Tfr1-mediated mechanisms of acquiring iron, erythroid cells depend exclusively on Tfr1-mediated iron uptake.2 Tfr1−/− mice display severe anemia and die before embryonic day 12.5. Since anemia occurred in animals with hematopoietic-specific inactivation of Stat5a/b, we hypothesized that there was a defect in iron transport into erythroid precursors. RNA expression and FACS analyses revealed a decrease in Tfr1 mRNA and surface protein levels, respectively, on mutant erythroid cells. In addition, we found that active STAT5A stimulates expression of Tfr1 and binds to GAS sites in the first intron of the Tfr1 gene. Functional intronic GAS sites have been identified in other genes, such as the IGF-1 gene, which is highly activated by STAT5A/B.17 Given the known function of STAT5A/B as a transcriptional regulator, these data strongly suggest that STAT5A/B up-regulates Tfr1 transcription. Erythroid cells must increase surface Tfr1 levels even as cellular iron content increases, because large amounts of iron are needed for hemoglobin production.22,23 Therefore, although we cannot exclude posttranscriptional regulation of Tfr1 mRNA stability by STAT5A/B, these data support a role for STAT5A/B in controlling Tfr1 transcription.
To determine whether the phenotypic differences observed in Stat5a/b mutant mice compared with controls are autonomous to the hematopoietic lineage or due to changes in other tissues potentially affected by the loss of STAT5A/B, we transplanted Stat5a/b−/− fetal liver cells into lethally irradiated hosts. We found that mice that received a transplant of Stat5a/b−/− cells displayed a phenotype that mimicked that of Stat5a/bf/f; TC mice. These mice had microcytic, hypochromic anemia, increased liver iron, serum iron, and transferrin saturation and decreased erythroid Tfr1 levels. We conclude that loss of STAT5A/B in hematopoietic cells is sufficient to induce these phenotypes.
In addition to erythroid iron deficiency, globin chain imbalance can be a primary cause of microcytosis. We analyzed globin mRNA levels in Stat5a/b−/− fetal liver Ter119-positive cells and found that expression of all but one globin gene was unimpaired (Table 3). Steady-state levels of Hbb-hb1 mRNA (encoding hemoglobin Z, beta-like embryonic chain) were reduced by 50%. We do not believe that decreased embryonic beta chain expression explains the microcytosis that we observed. Mutations in the gene encoding the divalent metal transporter 1 (DMT1) in humans also cause iron overload and hypochromic microcytic anemia.24 Although DMT1 mRNA levels in Stat5a/bf/f; TC mice were reduced by approximately 60% (data not shown), we were unable to locate GAS sites within putative regulatory sequences, suggesting that the reduced expression was a secondary event. However, the absence of bona fide GAS sides does not rule out that the Dmt1 gene is under the control of STAT5A/B.
Unlike the well-studied posttranscriptional regulation of Tfr1 mRNA, transcriptional regulation of Tfr1 remains less well characterized. It has been shown that various growth and differentiation factors can stimulate erythroid Tfr1 transcription.25 In this study, we identified STAT5A/B as critical for efficient expression of the Tfr1 gene in erythroid precursors. STAT5A/B is considered a cytokine-inducible modulator and does not necessarily control basal levels of transcription. An accepted role of STAT5A/B is in the induction of target genes in hormone-responsive tissues to achieve high levels of specific mRNAs rapidly.26–28 Similarly, Epo-induced stimulation of STAT5A/B activity would lead to the induction of erythroid Tfr1 transcription, which would allow sufficient iron to enter erythrocyte precursors for hemoglobin synthesis.
Production of the iron-regulatory hormone hepcidin is modulated in response to changes in iron stores and erythroid demand. Accelerated or ineffective erythropoiesis induces an unknown signal, “the erythroid regulator,” which leads to down-regulation of hepcidin expression in the liver, and consequently an increase in iron entering the system. Conversely, under conditions of iron overload, hepcidin expression is up-regulated by the “stores regulator” to prevent additional iron intake.29 Under some circumstances, mice that are anemic but iron overloaded may have coexisting signals to both down-regulate and up-regulate hepcidin production. In most characterized cases of anemia coupled with iron overload, such as transferrin-deficient mice19 and β-thalassemic mice,30 the erythroid signal is dominant and hepcidin production is markedly reduced. Therefore, we had expected hepcidin expression to be reduced in Stat5a/bf/f; TC mice. However, hepcidin expression was slightly increased in these mice, suggesting that the stores regulator is the dominant signal. As the genetic defect in Stat5a/bf/f; TC mice is restricted to hematopoietic cells, we hypothesize that the stores regulator dominates due to a deficiency in production or signaling of the erythroid regulator.
