

Abstract
The retinoblastoma gene product (RB) regulates cell cycle, quiescence and survival in a cell type and environment dependent manner. RB function is critical in the pulmonary epithelium as evidenced by nearly universal RB inactivation in lung cancer and increased lung cancer risk in persons with germline RB gene mutations. Lung carcinomas occur in the context of epithelial remodeling induced by cytotoxic damage. While the role of RB in development and normal organ homeostasis has been extensively studied, RB function in the context of cellular injury and repair has remained largely unexplored. In the current studies, the RB gene was selectively deleted in the respiratory epithelium of the mouse. Although RB was not required for establishing or maintaining quiescence during lung homeostasis, RB was essential for establishing quiescence during epithelial repair after injury. Notably, aberrant cell cycle progression was sustained for 9 months after injury in RB deficient lungs. Prenatal and postnatal RB ablation had similar effects providing evidence that timing of RB loss was not critical to the outcome, and that the injury induced phenotype was not secondary to compensatory alterations occurring during development. These data demonstrate that RB is essential for repair of the respiratory epithelium following cytotoxic damage, and support a critical unique role for RB in the context of epithelial remodeling after injury. Since human cancers are associated with chronic cellular damage, these findings have important new implications for RB mediated tumor suppression.
Keywords: RB, Cre-LoxP, Lung injury
INTRODUCTION
The retinoblastoma gene product (RB) is a critical cell cycle regulator and influences numerous cellular processes including cellular differentiation, survival, terminal cell cycle exit and maintenance of the post mitotic state (for review (1, 2)). The current model of RB function suggests a critical regulatory role in many, if not all, cell types. Consistent with this notion, RB is widely expressed during development and in adult tissues. However, germline RB gene ablation in the mouse results in embryonic lethality associated with organ specific defects predominantly in the nervous system, hematopoietic system and eye lens (3–5). Many of these phenotypes are secondary to inadequate placental function and represent non-cell autonomous RB functions. Accordingly, many of the neurological and erythroid abnormalities seen in conventional knockout mice are rescued when RB deficient fetuses are supplied with a RB sufficient placenta (6). Additionally, defective erythropoiesis in RB null embryos results, at least in part, from abnormalities in fetal liver macrophages rather than erythrocytes (7). Taken together, these data provide evidence that RB function is highly cell type specific, and that many cells do not require cell autonomous RB functions during development.
RB is inactivated in many, if not all, human cancers providing convincing evidence that RB is essential for tumor suppression (for review (8)). Over 80% of adult malignancies are carcinomas; i.e. malignancies that arise from epithelial cells. Surprisingly, ablation of RB in multiple epithelial cell types in the mouse leads to relatively mild phenotypes (9–15). This is due in part to functional redundancy and/or compensation by the other RB family proteins, p107 and p130. However, cellular response to loss of RB function is also highly dependent upon cellular context. RB ablation in the lung and prostate epithelium results in increased proliferation and apoptosis, whereas apoptosis does not accompany the aberrant proliferation observed in RB deficient skin (12, 14, 15). Hepatocyte specific RB ablation leads to aberrant S-phase entry that is associated with neither hyperplasia nor apoptosis (9). Finally, RB ablation targeted to the intestinal epithelium using the villin promoter causes no intestinal abnormalities (10). Thus, although RB is inactivated in carcinomas arising in multiple organs, epithelial cell response to RB loss is highly context specific.
Timing of RB ablation appears to play a critical role in determining phenotypic outcomes in vitro. Acute loss of RB in mouse embryo fibroblasts and keratinocytes in culture causes more severe cell cycle abnormalities than RB loss during development in vivo (14, 16). The physiologic relevance of temporally dependent effects of RB loss in culture remains unclear. Nonetheless, the data suggest that RB related phenotypes may be influenced by the precise timing of RB loss.
