

Abstract
Protein synthesis and its fidelity rely upon the aminoacyl-tRNA synthetases. Leucyl- (LeuRS), isoleucyl- (IleRS) and valyl- (ValRS) tRNA synthetases have evolved a discrete editing domain called CP1 that hydrolyzes the respective incorrectly misaminoacylated noncognate amino acids. Although active CP1 domain fragments have been isolated for IleRS and ValRS, previous reports suggested that the LeuRS CP1 domain required idiosyncratic adaptations to confer editing activity independent of the full-length enzyme. Herein, characterization of a series of rationally designed E. coli LeuRS fragments showed that the β-strands, which link the CP1 domain to the aminoacylation core of LeuRS are required for editing of mischarged tRNALeu. Hydrolytic activity was also enhanced by inclusion of short flexible peptides that have been called “hinges” at the end of both LeuRS β-strands. We propose that these long β-strand extensions of the LeuRS CP1 domain interact specifically with the tRNA for post-transfer editing of misaminoacylated amino acids.
The fidelity of protein synthesis relies upon the interpretation of genetic information by the aminoacyl-tRNA synthetases (aaRSs) (1, 2). Each of the aaRSs is responsible for aminoacylation of its cognate amino acid to a specific set of tRNA isoacceptor molecules. In a two-step reaction, the enzyme binds ATP and its cognate amino acid to form an activated aminoacyl-adenylate intermediate. In the second step, the amino acid moiety is transferred to the 3′ end of the tRNA.
Amino acids that are similar in size, shape, and/or polarity threaten enzyme specificity (3-6). For example, discrimination of amino acids that differ by one methyl group such as isoleucine and valine, was theoretically predicted to yield an error rate that was as high as 1 out of 5 (4). However, a number of aaRSs have evolved amino acid editing mechanisms to clear these types of mistakes (6). Separate active sites for aminoacylation and editing are employed in a ‘double sieve’ model to enhance amino acid selection and discrimination (7, 8). The first ‘coarse’ sieve contains the aminoacylation active site, which activates cognate amino acids along with structurally similar noncognate amino acids. The second ‘fine’ sieve is an editing active site, which hydrolyzes noncognate amino acids that are mischarged, but importantly, excludes the correctly charged amino acids.
Leucyl- (LeuRS), isoleucyl- (IleRS) and valyl- (ValRS) tRNA synthetases are homologous enzymes that belong to subclass IA of the aaRSs (9). These enzymes hydrolyze mischarged amino acids using a discrete domain called connective polypeptide 1 (CP1 (10)) (7, 11-13). The CP1 domain is connected to the canonical aminoacylation core via two β-strand linkers (14-16). The inserted protein domain splits the primary sequence of the Rossmann nucleotide binding fold that is responsible for aminoacylation into two halves (17).
The isolated CP1 domains of Escherichia coli IleRS and Bacillus stearothermophilus ValRS were cloned, expressed as active fusion proteins, and edited mischarged Val-tRNAIle and Thr-tRNAVal, respectively (18). Likewise, the recombinantly isolated CP1 domain of Aquifex aeolicus LeuRS, which is an αβ heterodimer, hydrolyzes mischarged Ile-tRNALeu independent of the main body of the enzyme (19). In contrast, CP1 domains that were isolated from E. coli LeuRS failed to deacylate mischarged Ile-tRNALeu (13, 19). Surprisingly however, the independent CP1 domain hydrolyzed the Ile-minihelixLeu (19). In this report, amino acid editing of the full-length mischarged tRNALeu by the isolated E. coli LeuRS CP1 domain required chimeric protein insertions or additions from the A. aeolicus LeuRS (19).
Based on the X-ray crystal structures and homology models of LeuRS (15, 20-24), we hypothesized that regions outside of the LeuRS CP1 domain were required for tRNA binding in the editing complex to confer hydrolysis. We created a series of CP1 domain fragments from E. coli LeuRS that included varied extensions of the N- and C-terminal β-strand ends. Isolation of each of these recombinant fragments identified active CP1 domain constructs that hydrolyzed mischarged Ile-tRNALeu. We propose that the LeuRS β-strands as well as flexible extensions of the β-strands that have been called “hinges” are important to tRNA binding in the amino acid editing complex of LeuRS.
EXPERIMENTAL PROCEDURES
Materials
Oligonucleotide primers were synthesized by Integrated DNA Technologies, Inc. (Coralville, IA) or Sigma Genosys (Woodlands, TX). The pET-15b vector was purchased from Novagen (San Diego, CA). Cloned Pfu DNA polymerase and dNTPs mix were acquired from Stratagene (La Jolla, CA). Restriction enzymes, (Dpn I, Nde I, and BamH I) and T4 DNA ligase were purchased from Promega (Madison, WI). Calf intestinal phosphatase (CIP) and Bst NI were obtained from New England Biolabs, Inc. (Beverly, MA). Tritium-labeled leucine and isoleucine were procured from Amersham Pharmacia Biotech (Piscataway, NJ). All DNA inserts and mutants were sequenced by either the UIUC core DNA technology services (Urbana, IL) or SeqWright (Houston, TX). Circular dichroism experiments were carried out as previously described (25).
Plasmid Construction and Mutagenesis
Genes encoding fragments of E. coli LeuRS CP1 domain sequences were amplified via the polymerase chain reaction (PCR) using 25 ng of plasmid p15EC3-1 (26) as a template and 125 ng each of a series of complementary forward (Fwd) and reverse (Rev) oligonucleotide primers that contained Nde I and BamH I restriction sites, respectively. These primers included: CP1-VV1: Fwd- 5′ GGAATTCCATATG GTT AAC GAC TAT GAC AAC 3′, Rev- 5′ CGGGATCCG TTA AAC GCC CAT CGC AGT CAG 3′; CP1-VV2: Fwd- 5′ GGAATTCCATATG GTG GAG ATC ACC TTC AAC 3′, Rev- 5′ CGGGATCCG TTA CAC TTT ACG CTC GCC AAC 3′; CP1-VL: Fwd- 5′ GGAATTCCATATG GTG GAG ATC ACC TTC AAC 3′, Rev- 5′ CGGGATCCG TTA CAG GCG GTA GTT CAC TTT 3′; CP1-RL: Fwd- 5′ GGAATTCCATATG CGT TCC GAA GGC GTG GAG 3′, Rev- 5′ CGGGATCCG TTA CAG GCG GTA GTT CAC TTT 3′ and CP1-VA: Fwd- 5′ GGAATTCCATATG GTT AAA ACC ATG CAG CGT 3′, Rev- 5′ CGGGATCCG TTA CGC GCC CCA GTA ACG CTG 3′. Each 50 μL PCR reaction mixture also contained 10 mM dNTPs and 2.5 U Pfu DNA polymerase (Stratagene) in commercially prepared buffer. The reaction mixtures were heated at 95 °C for 1 min, and then the DNA was amplified by PCR for 30 cycles under the following conditions: 95 °C for 30 sec, 55 °C for 45 sec, and 68 °C for 100 sec. The PCR fragments were gel-purified using a Qiaquick gel extraction kit-250 (Qiagen Inc. Valencia, CA) and cleaved with Nde I and BamH I restriction enzymes, followed by gel extraction again. The pET-15b vector also was cleaved with Nde I and BamH I, treated with CIP, and agarose gel-purified.
Each digested PCR fragment and the vector were ligated with T4 DNA ligase at 16 °C overnight. A 1 μL aliquot of the ligation was used to transform E. coli DH5α strain (Stratagene). The plasmid DNA was isolated using a Qiaprep spin mini prep kit-250 (Qiagen Inc.) from the overnight 3 mL culture of a single transformant. Plasmids expressing the E. coli LeuRS CP1 domain fragments were identified as follows: pBETeCP1-13-6 (CP1-VV1; Val236 to Val408), pBETeCP1-8-8 (CP1-VV2; Val230 to Val413), pBETeCP1-26-10 (CP1-VL; Val230 to Leu417), pBETeCP1-18-12 (CP1-RL; Arg226 to Leu417), and pBETeCP1-2-35 (CP1-VA; Val216 to Ala430).
