

Abstract
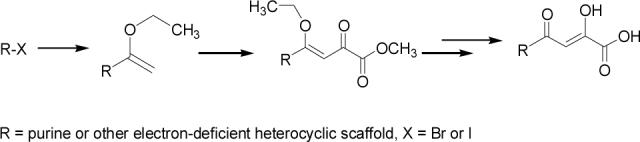
Claisen condensation, the key step in constructing the pharmacophore of aryl β-diketo acids (DKA) as integrase inhibitors, fails in certain cases of highly electron-deficient heterocycles such as purines. A general synthetic strategy to assemble the DKA motif on the purine scaffold has been accomplished. The synthetic sequence entails a palladium-catalyzed cross-coupling, a C-acylation involving a tandem addition/elimination reaction and a novel ferric ion-catalyzed selective hydrolysis of an enolic ether in the presence of a carboxylic acid ester.
The enzymes of the pol gene of the human immunodeficiency virus (HIV) have been identified as important viral targets for the discovery and development of anti-HIV therapeutic agents.1-3 HIV-1 integrase is one of the three enzymes of the pol gene that is critical for viral replication. It catalyzes biochemical steps that involve endonucleolytic cleavage (3'-processing) of the viral DNA in the cytoplasm and strand transfer (integration) of the tailored viral DNA into host cell DNA in the nucleus. While inhibitors of the two other key enzymes of the pol gene, reverse transcriptase and protease, have led to a number of clinically-approved agents that have had an enormous impact in the treatment of HIV infection,4-6 research efforts on inhibitors of HIV integrase have not resulted in a single FDA-approved drug.7-9 Because integrase is essential for HIV replication and has no human counterpart, it remains a significant target for the discovery of new anti-HIV agents.
Among the molecules with anti-HIV integrase inhibitory activity,10,11 DKA-containing inhibitors have emerged as significant potential anti-HIV drug candidates.12 The compound, S-1360 with a diketotriazole bioisostere of the DKA pharmacophore,13 L-731,988 with a pyrrole scaffold,14,15 and one of our compounds in which the DKA is constructed on a pyrimidine nucleobase scaffold,16 are among examples of potent inhibitors of integrase of this class of compounds.
However, there are some serious limitations in the synthetic methodology required to produce a structurally diverse family of new diketo acids that are of interest as HIV integrase inhibitors. This synthetic limitation is particularly problematic in certain cases for scaffolds that are highly electron deficient.17 Our initial attempts to introduce the DKA moiety on the 6- or 8-positions of purine scaffolds utilized the available literature strategy, i.e, a cross-Claisen condensation involving a purinyl ketone and an alkyloxalate. Literature methods show that the oxalylation of aryl ketones with dimethyl-, diethyl- or t-butyloxalates in the presence of NaH, NaOEt, or NaOMe affords the precursor of DKA, which, on alkaline or acidic hydrolysis, furnishes the desired DKA. The yields for the cross-Claisen condensations are in the range of 10−95 %, depending on the structure of the substrate and the reaction conditions (base, solvent, reaction temperature and time).18-21 However, all of these literature methods met with complete failure when applied to certain positions of purine or other electron-deficient heterocyclic scaffolds. In this Note, we report a new and reproducible synthetic strategy to assemble the DKA moiety on electron-deficient heterocyclic systems (Scheme 1).
SCHEME 1.
