

Abstract
Background
Rapid release testing reduces the waiting period for administration of time-sensitive cell therapy products. Current assay systems are labor intensive and time consuming. The Endosafe® Portable Test System (PTS™) is a chromogenic Limulus Amebocyte Lysate (LAL) portable endotoxin detection system which provides quantitative results in approximately 15 minutes. To evaluate Endosafe® performance, specifically with cell therapy products, side-by-side testing of traditional LAL systems, and the Endosafe® system was conducted at the Production Assistance for Cellular Therapies (PACT) facilities and the National Institutes of Health's Department of Transfusion Medicine.
Methods
Charles River Laboratories provided each center with a PTS™ reader and two commercially prepared lyophilized Reference Standard Endotoxin (RSE) vials. All samples tested with the Endosafe® system used 0.05 - 5.0 endotoxin units/mL (EU/mL) sensitivity cartridges provided by Charles River. Each vial was reconstituted with LAL water and tested in triplicate using the Endosafe® and in-house LAL methods. Subsequently, each center tested endotoxin content of standard dilutions of cell therapy products thus creating paired test results for each sample. Additionally, fabricated endotoxin-positive samples containing varying concentrations of endotoxin were prepared and shipped to all centers to perform blinded testing.
Results
Valid paired results, based on each center's LAL method and the Endosafe® system criteria were analyzed. Endotoxin detection between paired results was equivalent in most cases.
Discussion
The Endosafe® system provided reliable results with products typically produced in cell therapy manufacturing facilities, and would be an appropriate test on which to base the release of time-sensitive cell therapy products.
Keywords: Endotoxin, LAL, Endosafe® PTS™, Comparability
Introduction
Endotoxin is a complex molecule of polysaccharide, lipid A, and other cell wall components that is present in intact bacterial cells. The toxin exerts its effects by inducing pyrogenicity and increasing cell wall permeability. Clinically, endotoxin can result in diarrhea, septic shock, complement activation, marrow necrosis, and multiple other effects [1]. Testing for endotoxin has, therefore, become a standard release test for cellular and gene therapy products that are manufactured using in vitro culture or manipulation, and for certain non-homologous products that are to be administered without cryopreservation. The maximum acceptable level of endotoxin in these products is usually 5.0 EU/kg/dose [2].
The US Food and Drug Administration (FDA) has approved two methods for endotoxin testing, the rabbit pyrogen test described in 21 CFR 610.13, and the more widely used Limulus Amebocyte Lysate (LAL) method. When LAL was first introduced in the 1970s it was limited to the gel clot method, which employed multiple dilutions of the sample and controls. Today, kinetic methods utilizing microtiter plate readers are more common. Partly because of its sensitivity to technical variables, the LAL assay is notoriously demanding. Very careful set-up and pyrogen-free materials, including accessories and water, must be used. Creation of dilutions to construct standard curves and addition of control standard endotoxin spikes to test for possible inhibition or enhancement by the test sample, add another layer of complexity. These details influence the performance of the endotoxin assay. Failure of the assay for technical reasons and consequent repetition can delay infusion of non-cryopreserved products for 3-4 hours, potentially affecting product quality and patient well-being.
Recently, Charles River Laboratories introduced a Portable Test System (PTS™) that can assay endotoxin in approximately 15 minutes using an automated miniaturized LAL-based assay (Endosafe® PTS™ system.) The PTS™ cartridges are licensed and approved by the FDA for endotoxin testing. The Endosafe® PTS™ system is primarily intended to provide a rapid assay for detecting endotoxin in drugs, medical devices, biological products, and could, therefore, replace slower LAL assays currently in use for the release of cell therapy products.
Production Assistance for Cellular Therapies (PACT) is an NHLBI initiative to support the manufacture of cell therapy products. PACT is comprised of the Baylor College of Medicine, Center for Cell and Gene Therapy (CAGT); the University of Minnesota, Molecular and Cellular Therapeutics (MCT); and the University of Pittsburgh Cancer Institute (UPCI), with the EMMES Corporation serving as the administrative center [3]. In collaboration with the National Institutes of Health (NIH) Department of Transfusion Medicine (DTM), the PACT centers designed a study to evaluate the Endosafe® PTS™ system for the rapid detection of endotoxin in cell therapy products, and to compare it with the traditional LAL assays currently in use at the facilities. Due to the rare occurrence of endotoxin-positive cell therapy products, the study was not designed as a formal validation of the Endosafe® PTS™ system, but rather as a comparison with other widely used assays.