The CP1 wild-type fragments that were enzymatically active, CP1-VA and CP1-RL, were mutated to include T252A (VA-T252A and RL-T252A), T252Y (VA-T252Y and RL-T252Y), D345A (VA-D345A and RL-D345A), T247V (VA-T247V and RL-T247V), T248V (VA-T248V and RL-T248V), T247/8V (VA-T247V/T248V and RL- T247V/T248V), A293D (VA-A293D and RL-A293D), A293E (VA-A293E and RL-A293E), A293R (VA-A293R and RL-A293R) and A293K (VA-A293K and RL-A293K) by PCR-based site directed mutagenesis. The PCR reaction was carried out as described above except either pBETeCP1-2-35 or pBETeCP1-18-12 was included as template. The reaction mixtures were heated at 95 °C for 1 min, and then DNA was amplified by PCR for 25 cycles under the following conditions: 95 °C for 30 sec, 55 °C for 1 min, and 68 °C for 15 min. Each PCR reaction was digested with 20 U Dpn I and a 1 μL aliquot was used for transformation of E. coli DH5α.
Wild-type E. coli LeuRS plasmid (p15EC3-1 (26)) was mutated to include either A293E or A293R substitutions via PCR single-site mutagenesis using the following primers: A293E: Fwd- 5′ C ACC AAA GTT GCC GAA GAA GAA ATG GCG ACG ATG G 3′, Rev- 5′ C CAT CGT CGC CAT TTC TTC TTC GGC AAC TTT GGT G 3′ and A293R: Fwd- 5′ C ACC AAA GTT GCC GAA CGT GAA ATG GCG ACG ATG G 3′, Rev- 5′ C CAT CGT CGC CAT TTC ACG TTC GGC AAC TTT GGT G 3′. The mutant plasmids containing the full-length E. coli LeuRS A293E (pAMWp47) and A293R (pAMWp38) were sequenced by Lone Star Labs (Houston, TX).
Protein Purification
E. coli BL21 strain (Stratagene, La Jolla, CA) was transformed using mutant and wild-type plasmids that encoded fragments of the CP1 domain or full-length protein. A single transformant was transferred to a 3 mL LB culture with 100 μg/mL ampicillin (LB-Ap) and incubated at 37 °C overnight. A 500 mL culture of LB-Ap was inoculated with the entire overnight 3 mL culture and grown at 37 °C. When the OD600 was between 0.6 and 0.8, the culture was induced with 1 mM isopropyl-β-D-thiogalactopyranoside for 2 hours. The cells were then harvested at 6000 rpm for 15 min in an Avanti J-E centrifuge with a JLA-10.500 rotor (Beckman Coulter, Fullerton, CA). The pellet was resuspended in 9 mL HA1 buffer [20 mM Na2HPO4, 10 mM tris(hydroxymethyl) aminomethane (Tris) pH 8.0, 100 mM NaCl, and 5% glycerol] and sonicated at 40 amps for 2 min using a Sonics Vibra Cell Sonicator (Sonics, Newtown, CT). The lysate was collected after centrifugation at 12,000 rpm for 30 min at 4 °C. The lysate was combined with HIS-Select™ HF Nickel Affinity Resin (Sigma, St. Louis, MO) that was pre-equilibrated with HA1. The resin with lysate was mixed at 4 °C for 1 hour and centrifuged at low speed in a clinical centrifuge. The resin was then washed five times with 12 mL HA2 buffer [20 mM Na2HPO4, 10 mM Tris, pH 7.0, 500 mM NaCl, and 5% glycerol]. The six-histidine tagged protein was eluted with HA3 buffer [100 mM imidazole in HA1 buffer]. The protein was then concentrated using either a centricon-10 or centricon-50 apparatus (Millipore, Billerica, MA). A Bio-Rad Protein Assay (Bio-Rad Laboratories, Hercules, CA) was carried out to determine the final concentration of the proteins.
Preparation of tRNALeu
The plasmid ptDNAleu14 (450 μg) containing the gene of E. coli tRNALeuUAA (tRNALeu) (27) was digested overnight with 25 U Bst NI in a 1 mL reaction mixture at 60 °C and then used as a template for T7 RNA polymerase run-off transcription to generate tRNALeu (28). The six-histidine tagged T7 RNA polymerase protein was expressed from plasmid p6HRNAP, that was provided by the laboratory of Dr. K. Musier-Forsyth (University of Minnesota, MN) and purified as described (29). Each 1 mL transcription reaction contained 40 mM Tris, pH 8.0, 30 mM MgCl2, 5 mM dithiothreitol (DTT), 0.01 % Triton-X-100, 50 μg/mL bovine serum albumin (BSA), 7.5 mM of each NTP, 80 mg/mL polyethylene glycol (PEG8000), 0.04 U/μL RNase inhibitor (Eppendorf, Westbury, NY), 5 mM spermidine, 80 μg/mL template, 8 μg/mL pyrophosphatase (Sigma, St. Louis, MO) and 1 μM T7 RNA polymerase. The reaction mixture was incubated for 6 hours at 42 °C and the tRNA product purified on a 10 % polyacrylamide (19:1), 8 M urea denaturing gel via electrophoresis. After detection of the tRNALeu band by UV shadowing, the gel was excised, crushed and incubated overnight at 37 °C in 0.5 M NH4OAc and 1 mM ethylenediaminetetraacetic acid (EDTA) pH 8.0. The supernatant was collected and concentrated with butanol extractions to a 500 μL volume. The concentrated tRNALeu was then ethanol-precipitated using 0.5 mg/mL of glycogen. The pellet was washed twice with 70 % ethanol and dried, followed by resuspension in nuclease-free Milli-Q Ultrapure Water (Millipore, Billerica, MA). Purified tRNALeu was denatured at 80 °C for 1 min, followed by addition of 1 mM MgCl2 and quick cooling on ice. The concentration of the tRNA was determined at 260 nm using an extinction coefficient of 840,700 L/mol·cm (30).
Preparation of charged and mischarged tRNALeu
Purified tRNALeu was aminoacylated with either 20.5 μM [3H]-leucine (200 μCi/mL) in the presence of 1 μM E. coli wild type LeuRS for 1 hour or 20.5 μM [3H]-isoleucine (650 μCi/mL) with 1 μM of an editing-defective mutant of E. coli LeuRS for 3 hours. Each reaction included 60 mM Tris, pH 7.5, 10 mM MgCl2, 4 mM ATP, 1 mM DTT and 8 to 20 μM tRNALeu. The reactions were stopped by addition of 0.18 % acetic acid, followed by two equal volume extractions with phenol/chloroform/isoamyl alcohol (125:24:1; Fisher Biotech, Fair Lawn, NJ) that had been equilibrated to pH 4.3 (31). A one-half volume of 4.6 M NH4OAc, pH 5.0, was added to the aminoacylated tRNA, followed by ethanol precipitation. The dried pellet was resuspended in 10 mM KH2PO4, pH 5.0.
Charged and mischarged tRNALeu were quantitated as follows: Six 3 μL duplicate aliquots were transferred onto pads soaked with 5 % cold trichloroacetic acid (TCA). Three of the six pads were washed three times with 5 % TCA and once with 70 % cold ethanol followed by a 5 minute incubation in ether. The air-dried washed and unwashed pads were quantitated by scintillation counting using a Beckman LS 6000IC (Beckman Instruments, Inc., Fullerton, CA) to determine the total amount of free and charged [3H]-leucine or [3H]-isoleucine. The mischarged Ile-tRNALeu yield varied from 9 to 20 %, whereas the Leu-tRNALeu yields were near 100 %.
Post-transfer editing assays
The post-transfer editing activity was measured using a reaction mixture consisting of 60 mM Tris, pH 7.5, 10 mM MgCl2 and either [3H]-Leu-tRNALeu or [3H]-Ile-tRNALeu (12). The reactions were initiated by the addition of enzyme as indicated in the appropriate figure legend. Enzyme and substrate concentrations for kinetic analysis were optimized to capture initial velocities for post-transfer editing. Thus, a range of 0.1 μM to 1 μM mischarged Ile-tRNALeu concentrations were incorporated into enzymatic reactions for the full-length LeuRS and its mutants. Likewise, a range of 0.1 μM to 0.8 μM concentrations of mischarged Ile-tRNALeu transcript were used to measure kinetic parameters for CP1-VA and its mutants. The enzyme concentration for the wild-type LeuRS was 40 nM. The A293D and A293E mutant LeuRSs were 75 nM, while the A293R and A293K mutant LeuRSs were 100 nM. The enzyme concentrations used for kinetic analysis of the CP1-VA construct and mutations at the A293 site were as follows: wild type, 150 nM; VA-A293D and VA-A293E, 200 nM; VA-A293R and VA-A293K, 100 nM. At selected time intervals, reaction aliquots were quenched on pads pre-wetted with 5 % TCA. The pads were washed, dried, and quantitated as described above.