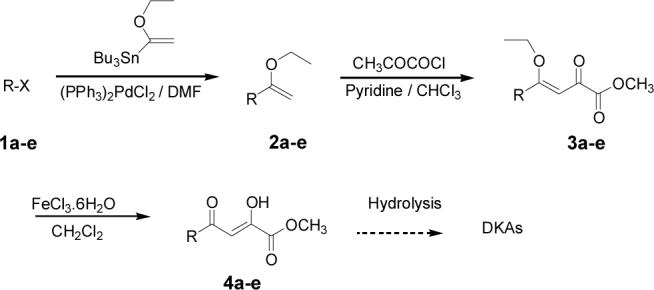
Representation of the General Synthetic Sequence for Assembling the DKA Pharmacophore
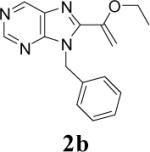
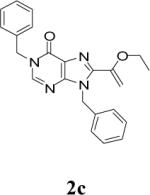
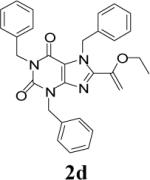
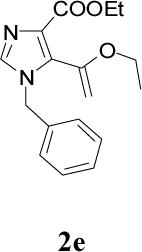
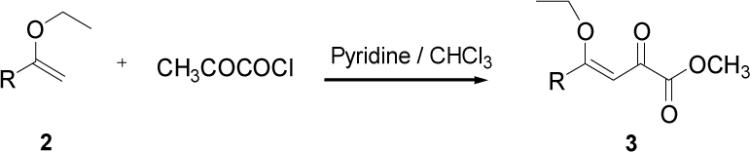
Appropriately functionalized haloheterocycles (1a-e), prepared by a previously developed methodology,22 were converted to the vinyl ethyl ethers (2a-e) by a Stille palladium-catalyzed cross-coupling23,24 in 47−97 % yield (Table 1). C-acylation25 of the vinyl ether intermediates, 2a-e, with methylchlorooxoacetate in presence of pyridine at 0 °C gave the keto esters, 3a-e, in 31−79 % yield (Table 2). This synthetic step proceeds through a tandem addition-elimination mechanism.26 Selective hydrolysis of the enolic ether functionality in 3a-e turned out to be unexpectedly challenging. The hydrolysis favored the ester functionality and then completely stopped at the carboxylic acid stage under all of the conditions that were attempted.27,28
Table 1.
Preparation of Vinyl Ethyl Ethers via Stille Coupling
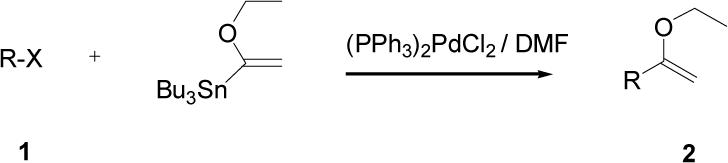 | ||||
|---|---|---|---|---|
| entry | reactant | product | time (h) | Yield (%) |
| 1 | 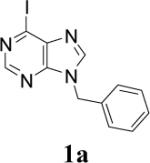 |
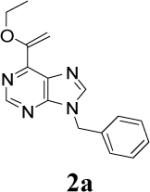 |
6 | 47 |
| 2 | 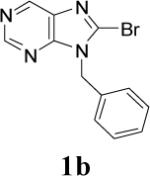 |
 |
48 | 60 |
| 3 | 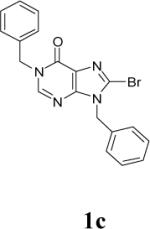 |
 |
22 | 88 |
| 4 | 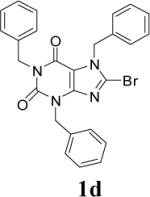 |
 |
8 | 75 |
| 5 | 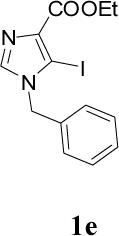 |
 |
12 | 97 |
Table 2.