Similar to Stat5a/bf/f; TC mice, Tfr1+/− mice exhibit decreased cell surface Tfr1 and have microcytic, hypochromic erythrocytes.2 However, unlike Stat5a/bf/f; TC mice, which have fewer red blood cells than wild-type controls, Tfr1+/− mice have a compensatory induction of erythropoiesis and an increased red blood cell count. Whereas Stat5a/bf/f; TC mice have decreased hemoglobin and hematocrit, Tfr1+/− mice display normal hemoglobin levels and hematocrits due to the increase in red blood cell number. It is an open question why Tfr1+/−, but not Stat5a/bf/f; TC, mice exhibit a compensatory increase in red blood cell production. In addition to decreased erythroid iron, Tfr1+/− mice have decreased hepatocyte iron due to the global reduction in Tfr1 expression. This contrasts with Stat5a/bf/f; TC mice, which have a hematopoietic-specific reduction in Tfr1 expression and have increased hepatocyte iron deposition.
In summary, this study provides evidence that STAT5A/B is an important transcriptional regulator of Tfr1 in erythroid cells. Transcriptional regulation of Tfr1 has not been fully characterized in these or other cells. It will be interesting to investigate the importance of STAT5A/B-driven Tfr1 transcription in nonerythroid cell types and to compare how STAT5A/B activity is stimulated in each context.
Supplementary Material
Acknowledgments
The authors thank Susanne Pechhold for cell sorting and Ann Dean for providing MEL cells. We also thank Toshio Kitamura for providing the constitutively active Stat5A construct and Weiping Chen and Maggie Cam for microarray analysis.
This study was supported by the intramural research programs of NIDDK and NIAID, NIH, by NIH R01 HL51057 (N.C.A.) and NIH T32 HL07623 (S.K.M.).
Footnotes
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: B.-M.Z., S.K.M., R.N., J.L., G.W.R., N.C.A., and L.H. designed experiments; B.-M.Z., S.K.M., R.N., J.L., Y.C., C.M., A.K., and G.W.R. executed experiments; B.-M.Z., S.K.M., J.L., Y.C., and C.M. analyzed data; B.-M.Z., S.K.M., R.N., J.L., A.K., G.W.R., N.C.A., and L.H. interpreted data; and B.-M.Z., S.K.M., G.W.R., N.C.A., and L.H. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Lothar Hennighausen or Bing-Mei Zhu, NIH/NIDDK, Bulding 8, Room 101, 8 Center Drive, Bethesda, MD 20892; e-mail: (L.H.) lotharh@mail.nih.gov or (B.-M.Z.) bingmei@mail.nih.gov.
References
- 1.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117:285–297. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- 2.Levy JE, Jin O, Fujiwara Y, Kuo F, Andrews NC. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat Genet. 1999;21:396–399. doi: 10.1038/7727. [DOI] [PubMed] [Google Scholar]
- 3.Andrews NC, Schmidt PJ. Iron homeostasis. Annu Rev Physiol. 2007;69:69–85. doi: 10.1146/annurev.physiol.69.031905.164337. [DOI] [PubMed] [Google Scholar]
- 4.Neubauer H, Cumano A, Muller M, Wu H, Huffstadt U, Pfeffer K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. 1998;93:397–409. doi: 10.1016/s0092-8674(00)81168-x. [DOI] [PubMed] [Google Scholar]
- 5.Parganas E, Wang D, Stravopodis D, et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998;93:385–395. doi: 10.1016/s0092-8674(00)81167-8. [DOI] [PubMed] [Google Scholar]
- 6.Wu H, Liu X, Jaenisch R, Lodish HF. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell. 1995;83:59–67. doi: 10.1016/0092-8674(95)90234-1. [DOI] [PubMed] [Google Scholar]
- 7.Teglund S, McKay C, Schuetz E, et al. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998;93:841–850. doi: 10.1016/s0092-8674(00)81444-0. [DOI] [PubMed] [Google Scholar]
- 8.Cui Y, Riedlinger G, Miyoshi K, et al. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol. 2004;24:8037–8047. doi: 10.1128/MCB.24.18.8037-8047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Socolovsky M, Fallon AE, Wang S, Brugnara C, Lodish HF. Fetal anemia and apoptosis of red cell progenitors in Stat5a-/-5b-/- mice: a direct role for Stat5 in Bcl-X(L) induction. Cell. 1999;98:181–191. doi: 10.1016/s0092-8674(00)81013-2. [DOI] [PubMed] [Google Scholar]