RESULTS
Design of enzymatically active isolated E. coli LeuRS CP1 domain fragments
We designed E. coli LeuRS fragments that contained the CP1 domain based on the X-ray crystal structure of T. thermophilus LeuRS (15, 22), the homology model of E. coli LeuRS (20) and primary sequence homology. The CP1 domain of T. thermophilus LeuRS was identified as the domain that extends from residues Ile224 to Tyr417, which corresponds to Ile224 and Tyr415 respectively, in E. coli LeuRS (15). These residues encompass the complete CP1 domain insertion and both the N- and C-terminal β-strand linkers. Molecular dynamics of the homology model of E. coli LeuRS (20), which was based on the crystal structure of the T. thermophilus LeuRS, suggested longer β-strand linkers to the main body. The N-terminal β-strand linker spans residues Arg226 to Val236 and the C-terminal β-strand extended from Val408 to Ala430 (Figure 1) (20). In addition, the co-crystal structure of T. thermophilus LeuRS (21) indicated two flexible motifs on the N- and C-terminal β-strand extensions that interact with tRNALeu and are involved in rotating the CP1 domain by about 35° (21). These flexible motifs which have been referred to as “hinges” are 220-QRNWIGRSEG (220-QRAWIGRSEG in T. thermophilus) and 411-RKVNYRL (413-GRVTYRL in T. thermophilus) on the N- and C-terminal β-strands, respectively (21). We hypothesized that inclusion of these flanking tRNALeu-binding extensions of the β-strand linkers would confer amino acid editing activity to the isolated CP1 domain of E. coli LeuRS.
Figure 1.

Primary and tertiary structure of LeuRS CP1 domain. (A) Homology model of E. coli LeuRS (20). The CP1 domain is shown in green and the canonical main body of LeuRS is gray. The N-terminal β-strand linker and β-strand extension are colored in red and orange, respectively. The C-terminal β-strand linker and β-strand extension are colored in blue and light blue, respectively. The N- and C-terminal end residues of different CP1 constructs are highlighted. This structure was generated using Viewerlite Version 5.0 (Accelrys Inc.) (B) Primary sequence alignment of the E. coli LeuRS CP1 domain flanking regions containing both the N- and C-terminal β-strand linkers and extensions. Residue numbers for the first and last amino acids are noted at the beginning and end of each primary sequence respectively. The number of residues in the CP1 domain between the N- and C-terminal β-strand sequences is indicated in parentheses. Arrows mark N-terminal residues including Val216, Arg226, Val230, Val236 and C-terminal residues including Val408, Val413, Leu417, Ala430 of different CP1 constructs. The N- and C-terminal β-strand linkers are indicated as red and blue arrows, respectively. The N- and C-terminal β-strand extensions that have been included in the CP1 domains are shown as orange and light blue bars, respectively. Pink boxes highlight dynamic motifs in the N- and C-terminal β-strand extensions that have been proposed to act as “hinges” (21). Residues of the conserved motif shaded in gray interact with the tRNA in the co-crystal structure model (21). Each of the CP1 domain constructs are indicated as follows: CP1-VV1, Val236 to Val408; CP1-VV2, Val230 to Val413; CP1-VL, Val230 to Leu417; CP1-RL, Arg226 to Leu417 and CP1-VA, Val216 to Ala430.
Five LeuRS CP1-containing fragments, which had varying lengths of N- and C-terminal extensions, were constructed and expressed recombinantly. As shown in Figure 1, CP1-VV1 (Val236 to Val408) completely lacked the β-strand linkers that connect the CP1 domain to the aminoacylation core. CP1-VV2 (Val230 to Val413) included portions of both of the β-strand linkers that were associated directly with the folded CP1 domain, while CP1-VL (Val230 to Leu417) extended the C-terminal β-strand linker to include the C-terminal flexible motif (411-RKVNYRL) that has been called a “hinge” (21). Both N- and C-terminal β-strand linkers were included in CP1-RL (Arg226 to Leu417), which encompass a portion of the N-terminal flexible motif and the complete C-terminal flexible motif (Figure 1). The largest construct, CP1-VA included the flexible hinge motifs on both the N- and C-terminal extensions of the CP1-domain (Figure 1). Multiple x-ray crystallography structures in different substrate-bound complexes showed that these extensions of the β-strand linkers interact with tRNALeu are dynamic and undergo conformational changes as the LeuRS transitions between the aminoacylation and editing complexes (15, 21, 23).
Each of the recombinant CP1 domain fragments were purified by affinity chromatography via a six-histidine N-terminal fusion and tested for post-transfer editing activity. The longest fragments, CP1-VA and CP1-RL of 215 and 192 amino acids respectively, were active in the hydrolysis of Ile-tRNALeu (Figure 2). The specific activities of CP1-VA and CP1-RL were respectively 64.8 U/mg and 5.0 U/mg compared to a specific activity of 340 U/mg for the full-length E. coli LeuRS. The shortest protein fragments, CP1-VV1, CP1-VV2 and CP1-VL were not functional in editing mischarged Ile-tRNALeu, even at high concentrations of 5 μM enzyme (Figure 2). This indicated that the β-strands and its extensions were important to the hydrolytic activity of the CP1 domain. These results contrast with a previous report where two longer 261 (Thr129 to Gly389) and 305 amino acid (Ala126 to Ala430) fragments of the E. coli CP1 domain failed to hydrolyze Ile-tRNALeu (13, 19). The N-terminal end of these constructs (13, 19) cuts into a portion of the canonical aminoacylation core that is responsible for aminoacylation activity. It is possible that the N-terminal ends of these long fragments did not fold properly and blocked essential interactions with the tRNA.
Figure 2.
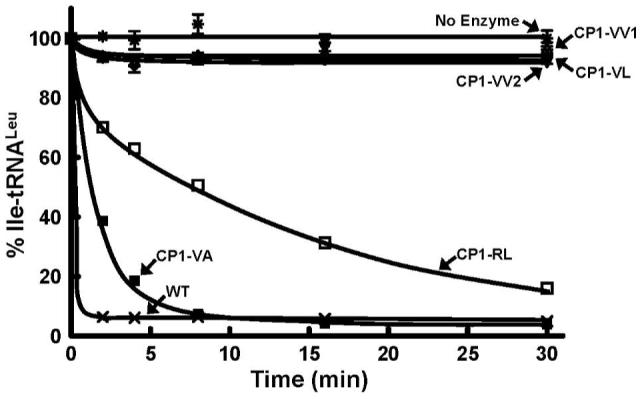
Hydrolytic post-transfer editing activity of isolated CP1 domain-containing fragments of E. coli LeuRS. Each editing reaction was carried out in the presence of 5 μM enzyme and 0.2 μM in vitro transcribed Ile-tRNALeu. Symbols represent activity by full-length LeuRS wild type (WT) and CP1-containing fragments as follows: wild type-WT (×), CP1-VA (■), CP1-RL (□), CP1-VV1 (△), CP1-VV2 (▽), CP1-VL (◇) and no enzyme (*).
We hypothesized that the β-strand extensions of the CP1 domain provide important interactions to facilitate tRNA binding. Since homogeneous pools of mischarged tRNALeu cannot be obtained in yields greater than 30% (32), we measured interactions of the active CP1 domain with pure uncharged tRNALeu. The KD for the CP1-VA and CP1-RL domain fragments with uncharged tRNA at 25 °C were 1.5 ± 0.5 μM and 1.5 ± 0.8 μM, respectively. At higher temperatures, the interactions could not be reproducibly measured. It is possible that the flexible β-strand extensions were more dynamic at higher temperatures and had higher dissociation rates. This would be consistent with other investigations that relied on peptide models to investigate weak protein-RNA interactions (33, 34), which might be reminiscent of tRNA interactions with the unconstrained CP1 β-strands. Analysis by circular dichroism (CD) showed that the inclusion of tRNA with CP1-VA (Figure 3) and CP1-RL (data not shown) improved the signal for the CD spectra of the protein. The CD spectra of the longest CP1-VA construct in the presence of uncharged tRNA indicated a loss of secondary structure at 37 °C compared to 25 °C (Figure 3). We propose that at least at lower temperatures (35), a significant population of the flexible β-strands and their extensions were ordered when tRNA was bound to the CP1 domain construct.
Figure 3.