RB is almost universally inactivated in lung cancers providing strong evidence that RB is a critical regulator in the pulmonary epithelium (for review (17)). Moreover, humans with RB germline mutations are at increased risk for developing lung cancer (18, 19). Lung carcinomas are associated with cigarette smoking in 80–85% of sporadic cases as well as in patients with germline RB mutations (18, 19). Additionally, chronic lung diseases characterized by continual epithelial remodeling are associated with an increased risk of lung cancer in the absence of smoking (for review (20, 21). Thus, like many other malignancies, lung cancer occurs in the context of chronic epithelial damage.
The present studies were designed to directly test whether Rb function is critical in the context of lung epithelial remodeling after injury. Although Rb was not required for establishing and maintaining epithelial cell quiescence during lung homeostasis, Rb was essential for establishing cellular quiescence in the context of epithelial regeneration after injury. Aberrant cell cycle progression in RB deficient lungs was sustained for at least 9 months after injury, whereas the epithelium in RB proficient lungs was quiescent two weeks after injury. The phenotype was similar regardless of whether RB loss occurred during development or in the postnatal lung. These studies demonstrate that RB is essential in the remodeling lung epithelium, and support a more critical role for RB in the setting of epithelial repair after injury as compared to lung homeostasis. Since human malignancies are commonly associated with chronic injury, these findings have important implications for RB mediated tumor suppression.
MATERIALS AND METHODS
Animal generation and treatment
Mice with RB deficient lung epithelium were generated by mating CC10-rtTA and tetO-Cre double transgenic mice to RBLoxP/LoxP mice and genotyped using tail and lung DNA as previously described (15). Primers RB-18 and RB-19 were used to differentiate the floxed (746 bp), wild type (678 bp) and recombined (321 bp) RB alleles. Thyroid stimulating hormone beta subunit (TSHbeta) was amplified as an endogenous internal control to verify template DNA quality and quantity using primers 5′TCCTCAAAGATGCTCATTAG3′ and 5′GTAACTCACTCATGCAAAGT3′ at an annealing temperature of 55°C for 35 cycles resulting in a 386 bp band. Gestational age was determined by detection of a vaginal plug (designated embryonic day E0.5). Prenatal RB ablation was induced by doxycycline (Sigma) administration to pregnant dams with a single intraperitoneal injection (125μg/0.5ml saline) on E0.5–E1.5 followed by maintenance on doxycycline in the drinking water (1.0 mg/ml) until birth. Postnatal RB ablation was induced in adult mice at 2–3 months of age by a single intraperitoneal doxycycline injection (125μg/0.5ml saline) followed by maintenance on doxycycline food (Modified Prolab RMH 1500 with 0.0625% Doxycycline, TestDiet/Purina Mills). Adult mice treated with naphthalene were fed doxycycline food 3 weeks prior to naphthalene treatment, and were maintained on doxycycline food until the time of sacrifice. Naphthalene (Sigma-Aldrich) was suspended in corn oil and administered to adult mice at 2–4 months of age by a single intraperitoneal injection (300 mg/kg). Animals were given BrdU (Amersham Biosciences) by intraperitoneal injection (2 mg/100gm) one hour prior to sacrifice.
Immunohistochemistry and TUNEL analysis
Tissues were fixed in 10% neutral buffered formalin and paraffin embedded. Morphology was assessed on hematoxylin and eosin stained sections. Immunohistochemistry and TUNEL analysis was performed on deparaffinized 5 micron sections after antigen retrieval in 10 mM citrate solution microwaved for seven minutes. Primary antibodies were diluted in 0.1% bovine serum albumin in phosphate buffered saline, applied to tissue sections and incubated overnight at 4°C using the following dilutions: Ki67 1:50 (Clone B56, BD PharMingen), phosphorylated (Ser10) Histone H3 1:1000 (US Biological), and CCSP 1:20,000 (kind gift from Steven Brody, Washington University, St. Louis, MO). Antibody staining was detected with Vectastain Elite ABC, M.O.M. Immunodetection and DAB Substrate Kits (Vector Laboratories, Inc.). For dual CCSP/BrdU immunolabeling, tissue sections were incubated with anti-CCSP antibody overnight followed by application of secondary antibody and ABC Elite Vectastain ABC-AP reagent (Vector Laboratories, Inc). Positive staining was detected with Vector Alkaline Phosphatase Blue Substrate Kit III (Vector Laboratories, Inc). Subsequent BrdU analysis was performed using Zymed BrdU Staining Kit (Zymed Laboratories, Inc.). Tissues were counterstained with hematoxylin or nuclear fast red. Counts represent evaluation of an average of 350 epithelial cells representing both proximal and distal conducting airways and at least two lung lobes per mouse. TUNEL analysis was performed using ApopTag Peroxiodase In Situ Apoptosis Detection Kit (Chemicon International). Percent positive cells were determined on samples blinded to genotype by locating a TUNEL positive cell and counting 100 cells surrounding the initially identified positive cell. Counts represent 200–400 epithelial cells per animal including both proximal and distal conducting airways and at least two lung lobes per mouse. Only cells that were TUNEL positive, showed morphologic features of apoptosis and remained attached to the basement membrane were counted. Statistical significance was determined by unpaired Student’s t-tests.