Tandem Addition-Elimination to Produce Keto Esters
 | ||||
|---|---|---|---|---|
| entry | reactant | product | time (h) | Yield (%) |
| 1 | 2a | 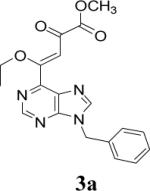 |
48 | 42 |
| 2 | 2b | 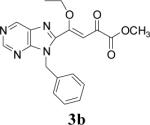 |
15 | 77 |
| 3 | 2c | 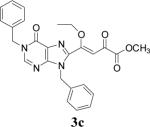 |
48 | 77 |
| 4 | 2d | 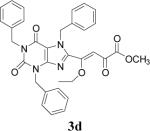 |
72 | 31 |
| 5 | 2e | 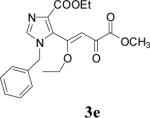 |
48 | 72 |
Because of the failure of the hydrolysis of 3a-e, a totally new method that could effect selective enol ether hydrolysis under mild conditions was sought. Although ferric chloride has been demonstrated to promote deprotection chemistry,29-31 its use as a hydrolytic agent for the cleavage of enol ethers has not been described. Thus, this report represents the first use of ferric chloride hexahydrate (FeCl3.6H2O) as an agent for the selective cleavage of enol ethers in the presence of an ester functionality. The DKA pharmacophores, 4a-e, were produced in 42−91 % yield (Table 3). As the solubility of FeCl3.6H2O in the reaction solvents, dichloromethane or dichloroethane, is low, it is possible that this reagent plays a catalytic role, and the hydrolysis step is preceded by metal complexation.32 Liberation of the free enols, 4a-e, from the iron complex was accomplished by treatment with 1 N HCl. The enols required purification by anion exchange (Sephadex) chromatography as they failed to elute from conventional columns due to their strong binding with silica gel and alumina. Finally, hydrolysis of the ester functionality of 4a–e was easily accomplished under basic conditions (NaOH, MeOH).17
Table 3.
Selective Hydrolysis of Enol Ether Functionality
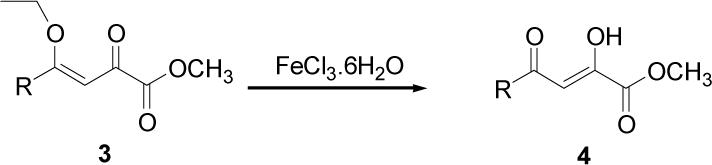 | ||||
|---|---|---|---|---|
| entry | reactant | product | time (h) | Yield (%) |
| 1 | 3a | 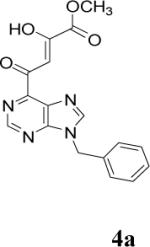 |
12 | 42 |
| 2 | 3b | 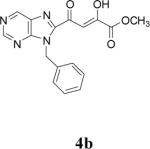 |
6 | 74 |
| 3 | 3c | 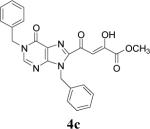 |
3 | 91 |
| 4 | 3d | 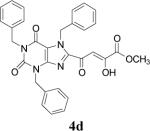 |
8 | 46 |
| 5 | 3e | 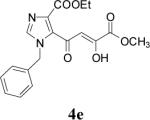 |
10 | 78 |
In summary, we have developed an efficient synthetic approach to assemble the DKA motif at defined positions of highly electron-deficient heterocycles such as purines. The method is applicable to a wide range of other electron-deficient heterocyclic scaffolds for which the Claisen condensation may be inefficient. A further advantage of this versatile approach, over the traditional Claisen condensation, is its degree of selectivity in tolerating functional groups such as additional ester units, which are susceptible to participation in cross-Claisen or Dieckmann condensations to produce undesired side products. The strategy described is of particular significance for providing a synthetic pathway for a diverse class of new HIV integrase inhibitors, which may also have biological potential as inhibitors of the replication of hepatitis C virus (HCV).33
Experimental Section
General Procedure for Stille Coupling
A mixture of halopurine 1a-e (2.8 mmol), bis(triphenylphosphine)palladium (II) chloride (0.02 mmol) and ethoxyvinyl(tributyl)tin (3.8 mmol) in dry DMF (50 mL) was heated under N2 at 75 °C for an appropriate time. DMF was distilled off and the resulting residue dissolved in EtOAc (50 mL) and filtered through a plug of celite. The solvent was removed and the residue obtained was purified by flash chromatography on silica gel to give the desired product.
General Procedure for C-acylation
To a stirred solution of ethoxy vinyl ether 2a-e (0.7 mmol) and pyridine (1.4 mmol) in dry chloroform (10 mL) at 0°C was added, under dropwise conditions, methyl chlorooxoacetate (1.4 mmol) in dry chloroform (5 mL). The reaction mixture was allowed to stand in the refrigerator at 0°C for an appropriate period of time and then washed with water and dried over anhydrous sodium sulfate. Removal of CHCl3 gave a yellowish syrup from which pure product was isolated by column chromatography on silica gel.