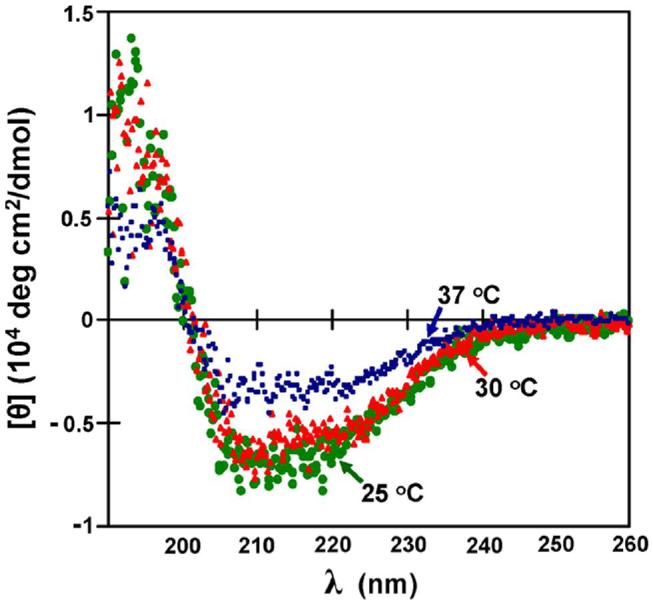
Circular dichroism spectrum of CP1-VA bound to tRNALeu. A 450 μL sample of 2 μM CP1-VA and 1 μM tRNALeu transcript in 5 mM KPi, pH 7.5, was measured using a 0.1-cm path cell in a Jasco J-720 spectropolarimeter. The temperature was controlled using a Fischer Scientific circulating water bath. Background signals from the cell and the buffer containing the tRNA were subtracted from each spectrum. The spectra measured at 25 °C (●), 30 °C (▲) and 37 °C (■) were colored in green, red and blue, respectively.
Mutations that had been shown to inactivate post-transfer editing activity of full-length LeuRS were introduced into each of the active CP1 domains. A universally conserved aspartic acid that anchors the amino moiety of the bound editing substrate in LeuRS, IleRS, and ValRS was changed to an alanine (22, 36). As found for the full-length enzyme, the D345A mutation in the CP1-VA and CP1-RL isolated domains abolished hydrolytic post-transfer editing activity (Figure 4).
Figure 4.
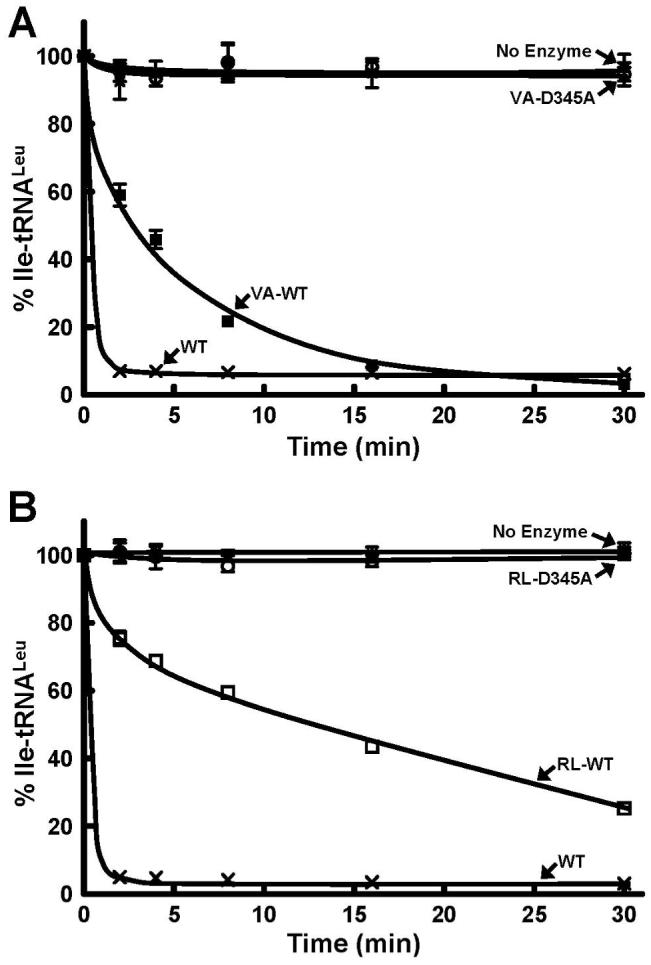
Hydrolytic post-transfer editing activity of wild type and D345A mutant E. coli LeuRS CP1 domains. Panels A and B refer to the editing activity of the mischarged Ile-tRNALeu by CP1-VA (1 μM) and CP1-RL (5 μM), respectively. Each hydrolytic editing reaction was carried out in the presence of 0.2 μM in vitro transcribed Ile-tRNALeu. Symbols represent activity by full-length LeuRS wild type-WT (×) and CP1-containing fragments as follows: VA-WT (■), RL-WT (□), D345A (○) and no enzyme (*).
Amino acid specificity and activity of the editing active site
The threonine-rich region, 247-TTRPDT within the CP1 domain of LeuRS and IleRS has been shown to be important for editing activity (12, 16, 37-40). A highly conserved Thr252 in LeuRS that blocks leucine substrate from binding efficiently in the amino acid binding pocket of the editing active site is an essential substrate discriminator (12, 38). The T252Y mutation occupies the amino acid binding pocket and blocks the binding of substrate to abolish editing activity (37, 39). Introduction of the T252Y mutation into CP1-VA and CP1-RL decreased hydrolysis of mischarged Ile-tRNALeu to background levels (Figure 5) similar to the full-length mutant LeuRSs (37, 39).
Figure 5.
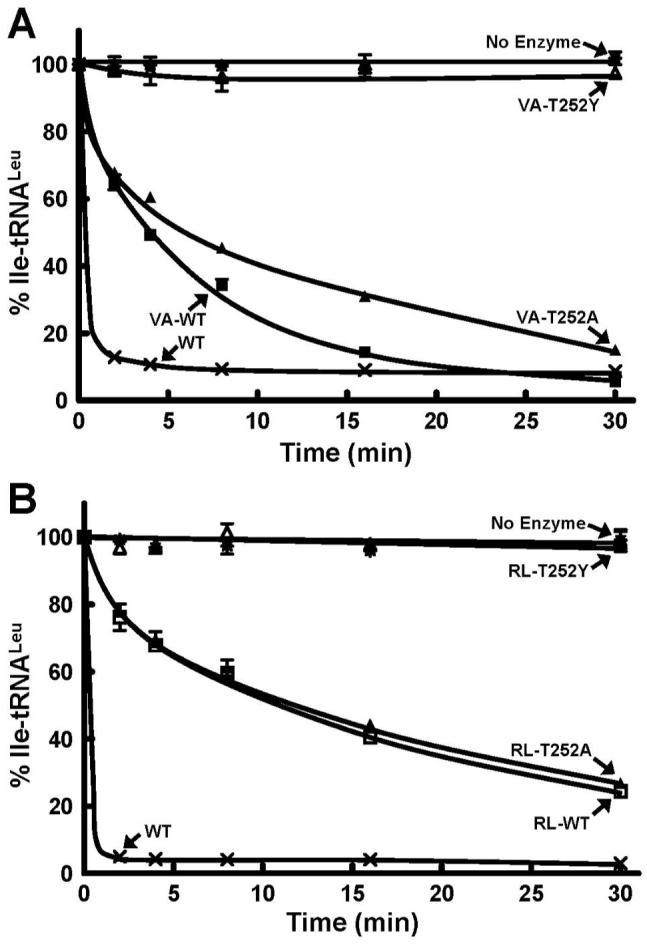
Hydrolytic post-transfer editing activity of wild type and T252 mutants of E. coli LeuRS CP1 domains. Panels A and B refer to the hydrolysis activity of the mischarged Ile-tRNALeu by CP1-VA (1 μM) and CP1-RL (5 μM), respectively. Each reaction was carried out in the presence of 0.2 μM in vitro transcribed Ile-tRNALeu. Symbols represent activity by full-length LeuRS wild type-WT (×) and CP1-containing fragments as follows: VA-WT (■), RL-WT (□), T252A (▲), T252Y (△) and no enzyme (*).
Mutation of Thr252 to alanine in the full-length LeuRS uncouples specificity and hydrolyzes correctly charged Leu-tRNALeu (12, 37, 38). Introduction of the T252A mutation into the most active CP1-VA domain conferred hydrolysis of Leu-tRNALeu (Figure 6) as found for the full-length T252A mutation. However, a T252A mutation in the smaller CP1-RL did not deacylate the Leu-tRNALeu, even at high concentrations of 5 μM enzyme (data not shown). Both of the T252A mutations within the CP1-RL and CP1-VA exhibited significant Ile-tRNALeu hydrolysis activity indicating that they have maintained competent hydrolytic editing active sites (Figure 5). We propose that the loss of interactions of the tRNA in the smaller truncations of the CP1-containing fragment combined with the more open amino acid binding pocket fail to adequately bind or accurately orient the correctly charged tRNALeu for hydrolysis by the CP1-RL-T252A mutant.