- 10.Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(-/-)5b(-/-) mice due to decreased survival of early erythroblasts. Blood. 2001;98:3261–3273. doi: 10.1182/blood.v98.12.3261. [DOI] [PubMed] [Google Scholar]
- 11.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 12.Sasaki A, Yasukawa H, Shouda T, Kitamura T, Dikic I, Yoshimura A. CIS3/SOCS-3 suppresses erythropoietin (EPO) signaling by binding the EPO receptor and JAK2. J Biol Chem. 2000;275:29338–29347. doi: 10.1074/jbc.M003456200. [DOI] [PubMed] [Google Scholar]
- 13.Torrance JD, Bothwell TH. New York, NY: Churchill Livingstone Press; 1980. Methods in Hematology: Iron. [Google Scholar]
- 14.Nemeth E, Rivera S, Gabayan V, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113:1271–1276. doi: 10.1172/JCI20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.National Center for Biotechnology Information. Gene Expression Omnibus. http://www.ncbi.nlm.nih.gov/geo/
- 16.Morinobu A, Kanno Y, O'Shea JJ. Discrete roles for histone acetylation in human T helper 1 cell-specific gene expression. J Biol Chem. 2004;279:40640–40646. doi: 10.1074/jbc.M407576200. [DOI] [PubMed] [Google Scholar]
- 17.Chia DJ, Ono M, Woelfle J, Schlesinger-Massart M, Jiang H, Rotwein P. Characterization of distinct Stat5b binding sites that mediate growth hormone-stimulated IGF-I gene transcription. J Biol Chem. 2006;281:3190–3197. doi: 10.1074/jbc.M510204200. [DOI] [PubMed] [Google Scholar]
- 18.Wagner KU, Wall RJ, St-Onge L, et al. Cre-mediated gene deletion in the mammary gland. Nucleic Acids Res. 1997;25:4323–4330. doi: 10.1093/nar/25.21.4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trenor CC, III, Campagna DR, Sellers VM, Andrews NC, Fleming MD. The molecular defect in hypotransferrinemic mice. Blood. 2000;96:1113–1118. [PubMed] [Google Scholar]
- 20.Onishi M, Nosaka T, Misawa K, et al. Identification and characterization of a constitutively active STAT5 mutant that promotes cell proliferation. Mol Cell Biol. 1998;18:3871–3879. doi: 10.1128/mcb.18.7.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wellcome Trust Sanger. Ensembl. http://www.ensembl.org.
- 22.Chan LN, Gerhardt EM. Transferrin receptor gene is hyperexpressed and transcriptionally regulated in differentiating erythroid cells. J Biol Chem. 1992;267:8254–8259. [PubMed] [Google Scholar]
- 23.Chan RY, Seiser C, Schulman HM, Kuhn LC, Ponka P. Regulation of transferrin receptor mRNA expression. Distinct regulatory features in erythroid cells. Eur J Biochem. 1994;220:683–692. doi: 10.1111/j.1432-1033.1994.tb18669.x. [DOI] [PubMed] [Google Scholar]
- 24.Pospisilova D, Mims MP, Nemeth E, Ganz T, Prchal JT. DMT1 mutation: response of anemia to darbepoetin administration and implications for iron homeostasis. Blood. 2006;108:404–405. doi: 10.1182/blood-2006-02-003962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lok CN, Loh TT. Regulation of transferrin function and expression: review and update. Biol Signals Recept. 1998;7:157–178. doi: 10.1159/000014542. [DOI] [PubMed] [Google Scholar]
- 26.Burdon T, Sankaran L, Wall RJ, Spencer M, Hennighausen L. Expression of a whey acidic protein transgene during mammary development: evidence for different mechanisms of regulation during pregnancy and lactation. J Biol Chem. 1991;266:6909–6914. [PubMed] [Google Scholar]
- 27.Pittius CW, Sankaran L, Topper YJ, Hennighausen L. Comparison of the regulation of the whey acidic protein gene with that of a hybrid gene containing the whey acidic protein gene promoter in transgenic mice. Mol Endocrinol. 1988;2:1027–1032. doi: 10.1210/mend-2-11-1027. [DOI] [PubMed] [Google Scholar]
- 28.Woelfle J, Chia DJ, Rotwein P. Mechanisms of growth hormone (GH) action: identification of conserved Stat5 binding sites that mediate GH-induced insulin-like growth factor-I gene activation. J Biol Chem. 2003;278:51261–51266. doi: 10.1074/jbc.M309486200. [DOI] [PubMed] [Google Scholar]
- 29.Finch C. Regulators of iron balance in humans. Blood. 1994;84:1697–1702. [PubMed] [Google Scholar]
- 30.Adamsky K, Weizer O, Amariglio N, et al. Decreased hepcidin mRNA expression in thalassemic mice. Br J Haematol. 2004;124:123–124. doi: 10.1046/j.1365-2141.2003.04734.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}