General Procedure for Selective Hydrolysis of Enol Ether
To a solution of ethoxy keto ester 3a-e (0.5 mmol) in dichloromethane or dichloroethane (30 mL) was added FeCl3.6H2O (0.9 mmol) and the reaction mixture stirred at reflux for the appropriate time. The solvents were removed and the residue dissolved in EtOAc (50 mL) and washed with 1 N HCl (50 mL) followed by water (50 mL). The organic layer was dried over anhydrous Na2SO4 and concentrated to give a yellow residue, which was purified by anion exchange chromatography (DEAE sephadex resin, CH3CN:H2O, 1:1) and crystallized.
Supplementary Material
FIGURE 1.
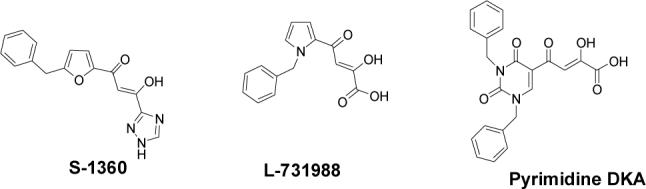
Structues of three β-diketo compounds that are inhibitors of HIV-1 integrase.
Acknowledgment
This project was supported by Grant No. RO1 AI 43181 from the National Institutes of Health (NIAID). The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
Supporting Information Available: 1H NMR, 13C NMR, HRMS data and physical constants of all the compounds synthesized. This material is available free of charge via the internet at http://pubs.acs.org.
REFERENCES
- 1.Fauci AS. Science. 1988;239:617. doi: 10.1126/science.3277274. [DOI] [PubMed] [Google Scholar]
- 2.Frankel AD, Young JAT. Annu. Rev. Biochem. 1998;67:1. doi: 10.1146/annurev.biochem.67.1.1. [DOI] [PubMed] [Google Scholar]
- 3.De Clercq E. Nature Rev. Drug Discovery. 2002;1:13. doi: 10.1038/nrd703. [DOI] [PubMed] [Google Scholar]
- 4.Johnson SC, Gerber JG. In: Advances in Internal Medicine. Schrier RW, Baxter JD, Dzau VJ, Fauci AS, editors. Vol. 45. Mosby; St. Louis: 2000. pp. 1–40. [PubMed] [Google Scholar]
- 5.Nair V, St. Clair MH, Reardon JE, Krasny HC, Hazen RJ, Paff MT, Boone LR, Tisdale M, Najera I, Dornsife RE, Averett DR, Borroto-Esoda K, Yale JL, Zimmerman TP, Rideout JL. Antimicrob Agents Chemother. 1995;39:1993. doi: 10.1128/aac.39.9.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Clercq E. J. Med. Chem. 2005;48:1297. doi: 10.1021/jm040158k. [DOI] [PubMed] [Google Scholar]
- 7.Pommier Y, Johnson AA, Marchand C. Nature Rev. Drug Discovery. 2005;4:236. doi: 10.1038/nrd1660. [DOI] [PubMed] [Google Scholar]
- 8.Nair V. Frontiers in Med. Chem. 2005;2:3. [Google Scholar]
- 9.Dayam R, Neamati N. Curr. Pharm. Design. 2003;9:1789. doi: 10.2174/1381612033454469. [DOI] [PubMed] [Google Scholar]
- 10.Nair V, Chi G. Rev. Med. Virol. 2007;17:277. doi: 10.1002/rmv.539. [DOI] [PubMed] [Google Scholar]
- 11.Maurin C, Bailly F, Cotelle P. Curr. Med. Chem. 2003;10:1795. doi: 10.2174/0929867033456981. [DOI] [PubMed] [Google Scholar]
- 12.Neamati N. Expert. Opin. Ther. Pat. 2002;12:709. doi: 10.1517/13543776.16.2.165. [DOI] [PubMed] [Google Scholar]
- 13.Billich A. Curr. Opin. Investig. Drugs. 2003;4:206. [PubMed] [Google Scholar]
- 14.Hazuda DJ, Felock P, Witmer M, Wolfe A, Stillmock K, Grobler JA, Espeseth A, Gabryelski L, Schleif W, Blau C, Miller MD. Science. 2000;287:646. doi: 10.1126/science.287.5453.646. [DOI] [PubMed] [Google Scholar]
- 15.Selnick HG, Hazuda DJ, Egbertson M, Guare JP, Wai JS, Young SD, Clark DL, Medina JC. 1999. PCT, WO 62513.