Methods
Participants
A total of five cell therapy-manufacturing laboratories at four facilities participated in this evaluation. These included the three PACT centers, Baylor College of Medicine CAGT, the University of Minnesota MCT, and the University of Pittsburgh, which had two participating laboratories, the Hematopoietic Stem Cell (HSC) Laboratory and the Immune Monitoring Cellular Products Laboratory (IMCPL). The NIH DTM also participated in the study. The EMMES Corporation, the Administrative Center for PACT, served as the coordinating center.
Endosafe® System
The Endosafe® PTS™ system consists of LAL reagent and endotoxin controls applied to a single use, polystyrene cartridge (Figure 1.) Cartridges with 0.01 - 1.0 EU/mL, 0.05 - 5.0 EU/mL, and 0.1-10EU/mL sensitivities are commercially available and are used to detect endotoxin levels from 0.01 to 10EU/mL [4]. The cartridges are potency tested, spike recovery is performed, and the calibration code is determined. The calibration code contains the cartridge test parameters that were determined during potency testing, as well as the archived curve for that batch of cartridges. The calibration code is then verified with an additional test cartridge. The cartridge utilizes kinetic chromogenic LAL formulations in a modified format, which measures color intensity proportional to endotoxin concentration. Each cartridge consists of two sample channels and two spiked channels, consistent with current FDA guidance for licensed quantitative LAL methods. Each reservoir contains a specific amount of LAL reagent, synthetic chromogenic substrate, control standard endotoxin (CSE), and buffers uniformly imbedded in the cartridge [4]. Formulation of new substrates with each run is, therefore, eliminated for the user. A technician inserts the disposable cartridge into the reader and dispenses 25μL of the prepared sample into the four reservoirs. Using an internal pump, the reader draws, mixes, and incubates the sample with the various reagents at programmed time intervals before transferring it to the optical chambers. The portable spectrophotometer then monitors the change in the optical density and calculates the endotoxin level based on the resulting kinetic values. Cartridges with 0.05 - 5.0 EU/mL sensitivity were used in this study. Samples need to have reaction times that can be plotted on the archived curve. Results greater than 5.0 EU/mL can be run using dilutions in order to obtain results that are within the linearity of the assay cartridge (0.05-5.0EU/mL). Results are automatically multiplied by the dilution factor entered into the Endosafe® system. With the correct dilution the unit achieves results in approximately 15 minutes [4]. Samples that are positive for endotoxin will react faster.
Figure 1. Endosafe® PTS™ System.

Laboratory staff inserting a disposable cartridge into the Endosafe® PTS™ System
Traditional Assay Methods
The PACT centers and the NIH DTM utilized either the Lonza kinetic-chromogenic (Kinetic QCL™ test system) (K.C.), kinetic-turbidimetric (PYROGENT®-5000) (K.T.), or gel clot (Limulus Amebocyte Lysate Pyrogent Plus) (G.C.) assay systems, all of which are acceptable endotoxin detection methods by FDA. These assays were performed following the standard operating procedures in place at each center.
Cell Therapy Product Sample Collection and Testing
A study was conducted prior to testing cell therapy products to verify performance and capability of the Endosafe® system compared to the traditional assay method at each center. Each laboratory reconstituted two commercially prepared lyophilized Reference Standard Endotoxin (RSE) vials (provided by Charles River), with LAL water, to each yield approximately 0.25 EU/mL and 0.5 EU/mL of endotoxin. Subsequently, various cell therapy products prepared at the participating centers (Table 1) were tested using the traditional endotoxin assay method in place at the center and the Endosafe® system. The majority of the samples were from clinical products (Table 1) that would be routinely tested for endotoxin at the participating facilities. Standard dilutions of 1:20 and 1:40 were used in the majority of our testing to ensure sampling procedures were consistent across all 4 centers.