Figure 6.
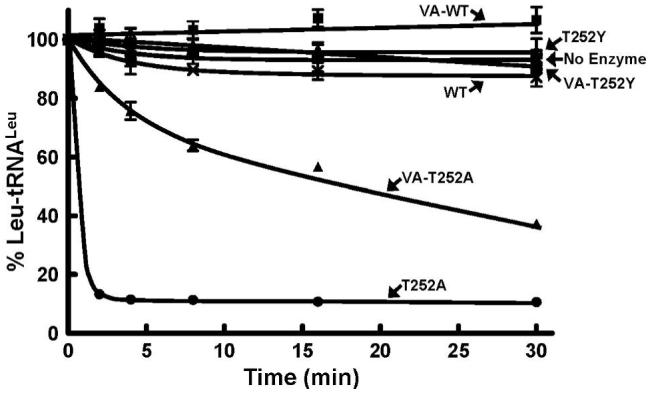
Hydrolytic post-transfer editing activity of cognate Leu-tRNALeu by wild type and mutant E. coli LeuRS CP1 domains. Each editing reaction was carried out in the presence of 5 μM enzyme and 0.5 μM in vitro transcribed Leu-tRNALeu. Editing activity of full-length LeuRS enzymes are represented as follows: Wild type-WT (×), T252A (●), T252Y (○). Symbols that represent the activity of CP1 domains are VA-WT (■), T252A (▲), T252Y (△) and no enzyme (*).
The neighboring Thr247 and Thr248 within this threonine-rich region of the CP1 domain interact with a post-transfer editing substrate analogue containing norvaline (Nva2AA) in the crystal structure of T. thermophilus LeuRS (22). Mutation in the full-length LeuRS required a double mutation of these two conserved threonines to significantly impair post-transfer editing (32). Single mutations of Thr247 and Thr248 to valine exhibited minimal effects on the full-length LeuRS. However, simultaneous mutation to the double valine variant in LeuRS abolished post-transfer editing activity (32). When these mutations were introduced into the CP1-VA and CP1-RL, the double mutant, T247V/T248V and both the single mutants, T247V and T248V completely abolished hydrolysis of Ile-tRNALeu (Figure 7). Thus, similar to the hydrolysis of Leu-tRNALeu by the T252A mutation in the full-length LeuRS and isolated CP1 domains, post-transfer editing activity of the neighboring threonine residues were sensitive to the loss of potential tRNA binding interactions with the missing aminoacylation core that is found in all class I tRNA synthetases. In addition, structural and biochemical work has shown that a unique C-terminal domain in E. coli LeuRS, which is also missing in the active isolated CP1 domains, is required in the full-length enzyme for editing activity (25).
Figure 7.
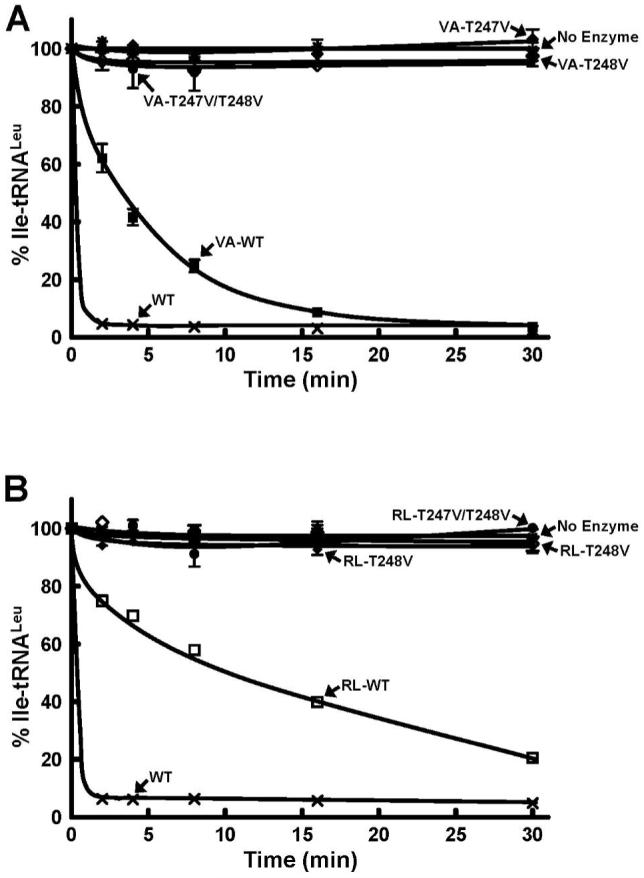
Hydrolytic post-transfer editing activity of wild type, T247V and T248V mutants of E. coli LeuRS CP1 domains. Panels A and B refer to the editing activity of the mischarged Ile-tRNALeu by CP1-VA (1 μM) and CP1-RL (5 μM), respectively. Each hydrolytic editing reaction was carried out with 0.2 μM in vitro transcribed Ile-tRNALeu. Symbols represent activity by full-length LeuRS wild type-WT (×), VA-WT (■), RL-WT (□), T247V (◆), T248V (◇), T247V/T248V (●) and no enzyme (*).
The mobile Ala293-containing peptide influences post-transfer editing activity
The five crystal structures (15, 21-24) and the homology model (20) that are available for LeuRS in different substrate-bound complexes show different conformations for the Ala293-containing peptide. It is clear that this dynamic region of LeuRS is important to one or more functions of the enzyme, but its mechanism and precise role remains unclear. Docking investigations using the homology model of E. coli LeuRS suggest that this region can act as another amino acid binding site (20). The flexible loop within the CP1 domain has also been proposed to play a role in translocation or selective release of the adenylate intermediate for pre-transfer editing (41). It is also possible that it is important to post-transfer editing. We hypothesized that this dynamic peptide, which spans residues 290-300 within the CP1 domain, assumes different conformations as LeuRS carries out aminoacylation, editing, and also translocation of the tRNA from the aminoacylation to the editing site. The downstream neighboring region of Arg295 (Ala297 to Lys302) in the T. thermophilus co-crystal structure which corresponds to Ala293 in E. coli LeuRS is disordered and could also be involved in these multiple steps during enzyme catalysis (21).
The Ala293 site is semi-conserved. As Figure 8A shows, in many LeuRS enzymes a positively charged arginine or lysine residue often replaces the alanine. Introduction of a lysine or aspartic acid exhibited minimal effects on the overall aminoacylation activity (41). We carried out a similar mutational analysis of Ala293 in CP1-VA and CP1-RL. When Ala293 was mutated to a negatively charged aspartic acid (A293D) or glutamic acid (A293E) in CP1-VA, the post-transfer editing activity was similar to the wild type isolated domain (Figure 8B & C). Likewise, CP1-RL-A293D (Figure 8D) and CP1-RL-A293E (data not shown) mutant activities were similar to the wild type CP1 domain (Figure 8D). This is consistent with the full-length LeuRS A293D mutations (41). However, the post-transfer editing activity of CP1-VA was enhanced when substituted by a positively charged arginine (A293R) or lysine (A293K) (Figure 8B & C). Measurement of the kinetic parameters showed that the small increase in relative activity was due to a modest increase in the kcat (Table 1). The A293R (Figure 8D) and A293K (data not shown) mutations in CP1-RL also enhanced the amino acid editing activity.
Figure 8.