β-galactosidase staining
In situ staining for β-galactosidase activity was performed on frozen tissue sections as previously described (15).
RESULTS
Quiescence is established and maintained in adult RB deficient lung epithelium
A conditional RB knockout model was utilized to target RB ablation to the lung epithelium (15). Double transgenic mice bearing the reverse tetracycline responsive transactivator under control of the rat Clara cell 10kD protein (CC10)/Scgb1a1 gene promoter, and Cre recombinase (Cre) under control of the tet operator and a minimal CMV promoter were bred into a RbLoxP/LoxP background (Fig. 1). Previous studies demonstrated that Cre mediated recombination is epithelial specific and occurs in the vast majority of epithelial cells throughout the conducting airway in a doxycycline dependent manner (15). The rat CC10 promoter differs slightly from the endogenous mouse promoter and therefore Cre recombination expression, and thus RB ablation, is not restricted to Clara cells.
Figure 1. RB ablation results in sustained epithelial proliferation after injury.

Inducible RB ablation was targeted to the lung epithelium by mating CC10-rtTA and tetO-Cre double transgenic mice with RBLoxP/LoxP mice (A). Pregnant dams were treated with doxycycline (circles) which activates rtTA (arches) expressed under control of the lung epithelial specific promoter. Activated rtTA induces Cre expression leading to recombination at LoxP sites flanking exon 19 in the RB gene locus. Hematoxylin and eosin stained sections of RB ablated and control adult lungs show similar overall morphology (B). Immunohistochemical analysis for Ki67 in RB ablated and control adult lungs from mice treated with doxycycline throughout gestation (C). The percent Ki67 positive cells (arrows) was comparable in RB ablated and control lungs before naphthalene treatment (Day 0) and at day 4 after injury (Day 4). A statistically significant increase in Ki67 positive cells was noted on day 4 as compared to day 0 in RB ablated (p=0.009) and control (p=0.011) lungs. In contrast, the percent Ki67 positive cells was significantly increased in RB ablated lungs versus controls on day 14 after injury (Day 14). Quantitative analysis is presented as average + SEM (D, *p=0.01). Data is representative of 5–7 animals per time point. br=bronchiole; Original magnification: 1000×
We previously reported that RB ablation in the lung epithelium causes epithelial hypercellularity with increased proliferation and apoptosis at birth (15). Adult lungs showed increased neuroendocrine cells but lacked the morphologic features of hyperplasia and apoptosis present at birth suggesting that the majority of RB deficient epithelial cells were capable of compensating for loss of RB function postnatally (Fig. 1 and (15)). To directly determine whether RB deficient epithelial cells in the adult lung entered quiescence, lungs from double transgenic mice were analyzed for the proliferation marker Ki67. Ki67 is expressed in all phases of the cell cycle except G0 and thus marks non-quiescent cells (22). Controls for this and subsequent experiments consisted of littermates lacking one or both transgenes required for RB ablation. Ki67 expression was similar in RB ablated and control lungs (Fig. 1) providing evidence that RB function is not essential for establishing and maintaining quiescence in the mature respiratory epithelium despite the marked cell cycle abnormalities present at birth.