Table 1. Cell Therapy Products.
| Cell therapy products tested in this study that are routinely tested for endotoxin |
|
|
|
|
|
| Cell therapy products tested in this study that are not routinely tested for endotoxin |
|
|
|
Recognizing that most cell therapy products will be negative for endotoxin, samples that were positive for endotoxin (fabricated positives) were prepared at the MCT. These consisted of either LAL water (7 samples) or umbilical cord blood (UCB)-derived T-regulatory cells (2 samples) spiked with known quantities of E. coli. These samples were shipped frozen on dry ice via overnight express delivery from the submitting center to the other participants. They were stored at -80°C prior to thawing and analysis and were tested in a blinded manner at each site using 1:1(undiluted), 1:10, 1:20, and 1:40 dilutions. Repeat testing, if required, was performed on samples that had been stored overnight at 4°C.
Data analysis
All data were uploaded from each site to the EMMES Corporation where the results were compiled and analyzed. A test result was considered valid based on the % spike recovery and % coefficient of variation (%CV) parameters falling within the acceptance criteria established by both the Endosafe® PTS™ system and the traditional LAL methods manufacturers. Spike recovery values were considered valid if the results were between 50 and 200% for the Endosafe® and BioWhittaker turbidimetric data, and between 50 and 150% for the Lonza kinetic chromogenic data. Sample Coefficient of Variation values were considered valid if the results were less than 10% for all of the in-house data and less than 25% for the Endosafe® data. To ensure consistency in reporting results among the centers, valid test results were qualitatively separated into three categories: “Negative”, “Positive”, and “Borderline Positive.” Each category was defined and assigned a corresponding numerical value or range. The category assignments were based on further actions that the centers would implement when a result fell within a certain numerical range prior to releasing a product. Samples containing <2.0 EU/mL were classified as negative with no additional action required before a product would be released. Samples containing >2.0 EU/mL but <5.0 EU/mL were classified as borderline positive and would require additional action (e.g., confirmatory testing) before releasing a product. Samples containing >5.0 EU/mL were classified as positive and would require confirmatory testing and further evaluation before releasing a product. In the case of “borderline” or “positive” results, the standard procedure would be to calculate the infused EU/kg recipient weight/dose to determine whether it would be within specifications.
Results
The two pre-study (reconstituted RSE samples) samples were tested in triplicate at each facility. The results obtained from these standards are illustrated in Figure 2. The 30 paired pre-study results obtained represent the two reconstituted RSE vials each tested at a 1:1(undiluted) dilution as recommended by Charles River. Overall, both the Endosafe® PTS™ and the traditional LAL assay systems yielded the expected endotoxin levels of 0.25 EU/mL and 0.5 EU/mL, thereby verifying both systems' performance and sensitivity in detecting endotoxin (Figures 2a and 2b.) These results, however, would be classified as negative (since they are <2 EU/mL) based on criteria for reporting positive endotoxin results when using traditional assay systems for clinical grade products. The results from the remaining samples are as follows. Of the 178 total samples tested (fabricated positives and cell therapy products), a total of 139 paired samples (samples tested by both the traditional and Endosafe® assays) yielded valid results from both assays (Figure 3.) Valid results were based on acceptance criteria; i.e. % spike recovery and % CV, by each individual test system.
Figure 2. Results of two reconstituted (RSE) standards (2a/2b).

Each set of laboratory columns represents triplicate runs of the same RSE standard sample using the Endosafe® test system and the in-house method at the participating laboratories. The expected value for RSE sample number one (Figure 2a) is 0.25 with an expected endotoxin range (EER) of 0.125 EU/mL – 0.50 EU/mL representing the 2-fold margin of error represented by the gray shading. The expected value for RSE sample number two (Figure 2b) is 0.5 with an EER range of 0.25 EU/mL – 1.0 EU/mL representing the 2-fold margin of error represented by the gray shading. All testing on the Endosafe® was performed using 0.05 - 5.0EU/mL sensitivity cartridges. Legend: KC - Kinetic-Chromogenic, KT - Kinetic-Turbidimetric, GC - Gel Clot, EER - expected endotoxin range
Figure 3. Test Results Breakdown.