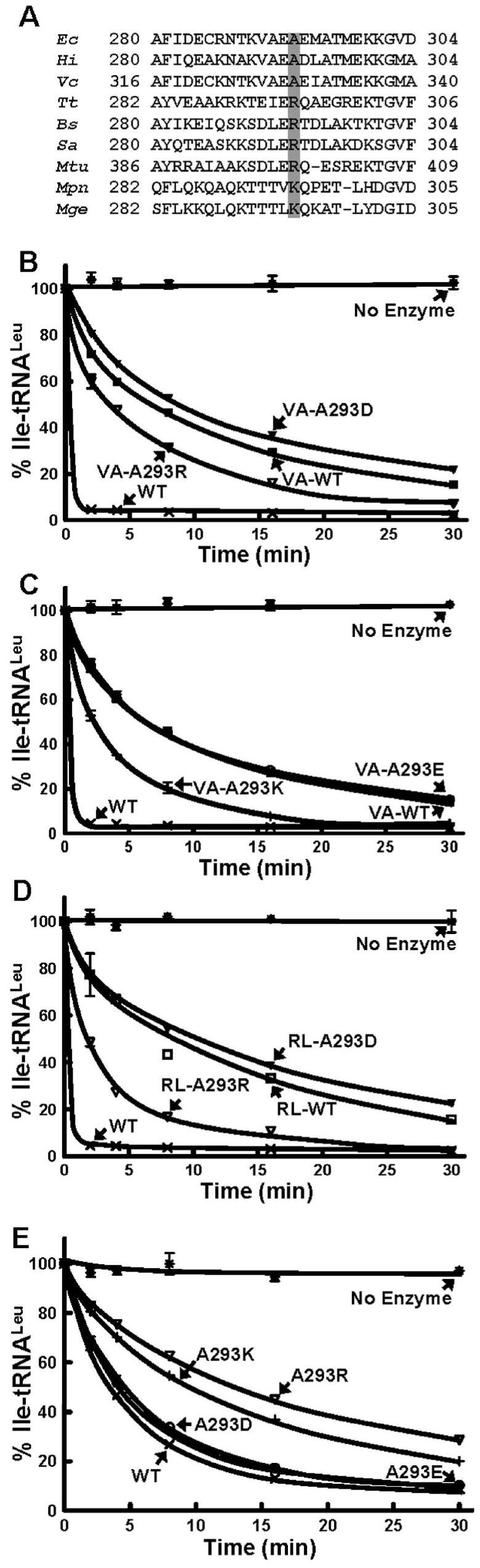
Hydrolytic post-transfer editing activity of wild type and Ala293-based mutations in the CP1 domains. (A) Primary sequence alignment of the Ala293 region of the CP1 domains. The semi-conserved residues shaded in gray at the Ala293 position are either arginine or lysine in most species. (B & C) Editing activity by 1 μM of CP1-VA and its Ala293 LeuRS mutants. (D) Editing activity by 5 μM of CP1-RL and its Ala293 LeuRS mutants. (E) Editing activity by 20 nM of full-length wild-type and Ala293 mutant LeuRSs. All editing reactions were carried out in the presence of 0.3 μM mis-charged Ile-tRNALeu. Symbols represent activity by full-length WT (×), VA-WT (■), RL-WT (□), A293D (▼), A293R (▽), A293E (○), A293K (+) and no enzyme (*).
Table 1.
Apparent kinetic parameters for post-transfer editing activity of Ile-tRNALeu by wild-type and A293 mutant LeuRSs and the isolated CP1-VA domain
| Mutations | KM (μM)a | kcat (s-1) | kcat/KM (μM-1s-1) | Relative kcat/KM |
|---|---|---|---|---|
| Wild-type | 0.14 ± 0.02 | 0.05 ± 0.01 | 0.32 ± 0.02 | 1 |
| A293D | 0.21 ± 0.10 | 0.05 ± 0.02 | 0.22 ± 0.05 | 0.7 |
| A293E | 0.17 ± 0.06 | 0.04 ± 0.005 | 0.22 ± 0.09 | 0.7 |
| A293R | 0.18 ± 0.06 | 0.01 ± 0.006 | 0.06 ± 0.03 | 0.2 |
| A293K | 0.27 ± 0.17 | 0.02 ± 0.01 | 0.08 ± 0.02 | 0.3 |
| CP1-VA | 0.66 ± 0.3 | 0.0015 ± 0.0008 | 0.0021 ± 0.0003 | 0.007 |
| VA-A293D | 0.89 ± 0.5 | 0.0012 ± 0.0003 | 0.0014 ± 0.0007 | 0.005 |
| VA-A293E | 2.0 ± 0.8 | 0.004 ± 0.002 | 0.0019 ± 0.0002 | 0.006 |
| VA-A293R | 1.9 ± 1.0 | 0.006 ± 0.003 | 0.0033 ± 0.0007 | 0.01 |
| VA-A293K | 1.9 ± 0.9 | 0.009 ± 0.003 | 0.0047 ± 0.0005 | 0.01 |
The measured KM is an apparent value.
We also tested these charged amino acid substitutions at the Ala293 position for their effect on post-transfer editing activity in the full-length LeuRS. Hydrolysis activity of E. coli LeuRS A293R and A293K were lower than the wild-type LeuRS (Figure 8E), which contrasts to the effect of these mutations in the isolated CP1 domains. Again, these alterations in activity were primarily due to small changes in the kcat (Table 1). The negatively charged A293D and A293E LeuRS hydrolytic activities were similar to the wild-type (Figure 8E) as found for these mutations in the isolated CP1 domains. Although these effects of Ala293 mutants in the isolated CP1 and the full-length LeuRS were not large, they were reproducible. We hypothesize that this region in the CP1-VA and CP1-RL may interact with the mischarged tRNALeu during post-transfer editing to facilitate hydrolysis. However, this interaction appears to be hindered in the context of the full-length protein. It has been proposed that the Ala293-containing peptide of the CP1 domain interacts with the surface of the canonical LeuRS core via a peptide that contains a conserved lysine (Lys186) (41). Thus, it is possible in the context of the full-length E. coli LeuRS, introduction of a positive charge at Ala293 might block the Lys186-dependent interaction (41) with the main body.
DISCUSSION
LeuRS, IleRS and ValRS contain homologous CP1 domains, which are responsible for editing incorrectly charged non-cognate amino acids. The isolated CP1 domain of E. coli IleRS (275 amino acids) and B. stearothermophilus ValRS (202 amino acids) hydrolyze mischarged Val-tRNAIle and Thr-tRNAVal, respectively (18). Likewise, the isolated CP1 domain of A. aeolicus LeuRS (332 amino acids) edits mischarged Ile-tRNALeu (19). Previous attempts to isolate an active CP1 domain of E. coli LeuRS that was independent from the main body were unsuccessful (13, 19). These included very long CP1 domain extensions of approximately 100 amino acids which contained portions of the LeuRS aminoacylation core (13, 19). We have shown here that absence of the β-strand linkers also yields an inactive CP1 domain editing active site. This contrasts with IleRS and ValRS (18) as well as the ancient A. aeolicus LeuRS (19).
The co-crystal structure of the LeuRS complex for T. thermophilus (21) suggest that two short flexible “hinge” motifs on both the N- and C-terminal β-strand extensions interact with the single-stranded 3′ end of the tRNA. In addition, these two flexible “hinge” motifs undergo conformational changes upon rotation of the CP1 domain by about 35° from the position observed in the uncomplexed LeuRS structure (21). Thus, these peptide regions are dynamic in their interactions with the tRNA as the enzyme shifts between the aminoacylation, editing, and exit complexes of tRNA-bound LeuRS during catalysis (21). Temperature-dependent CD results and equilibrium binding measurements suggest that the RNA-protein complex, where the active CP1 domain fragment is bound to tRNA, is more stable at lower temperatures. In addition, mutational analysis indicates that the β-strand peptide extensions, which include the hinge region, enhance specificity of the amino acid editing reaction. Thus, we propose that the activity as well as specificity of the LeuRS CP1 domain is dependent on distal RNA-protein interactions that occur with the flexible β-strand extensions.
The two “hinge” motifs on the N- and C-terminal extensions of the CP1 domain in T. thermophilus LeuRS (21) correlate to 220-QRNWIGRSEG and 411-RKVNYRL, respectively in E. coli LeuRS. The homology model of E. coli LeuRS showed that the residues, Gln220 to Gly225 of the N-terminal motif comprise part of an α-helix in the main body of the LeuRS and Arg226 to Gly229 are part of the N-terminal β-strand linker (20). X-ray crystal structures of T. thermophilus LeuRS suggest that the conserved Ser227 of the N-terminal β-strand dynamic motif in E. coli LeuRS interacts with the C74 ribose of tRNA when the post-transfer editing substrate analogue binds in the editing active site (21).
The homology model (20) indicates that the C-terminal flexible motif (411-RKVNYRL) exists as a part of the C-terminal β-strand linker. The Arg418 of T. thermophilus LeuRS, which corresponds to Arg416 in E. coli LeuRS, interacts with the A73 discriminator base of the tRNA in the exit conformation (21). In addition, the model superimposing the co-crystal structures of T. thermophilus and P. horikoshii LeuRS suggests that the conserved arginine-rich motif 418-RLRDWLISRQRYWG of T. thermophilus (416- RLRDWGVSRQRYWG in E. coli LeuRS) interacts with the 3′ end of the tRNA in the aminoacylation conformation (21, 23). This model also predicted that several basic arginine residues within this conserved region, are important for tRNA binding (21). For example, the main chain carboxyl group of Arg420 (Arg418 in E. coli) forms a hydrogen bond with N6 of A73 of the tRNA (21). Likewise, the side chain of Arg426 (Arg424 in E. coli) interacts via a hydrogen bond with the phosphate linker between C74 and C75 of the tRNA (21). In yeast mitochondrial LeuRS, mutational analysis also showed that this peptide that extends from the C-terminal β-strand is important to aminoacylation (42).