- 16.Nair V, Chi G, Ptak R, Neamati N. J. Med. Chem. 2006;49:445. doi: 10.1021/jm0508890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nair V, Uchil V, Neamati N. Bioorg. Med. Chem. Lett. 2006;16:1920. doi: 10.1016/j.bmcl.2005.12.093. [DOI] [PubMed] [Google Scholar]
- 18.Williams HWR, Eicher E, Randell WC, Rooney CS, Cragoe EJ, Jr., Streeter KB, Schwam H, Michelson SR, Patchett AA, Taub D. J. Med. Chem. 1983;26:1196. doi: 10.1021/jm00362a020. [DOI] [PubMed] [Google Scholar]
- 19.Pais GCG, Zhang X, Marchand C, Neamati N, Cowansage K, Svarovskaia ES, Pathak VK, Tang Y, Nicklaus M, Pommier Y, Burke TR. J. Med. Chem. 2002;45:3184. doi: 10.1021/jm020037p. [DOI] [PubMed] [Google Scholar]
- 20.Yuan J, Gulianello M, De Lombaert S, Brodbeck R, Kieltyka A, Hodgetts K. J. Bioorg. Med. Chem. Lett. 2002;12:2133. doi: 10.1016/s0960-894x(02)00358-x. [DOI] [PubMed] [Google Scholar]
- 21.Jiang X-H, Song L-D, Long Y-Q. J. Org. Chem. 2003;68:7555. doi: 10.1021/jo034903x. [DOI] [PubMed] [Google Scholar]
- 22.Nair V, Young DA, De Silvia R. J. Org. Chem. 1987;52:1344. [Google Scholar]
- 23.Nair V, Turner GA, Buenger GS, Chamberlain SD. J. Org. Chem. 1988;53:3051. [Google Scholar]
- 24.Gundersen L-L, Bakkestuen AK, Aasen JA, Øverås H, Rise F. Tetrahedron. 1994;50:9743. [Google Scholar]
- 25.Tietze L-F, Meier H, Foss E. Synthesis. 1988:274. [Google Scholar]
- 26.March J. Advanced Organic Chemistry, Reactions, Mechanisms and Structures. 4th ed. John Wiley & Sons; New York: 1999. p. 599. [Google Scholar]
- 27.Greene TW, Wuts PGM. Protective Groups in Organic Synthesis. 3rd ed. John Wiley & Sons; New York: 1999. [Google Scholar]
- 28.March J. Advanced Organic Chemistry, Reactions, Mechanisms and Structures. 4th ed. John Wiley & Sons; New York: 1999. p. 375. [Google Scholar]
- 29.Fadel A, Yefsah R, Salaun J. Synthesis. 1987:37. [Google Scholar]
- 30.Ikemoto N, Kim OK, Lo L-C, Satyanarayana V, Chang M, Nakanishi K. Tetrahedron Lett. 1992;33:4295. [Google Scholar]
- 31.Kim KS, Song YH, Lee BH, Hahn CS. J. Org. Chem. 1986;51:404. [Google Scholar]
- 32.Sen SE, Roach SL, Boggs JK, Ewing GJ, Magrath J. J. Org. Chem. 1997;62:6684. [Google Scholar]
- 33.Summa V, Petrocchi A, Pace P, Matassa VG, De Francesco R, Altamura S, Tomei L, Koch U, Neuner P. J. Med. Chem. 2004;47:14. doi: 10.1021/jm0342109. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.