Method legend: K.C. - Kinetic-Chromogenic, K.T. - Kinetic-Turbidimetric, G.C. - Gel Clot, CV - Coefficient of variation, FPS - Fabricated Positive Sample, RSE - Reference Standard Endotoxin
Forty-five of the 139 paired samples yielding valid results were obtained from the cell therapy samples, and represented 16 different product types tested at standard dilutions. The 64 paired samples that yielded valid results from the fabricated positive samples represent 9 separate samples that were tested at all of the laboratories. Each sample was tested at 1:1(undiluted) and 1:10 dilutions, in addition to the typically recommended 1:20 and 1:40 dilutions, in order to achieve accurate endotoxin recoveries from borderline samples. Further confirmatory testing would normally be performed on “positive” or “borderline positive” samples. It is unlikely that cell products prepared for release for patient administration would be initially tested at 1:1(undiluted) and 1:10 dilutions under normal testing practices.
Of the 39 paired invalid test results, 34 were from standard cell therapy products and 5 from fabricated positive samples. The most common reason for test failure was an out of range spike recovery value on the Endosafe® test system, which occurred in 30/39 samples (76.9%). Four paired results (4/39, 10.3%) were excluded from analysis due to an invalid spike recovery obtained from traditional assay systems. Another four paired results (4/39, 10.3%) were eliminated due to a combination of an invalid % spike recovery from both the Endosafe® and traditional methods (2 pairs), and an invalid % spike recovery and sample %CV (2 pairs) on the Endosafe® system. The remaining sample (1/39, 2.5%) was excluded due to an invalid sample %CV on the Endosafe® system (Figure 3.) Samples derived from dendritic cell preparations accounted for 13/34 (38%) of the invalid results. An invalid % spike recovery accounted for 12/13 (92.3%) of the failed runs with the other sample (1/13, 7.7%) being excluded from analysis due to an invalid sample %CV. The majority of invalid results were based on inadequate % spike recoveries. Typically this is due to enhancement or inhibition, and occurs with both traditional LAL and Endosafe® test systems; however, the exact reason for inadequate % spike recovery was not investigated in this study. It was not determined if enhancement and/or inhibition reactions were due to high protein content, red blood cells, or other contaminants in the test samples. Results from such a determination could ultimately reduce the need for repeat testing but performing these assays was beyond the scope of this study. Repeat testing was not required in this study but was performed in some of the 39 cases of failed paired samples. Specifically, 10 specimens which failed with the Endosafe® system and contributed to the 39 failed paired results were repeated. Six of the 10 specimens failed again with repeat testing at the same dilution, 2 specimens passed with repeat testing at the same dilution and 2 specimens passed with increasing the dilution.
The concordance between results obtained using traditional assay systems and the Endosafe® system is shown in Table 2. There was overall agreement between the qualitative results obtained with the two systems in 115/139 (82.7%) paired samples. For the 30-paired pre-study samples that consisted of an RSE preparation of endotoxin, there was 100% concordance between the Endosafe® and traditional LAL testing methods.
Table 2. Concordance between Qualitative Results obtained using Traditional vs. the Endosafe® test systems.
| Results | Pre-Study Samples
(N) |
Standard Cell Therapy Samples
(N) |
Fabricated Positive Samples
(N) |
Overall Total Paired Samples by Results
(N) |
|---|---|---|---|---|
| Endosafe® - Positive/Borderline
Traditional - Positive/Borderline |
0 | 2 | 18 | 20/139 (14.4%) |
| Endosafe® - Negative
Traditional - Negative |
30 | 35 | 30 | 95/139 (68.3%) |
|
Total N of Paired Samples
(% Concordance within sample types) |
30/30 (100%) | 37/45 (82%) | 48/64 (75%) | 115/139 (82.7%) |
N = Number
Negative = <2.0EU/mL
Positive = >5.0 EU/mL
Borderline = >2.0 EU/mL - <5.0 EU/mL
For 45 paired cell therapy samples, there was an 82 % (37/45) concordance between Endosafe® and the traditional testing methods. The 64-paired fabricated positive samples yielded a 75% (48/64) concordance (Table 2.) The discordance between results obtained using traditional assay systems and the Endosafe® system is shown in Table 3. A total of 24 (16 fabricated positives and 8 cell therapy products) of the 139 paired sample results yielded an overall 17.3% discordance. The breakdown of the 24 paired sample results is as follows. In 18/139(16 fabricated positives, 2 cell therapy products) paired samples, the traditional assay produced a “positive” or “borderline” result while the Endosafe® gave a “negative” (<2 EU/mL) result. In the remaining 6/139 (all cell therapy products) paired samples, the Endosafe® result was “borderline” or “positive” while the traditional assay result was “negative.” These 6 samples were from dendritic cell tumor vaccines, peripheral blood (PB) stem cells, and natural killer (NK) [1 UCB-derived CD3-depleted product and 1 PB-derived CD3-depleted product] cell products. Conversely, Endosafe® was negative in 2 cases where the traditional method was borderline positive (Table 3.) These two samples were both from NK cell preparations with one being a PB-derived CD3-depleted/CD56-enriched and the other being an UCB-derived CD3-depleted product. In the fabricated positive samples, there were no cases of a negative traditional test result with a positive or borderline positive Endosafe® result.