Based on multiple X-ray crystal structures, we hypothesized that regions in the β-strand extensions of the enzyme might provide important interactions with the tRNA to bind and orient its mischarged 3′ end for post-transfer editing. We created a series of E. coli LeuRS fragments to identify sites outside of the CP1 domain that contribute to amino acid editing. Our shortest active fragment (CP1-RL; Arg226 to Leu417) included both the N- and C-terminal β-strand linkers in addition to the CP1 domain. The N-terminal β-strand linker residues of CP1-RL overlap with the N-terminal “hinge” motif and the C-terminal β-strand linker includes the dynamic C-terminal “hinge” motif 411-RKVNYRL (21) suggesting that it is important in interacting with the tRNA in the editing complex as the crystal structure predicts. The requirement for the C-terminal motif is also consistent with a previously reported E. coli LeuRS isolated CP1 fragment (Thr129 to Gly389) that was inactive and is missing all seven of the amino acids that comprise the dynamic C-terminal extension (13).
Significantly, extension of the β-strand ends to include both the N- and C-terminal flexible “hinge” motif residues that interact with tRNA during post-transfer editing, generated an active isolated CP1 domain that was more reminiscent of the specificity of the full-length LeuRS. A conserved Thr252 residue blocks charged leucine from binding to the editing active site in LeuRS (12, 22, 38). The T252A mutation that uncouples specificity in the full-length LeuRS required both extensions of the β-strands in the CP1-VA construct to confer hydrolysis of Leu-tRNALeu.
Our mutational analysis of CP1-VA and CP1-RL included substitution of the semi-conserved Ala293 to both positive and negatively charged residues. We have previously proposed that in the context of the full-length protein, the Ala293 site might be more important for interacting with the main body of LeuRS (41). Introducing a positive charge at this position could repel a putative Lys186-mediated interaction that orients the main body and CP1 domain (41) and could account for a small decrease in the kcat during post-transfer editing. In contrast, in the isolated CP1 domains, this region appears to be utilized for interactions with the tRNA. In this case, positive charges at the Ala293 site in the CP1 domain fragments (A293R and A293K) would be expected to promote hydrolysis because of increased tRNA binding. Kinetic measurements for the VA-A293K and VA-A293R mutations showed that a small increase in KM for these mutants was offset by a lower kcat to increase kcat/KM. Thus, we hypothesize that introduction of these positively charged residues into the CP1 domain fragment provide additional electrostatic contributions to aid RNA-protein interactions, and increase the efficiency of hydrolysis during amino acid editing.
Previously, fusion of the tRNA binding β-subunit of A. aeolicus to the CP1 domain fragment of E. coli LeuRS that spanned Ala126 to Ala430, also conferred hydrolysis of Ile-tRNALeu (19). In addition, the insertion of an idiosyncratic 20-amino acid motif from the CP1 domain of A. aeolicus stimulated editing activity of the isolated CP1 domain of E. coli LeuRS (19). It was hypothesized that the β-subunit or 20 amino acid peptide from A. aeolicus, which is rich in lysine and arginine residues, might stabilize interactions between the isolated CP1 domain and the mischarged tRNALeu substrate (19). This might be true in the case of the chimeric enzyme, but our investigation shows that the native E. coli LeuRS CP1 domain can be activated by simply including its own N- and C-terminal β-strand linker extensions. This is consistent with X-ray crystal structures that show interactions with the tRNA (21, 23). Since the ancient A. aeolicus LeuRS appears to be a one-of-a-kind αβ LeuRS heterodimer that originates from a split gene, we propose that our results with the E. coli enzyme is a better prototype for the LeuRSs.
Overall, these results demonstrate that an isolated fragment of LeuRS, which contains the editing active site, is capable of hydrolytic activity independent of the full-length LeuRS. Significantly however, portions of the flanking regions from the main body are required for this activity. We hypothesize that these flanking regions of the N- and C-terminal β-strands, which contain dynamic peptides that have been labeled as “hinge motifs” (21), are important to tRNA interactions in the post-transfer editing complex. These RNA-protein interactions that originate in the canonical core of LeuRS, which are somewhat distal to the editing active site, clearly orient and stabilize the end of the charged tRNA for hydrolysis.
ACKNOWLEDGEMENTS
We thank an anonymous reviewer for insightful comments on the role of the CP1 domain’s extended β-strands for amino acid editing.
Footnotes
This work was supported by the National Institutes of Health (Grant GM63789) and The Robert A. Welch Foundation (E-1404)
- tRNA
- transfer RNA
- LeuRS
- leucyl-tRNA synthetase
- IleRS
- isoleucyl-tRNA synthetase
- ValRS
- valyl-tRNA synthetase
- CP1
- connective polypeptide 1
- CD
- circular dichroism
REFERENCES
- 1.Martinis SA, Plateau P, Cavarelli J, Florentz C. Aminoacyl-tRNA synthetases: a family of expanding functions. EMBO J. 1999;18:4591–4596. doi: 10.1093/emboj/18.17.4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martinis SA, Plateau P, Cavarelli J, Florentz C. Aminoacyl-tRNA synthetases: a new image for a classical family. Biochimie. 1999;81:683–700. doi: 10.1016/s0300-9084(99)80126-6. [DOI] [PubMed] [Google Scholar]
- 3.Fersht AR. Editing mechanisms in protein synthesis. Rejection of valine by the isoleucyl-tRNA synthetase. Biochemistry. 1977;16:1025–1030. doi: 10.1021/bi00624a034. [DOI] [PubMed] [Google Scholar]
- 4.Pauling L. Festschrift für Prof. Dr. Arthur Stöll. Birkhauser Verlag; Basel, Switzerland: 1958. The probability of errors in the process of syntheis of protein molecules; pp. 597–602. [Google Scholar]
- 5.Englisch S, Englisch U, von der Haar F, Cramer F. The proofreading of hydroxy analogues of leucine and isoleucine by leucyl-tRNA synthetases from E. coli and yeast. Nucleic Acids Res. 1986;14:7529–7539. doi: 10.1093/nar/14.19.7529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hendrickson TL, Schimmel P. Transfer RNA-dependent amino acid discrimination by aminoacyl-tRNA synthetases. In: Lapointe J, Brakier-Gingras L, editors. Translation Mechanisms. Kluwer Academic / Plenum Pubs; New York: 2003. pp. 34–64. [Google Scholar]
- 7.Fersht AR, Dingwall C. Evidence for the double-sieve editing mechanism in protein synthesis. Steric exclusion of isoleucine by valyl-tRNA synthetases. Biochemistry. 1979;18:2627–2631. doi: 10.1021/bi00579a030. [DOI] [PubMed] [Google Scholar]
- 8.Fersht AR. Sieves in sequence. Science. 1998;280:541. doi: 10.1126/science.280.5363.541. [DOI] [PubMed] [Google Scholar]
- 9.O’ Donoghue P, Luthey-Schulten ZA. On the evolution of structure in aminoacyl-tRNA synthetases. Microbiol Mol Biol Rev. 2003;67:550–573. doi: 10.1128/MMBR.67.4.550-573.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Starzyk RM, Webster TA, Schimmel P. Evidence for dispensable sequences inserted into a nucleotide fold. Science. 1987;237:1614–1618. doi: 10.1126/science.3306924. [DOI] [PubMed] [Google Scholar]
- 11.Lin L, Schimmel P. Mutational analysis suggests the same design for editing activities of two tRNA synthetases. Biochemistry. 1996;35:5596–5601. doi: 10.1021/bi960011y. [DOI] [PubMed] [Google Scholar]