Table 3. Discordance between Qualitative Results obtained using Traditional vs. the Endosafe® test systems.
| Results | Pre-Study Samples
(N) |
Standard Cell Therapy Samples
(N) |
Fabricated Positive Samples
(N) |
Overall Total Paired Samples by Results
(N) |
|---|---|---|---|---|
| Endosafe® - Negative
Traditional - Positive/Borderline |
0 | 2 | 16 | 18/139 (13%) |
| Endosafe® - Positive/Borderline
Traditional - Negative |
0 | 6 | 0 | 6/139 (4.3%) |
| Endosafe® - Positive
Traditional - Negative |
0 | 0 | 0 | 0/139 (0%) |
| Total N of Paired Samples (% Discordance within sample types) | 0 (0%) | 8/45 (17.8%) | 16/64 (25%) | 24/139 (17.3%) |
N = Number
Negative = <2.0 EU/mL
Positive = >5.0 EU/mL
Borderline = >2.0 EU/mL - <5.0 EU/mL
When comparing the results of the fabricated positive samples obtained from both the traditional methods and Endosafe®, the majority of Endosafe® results yielded values consistently lower than the traditional results, regardless at what dilution the fabricated positive samples were tested. (Figure 4a and 4b) It should be noted that 1) in order to achieve borderline endotoxin recoveries, these samples were run at 1:1(undiluted) and 1:10 dilutions which are not typically performed, in addition to the recommended 1:20 and 1:40 dilutions for cell therapy products; and 2) further testing would normally be performed on “positive” or “borderline positive” samples before release of the product.
Figure 4. Graphical Comparison of Fabricated Positive Samples.
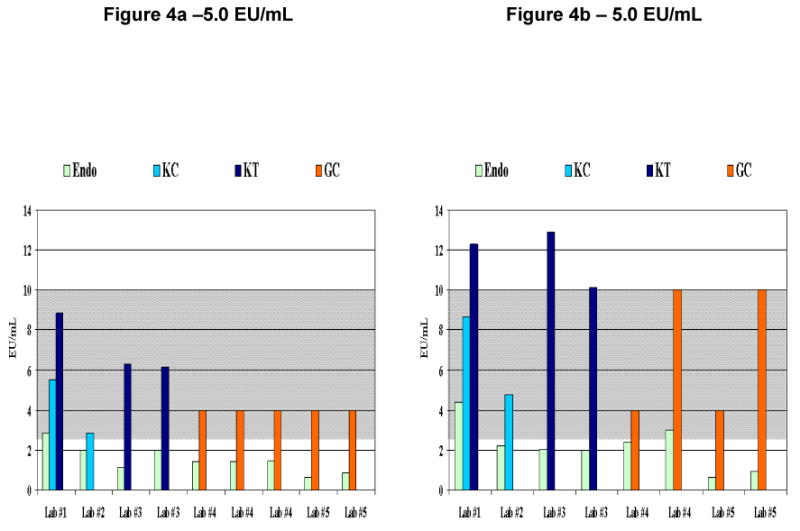
Figures 4a represent runs from one fabricated positive sample and figure 4b represent runs from a second fabricated positive sample using the Endosafe® test system and 0.05 –5.0 EU/mL sensitivity cartridges and the in-house method at the participating laboratories. The expected value for each of the positive samples (Figures 4a and 4b) is 5.0 with an expected endotoxin range (EER) of 2.5 EU/mL 10 EU/mL representing the 2-fold margin of error shown in gray shading. These samples were chosen from the pool of fabricated positive samples since they were representative of how a true positive sample might appear in laboratory analysis. Legend for 4a: Endo - Endosafe®, KC - Kinetic-Chromogenic, KT - Kinetic-Turbidimetric, GC - Gel Clot, EER - expected endotoxin range = 2.5 EU/mL-10 EU/mL, Lab #1- Endosafe® run at 1:10 dilution, Lab #1- KC run at 1:10 dilution, Lab #1- KT run at 1:10 dilution, Lab #2 - Endosafe®, and KC runs at 1:40 dilution, Lab #3 - Endosafe® run at 1:10 and 1:40 dilutions, Lab #3 - KT run at 1:20 and 1:40 dilutions, Lab #4 - Endosafe® runs at 1:1(undiluted) and 1:20 dilutions, Lab #4 - GC run using serial dilutions, Lab #5 - Endosafe® run at 1:1(undiluted) and 1:10 dilutions, Lab #5 - GC run using serial dilutions, Legend for 4b: Endo - Endosafe®, KC - Kinetic-Chromogenic, KT - Kinetic-Turbidimetric, GC - Gel Clot, EER - expected endotoxin range = 2.5 EU/mL-10 EU/mL, Lab #1- Endosafe® run at 1:10 dilution, Lab #1- KC run at 1:1(undiluted), Lab #1 - KT run at 1:10 dilution, Lab #2 - Endosafe® and KC runs at 1:40 dilution, Lab #3 - Endosafe® run at 1:10 and 1:40 dilutions, Lab #3 - KT run at and 1:20 and 1:40 dilutions, Lab #4 - Endosafe® run at 1:10 dilution, Lab #4 - GC run using serial dilutions, Lab #5 - Endosafe® run at 1:10 dilution, Lab #5 - GC run using serial dilutions
Conclusions
Endotoxin testing is generally required as part of the release criteria for cell therapy products where cell culture for greater than 72 hours has occurred and in some cases when the product has been manipulated and is to be used without cryopreservation. Traditional methods, including kinetic-chromogenic, kinetic-turbidimetric, and gel clot Limulus Amebocyte Lysate assay systems, have been routinely performed in most cell therapy laboratories, and are accepted by the FDA as meeting the regulatory requirements. All of these methods take a significant amount of time to perform (i.e., several hours) and become problematic if time-sensitive cell therapy products are being administered.
Charles River has developed a rapid endotoxin testing system to address these issues. The PACT group in collaboration with the NIH DTM compared the Endosafe® system with traditional methods of endotoxin testing using a variety of cell therapy products. Recognizing that most cell therapy product endotoxin tests are negative, additional testing was performed with centrally prepared fabricated endotoxin-positive specimens that were tested at each site in a blinded manner.
The results obtained should be viewed in the context of the variability inherent in LAL assays when products are being administered. Since the inception of the assay as an alternative to Rabbit Pyrogen Testing, it has been subject to a +/- 2-fold margin of error. The original gel-clot test was a semi-quantitative assay, in which the user's results were either positive (≥ LAL sensitivity) or negative (< LAL sensitivity.) To quantify endotoxin in a sample, the analyst ran serial 2-fold dilutions looking for the point at which one dilution was positive and the next 2-fold was negative.
We now have kinetic test methods that use defined standard curves for calculation of endotoxin levels, making the assay quantitative rather than semi-quantitative; however, the two-fold margin of error for all LAL assays still applies. This is due to the biological nature of both LAL and bacterial endotoxin, and takes into account the sensitive nature of the test, which is susceptible to differences among analysts, samples, and standard preparations, as well as the accessory materials used. While samples, which contain no endotoxin, should test clean under all circumstances, when samples contain endotoxin it is not uncommon to obtain different values especially across different analysts, laboratories, test methods and vendors. The FDA and United States Pharmacopeia (USP) recognize these differences and the limitations of LAL tests and the +/- 2-fold margin of error that is acceptable in the assay result is reflected in the 50-200% range of spike recovery in kinetic assays.
The results obtained in the present study demonstrated that the Endosafe® system performed well with the products typically produced in cell therapy manufacturing facilities. As expected, the results were primarily negative in both systems. The Endosafe® system provided reliable results in this setting, and would be an appropriate test on which to base the release of time-sensitive cell therapy products.