- 12.Mursinna RS, Lincecum TL, Jr., Martinis SA. A conserved threonine within Escherichia coli leucyl-tRNA synthetase prevents hydrolytic editing of leucyl-tRNALeu. Biochemistry. 2001;40:5376–5381. doi: 10.1021/bi002915w. [DOI] [PubMed] [Google Scholar]
- 13.Chen JF, Guo NN, Li T, Wang ED, Wang YL. CP1 domain in Escherichia coli leucyl-tRNA synthetase is crucial for its editing function. Biochemistry. 2000;39:6726–6731. doi: 10.1021/bi000108r. [DOI] [PubMed] [Google Scholar]
- 14.Silvian LF, Wang J, Steitz TA. Insights into editing from an Ile-tRNA synthetase structure with tRNAIle and mupirocin. Science. 1999;285:1074–1077. [PubMed] [Google Scholar]
- 15.Cusack S, Yaremchuk A, Tukalo M. The 2 Å crystal structure of leucyl-tRNA synthetase and its complex with a leucyl-adenylate analogue. EMBO J. 2000;19:2351–2361. doi: 10.1093/emboj/19.10.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nureki O, Vassylyev DG, Tateno M, Shimada A, Nakama T, Fukai S, Konno M, Hendrickson TL, Schimmel P, Yokoyama S. Enzyme structure with two catalytic sites for double-sieve selection of substrate. Science. 1998;280:578–582. doi: 10.1126/science.280.5363.578. [DOI] [PubMed] [Google Scholar]
- 17.Hou YM, Shiba K, Mottes C, Schimmel P. Sequence determination and modeling of structural motifs for the smallest monomeric aminoacyl-tRNA synthetase. Proc Natl Acad Sci U S A. 1991;88:976–980. doi: 10.1073/pnas.88.3.976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin L, Hale SP, Schimmel P. Aminoacylation error correction. Nature. 1996;384:33–34. doi: 10.1038/384033b0. [DOI] [PubMed] [Google Scholar]
- 19.Zhao MW, Zhu B, Hao R, Xu MG, Eriani G, Wang ED. Leucyl-tRNA synthetase from the ancestral bacterium Aquifex aeolicus contains relics of synthetase evolution. EMBO J. 2005;24:1430–1439. doi: 10.1038/sj.emboj.7600618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee KW, Briggs JM. Molecular modeling study of the editing active site of Escherichia coli leucyl-tRNA synthetase: two amino acid binding sites in the editing domain. Proteins: Struct., Funct., Genet. 2004;54:693–704. doi: 10.1002/prot.10300. [DOI] [PubMed] [Google Scholar]
- 21.Tukalo M, Yaremchuk A, Fukunaga R, Yokoyama S, Cusack S. The crystal structure of leucyl-tRNA synthetase complexed with tRNALeu in the post-transfer-editing conformation. Nat Struct Mol Biol. 2005;12:923–930. doi: 10.1038/nsmb986. [DOI] [PubMed] [Google Scholar]
- 22.Lincecum TL, Jr., Tukalo M, Yaremchuk A, Mursinna RS, Williams AM, Sproat BS, Van Den Eynde W, Link A, Van Calenbergh S, Grøtli M, Martinis SA, Cusack S. Structural and mechanistic basis of pre- and post-transfer editing by leucyl-tRNA synthetase. Mol Cell. 2003;11:951–963. doi: 10.1016/s1097-2765(03)00098-4. [DOI] [PubMed] [Google Scholar]
- 23.Fukunaga R, Yokoyama S. Aminoacylation complex structures of leucyl-tRNA synthetase and tRNALeu reveal two modes of discriminator-base recognition. Nat Struct Mol Biol. 2005;12:915–922. doi: 10.1038/nsmb985. [DOI] [PubMed] [Google Scholar]
- 24.Liu Y, Liao J, Zhu B, Wang ED, Ding J. Crystal structures of the editing domain of Escherichia coli leucyl-tRNA synthetase and its complexes with Met and Ile reveal a lock-and-key mechanism for amino acid discrimination. Biochem J. 2006;394:399–407. doi: 10.1042/BJ20051249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hsu JL, Rho SB, Vannella KM, Martinis SA. Functional divergence of a unique C-terminal domain of leucyl-tRNA synthetase to accommodate its splicing and aminoacylation roles. J Biol Chem. 2006;281:23075–23082. doi: 10.1074/jbc.M601606200. [DOI] [PubMed] [Google Scholar]
- 26.Martinis SA, Fox GE. Non-standard amino acid recognition by Escherichia coli leucyl-tRNA synthetase. Nucleic Acids Symp Ser. 1997;36:125–128. [PubMed] [Google Scholar]
- 27.Tocchini-Valentini G, Saks ME, Abelson J. tRNA leucine identity and recognition sets. J Mol Biol. 2000;298:779–793. doi: 10.1006/jmbi.2000.3694. [DOI] [PubMed] [Google Scholar]
- 28.Sampson JR, Uhlenbeck OC. Biochemical and physical characterization of an unmodified yeast phenylalanine transfer RNA transcribed in vitro. Proc Natl Acad Sci U S A. 1988;85:1033–1037. doi: 10.1073/pnas.85.4.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ellinger T, Ehricht R. Single-step purification of T7 RNA polymerase with a 6-histidine tag. Biotechniques. 1998;24:718–720. doi: 10.2144/98245bm03. [DOI] [PubMed] [Google Scholar]
- 30.Puglisi JD, Tinoco I., Jr. Absorbance melting curves of RNA. Methods Enzymol. 1989;180:304–325. doi: 10.1016/0076-6879(89)80108-9. [DOI] [PubMed] [Google Scholar]
- 31.Schreier AA, Schimmel PR. Transfer ribonucleic acid synthetase catalyzed deacylation of aminoacyl transfer ribonucleic acid in the absence of adenosine monophosphate and pyrophosphate. Biochemistry. 1972;11:1582–1589. doi: 10.1021/bi00759a006. [DOI] [PubMed] [Google Scholar]
- 32.Zhai Y, Martinis SA. Two conserved threonines collaborate in the Escherichia coli leucyl-tRNA synthetase amino acid editing mechanism. Biochemistry. 2005;44:15437–15443. doi: 10.1021/bi0514461. [DOI] [PubMed] [Google Scholar]
- 33.Long KS, Crothers DM. Interaction of human immunodeficiency virus type 1 Tat-derived peptides with TAR RNA. Biochemistry. 1995;34:8885–8895. doi: 10.1021/bi00027a041. [DOI] [PubMed] [Google Scholar]
- 34.Weeks KM, Crothers DM. RNA binding assays for Tat-derived peptides: implications for specificity. Biochemistry. 1992;31:10281–10287. doi: 10.1021/bi00157a015. [DOI] [PubMed] [Google Scholar]
- 35.Smith CA, Chen L, Frankel AD. Using peptides as models of RNA-protein interactions. Methods Enzymol. 2000;318:423–438. doi: 10.1016/s0076-6879(00)18067-x. [DOI] [PubMed] [Google Scholar]
- 36.Bishop AC, Nomanbhoy TK, Schimmel P. Blocking site-to-site translocation of a misactivated amino acid by mutation of a class I tRNA synthetase. Proc Natl Acad Sci U S A. 2002;99:585–590. doi: 10.1073/pnas.012611299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mursinna RS, Martinis SA. Rational design to block amino acid editing of a tRNA synthetase. J Am Chem Soc. 2002;124:7286–7287. doi: 10.1021/ja025879s. [DOI] [PubMed] [Google Scholar]
- 38.Mursinna RS, Lee KW, Briggs JM, Martinis SA. Molecular dissection of a critical specificity determinant within the amino acid editing domain of leucyl-tRNA synthetase. Biochemistry. 2004;43:155–165. doi: 10.1021/bi034919h. [DOI] [PubMed] [Google Scholar]
- 39.Tang Y, Tirrell DA. Attenuation of the editing activity of the Escherichia coli leucyl-tRNA synthetase allows incorporation of novel amino acids into proteins in vivo. Biochemistry. 2002;41:10635–10645. doi: 10.1021/bi026130x. [DOI] [PubMed] [Google Scholar]
- 40.Xu MG, Li J, Du X, Wang ED. Groups on the side chain of T252 in Escherichia coli leucyl-tRNA synthetase are important for discrimination of amino acids and cell viability. Biochem Biophys Res Commun. 2004;318:11–16. doi: 10.1016/j.bbrc.2004.03.180. [DOI] [PubMed] [Google Scholar]
- 41.Williams AM, Martinis SA. Mutational unmasking of a tRNA-dependent pathway for preventing genetic code ambiguity. Proc Natl Acad Sci U S A. 2006;103:3586–3591. doi: 10.1073/pnas.0507362103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nawaz MH, Pang YL, Martinis SA. Molecular and functional dissection of a putative RNA-binding region in yeast mitochondrial leucyl-tRNA synthetase. J Mol Biol. 2007;367:384–394. doi: 10.1016/j.jmb.2006.12.051. [DOI] [PubMed] [Google Scholar]