To challenge the system, fabricated positives were created to test endotoxin concentrations that would produce potentially ambiguous results and could result in differences between results from the Endosafe® system and the traditional methods. Of particular concern are those results that were negative (<2 EU/mL) in the Endosafe® system and either >2 but <5 EU/mL (Borderline Positive) or >5 EU/mL (Positive) in the traditional systems. Of the 18 paired results that fell into this category, 16 paired positive results were generated from the 9 fabricated positive samples. The remaining two paired samples produced negative (<2.0 EU/mL) results with Endosafe® and borderline positive (>2 but <5 EU/mL) results with the traditional methods. The two samples were standard cell therapy products: NK cells/UCB-derived CD3-depleted and NK cells/PB-derived CD3-depleted/CD56-enriched. Under normal circumstances we would expect that borderline or positive specimens would require further confirmatory testing prior to release. Basing release solely on the Endosafe® system results, these elevated endotoxin results would have been missed. This highlights the need to collect additional data to determine whether this would be an issue for cellular products.
The traditional test systems have the capability to analyze multiple dilutions of a given sample simultaneously while the Endosafe® system used in the current study can only analyze one test sample at a time. A multi-specimen test system is being developed by Charles River and when available, would further enhance the functionality of this rapid release test system. Based on the possibility of enhancement/inhibition reactions, further diluting of a specimen may increase the likelihood of a valid result and in a multi cartridge (Endosafe® MSC) system, where as many as 5 individual samples or dilutions of a single sample could be tested simultaneously [5]. The Endosafe® system was rapid (approximately 15 minutes compared to several hours for traditional assays), easy to use and posed few technical challenges. Training of staff was fast and straightforward in comparison to that required for other LAL assays. It is facilitated by prompts that are issued by the device from sample initiation and set up, through obtaining the quantitative result.
The advantages of the Endosafe® system lie in its speed and ease of use for testing single samples. This makes it particularly useful for release testing of non-cryopreserved samples where the aim is to prepare and administer the product within a short time frame. It is also useful as a back-up system when problems are encountered with traditional test systems, and repeat testing would further prolong the holding period before product administration, with potential adverse effects on the condition of the cells. The system is sensitive to the factors that adversely affect traditional LAL assays, e.g. high protein levels, RBCs, cellular debris etc., but the speed of testing makes it easier to perform multiple assays to address these problems. Inhibition/enhancement assays should be considered if specimens that exhibit known inferences are used in validating the system. Facilities should, however, validate the system against the particular approved method that they are currently using and determine that this validation is acceptable to regulatory agencies.
Supplementary Material
Acknowledgments
This work was supported in part by PACT (NHLBI contract #s N01HB37163, N01HB37164, N01HB37165, and N01HB37166.) The authors would like to thank the staff at Baylor College of Medicine, University of Minnesota, the University of Pittsburgh, and the NIH DTM for sample collection and testing. In addition, we would like to thank Charles River for providing the Endosafe® PTS™ kits and 0.05 - 5.0 EU/mL sensitivity cartridges and the EMMES Corporation for advice on the statistical analysis of the data.
This project was supported by NHLBI contract #s N01-HB-37163, N01-HB-37164, N01-HB-37165, and N01-HB-37166 from the National Heart Lung and Blood Institute.
References
- 1.Opal Steven M. The host response to endotoxin, antilipopolysaccharide strategies, and the management of severe sepsis. International Journal of Medical Microbiology (IJMM) 2007;297:365–377. doi: 10.1016/j.ijmm.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 2.Guideline on Validation of the Limulus Amebocyte Lysate Test as an End Product Endotoxin Test for Human and Animal Parenteral Drugs, Biological products, and Medical Devices. DHHS, PHS, and FDA; Dec, 1987. [Google Scholar]
- 3.Mondoro T, Thomas JW, Henslee-Downey PJ, Peterson CM. NHLBI plans for the promise of cell-based therapies. Cytotherapy. 2005;7:317–27. doi: 10.1080/14653240500237741. [DOI] [PubMed] [Google Scholar]
- 4.Endosafe®. PTS: A Portable, Kinetic Chromogenic LAL Test System. Charles River Laboratories International, Inc.; 2007. [Google Scholar]
- 5.Endosafe®. MCS System. Charles River Laboratories International, Inc.; 2007. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.