RB is critical for establishing quiescence after injury
Mice with RB deficient lungs were exposed to naphthalene to directly test whether RB function is critical during epithelial remodeling after injury. Cytotoxic damage induced by naphthalene is targeted to the lung epithelium because pulmonary epithelial cells contain high concentrations of the specific P-450 isoenzyme, CYP2F2, required for metabolizing naphthalene to its toxic metabolite (23). Temporal and morphologic characteristics of naphthalene induced injury and subsequent repair are well characterized (24, 25). Briefly, diffuse epithelial damage occurs within the first 24 hours after naphthalene administration. Thereafter the denuded airways are repopulated by naphthalene resistant cells. Cellular proliferation peaks on day 2–4, and the repair process is largely complete two weeks after injury.
Adult mice were treated with a single naphthalene injection at a dose known to induce epithelial injury throughout the conducting airways (26). Epithelial damage occurred in both proximal and distal conducting airways as confirmed morphologically (data not shown). Some death was observed within the first two weeks after treatment; however, RB ablated and control mice were similarly affected (21% (21/102) versus 26% (15/58), respectively). RB ablated and control lung epithelium was largely quiescent prior to treatment (Fig. 1). On day 4 after injury, epithelial proliferation was significantly increased over baseline levels in RB ablated (26% ± 5% vs. 3% ± 1%, p=0.002) and control lungs (21% ± 4% vs. 2% ± 0.5%, p=0.002) (Fig. 1). Increased proliferation was noted in both the proximal (30% ± 7% and 20% ± 5%, RB ablated and controls respectively, p=0.30) and distal (25% ± 4% and 23% ± 4%, respectively, p=0.65) conducting airways providing evidence that epithelial remodeling occurred throughout the airway. Remarkably, Ki67 expression was sustained in RB ablated lungs on day 14 after injury whereas proliferation in control lungs returned to baseline levels (Fig. 1). Thus, RB is essential for establishing quiescence following naphthalene induced injury.
Loss of RB function results in sustained cell cycle progression after injury
RB blocks cells in the G1 to S phase transition of the cell cycle (for review (1, 8)). Therefore, an expected consequence of RB loss is aberrant entry into S phase. As expected, the proportion of epithelial cells in S-phase was <1% in both control and RB ablated lungs on day 0 and significantly increased by day 4 after injury (Fig. 2 and data not shown). A modest increase in BrdU positive epithelial cells was noted in RB ablated lungs on day 0 as compared to controls (Fig. 2). The physiologic significance of this increase is unknown since BrdU positive cells represent <1% of the epithelial cells on day 0, and no concomitant increase in Ki67 expression (Fig. 1) or mitotic cells (Fig. 2) was detected at this time point. Given that RB null fibroblasts have shortened G1 and extended S phases, this finding could reflect a slightly different cell cycle profile in the RB ablated versus control lungs (27, 28). On day 14 after injury, however, BrdU incorporation was markedly increased in RB ablated lungs as compared to controls (Fig. 2) demonstrating that RB deficiency results in aberrant progression into S-phase.
Figure 2. Aberrantly proliferating cells in RB null lungs progress into S-phase and mitosis.

Immunohistochemical analysis for BrdU incorporation (A) and PH3 (B) in RB ablated and control adult lungs from mice treated with doxycycline throughout gestation. Statistically significant increases in BrdU and PH3 positive cells (arrows) were noted in RB ablated versus control lungs on day 14 after injury (*p=0.001 and *p=0.005, respectively). A minimal increase in percent BrdU positive cells was seen on day 0 in RB ablated versus control lungs (*p=0.02), but no significant increase was seen in percent PH3 positive cells on day 0 (p=0.39). Quantitative analysis is presented as average + SEM. Data is representative of 6–7 animals per time point. Double label immunohistochemical analysis for BrdU (brown nuclear stain, open arrowhead) and CCSP (blue cytoplasmic stain, closed arrowhead) in RB ablated and control adult lungs from mice treated with doxycycline throughout gestation (C). The majority of BrdU incorporating cells were CCSP positive (arrow) in the RB ablated and control lungs on day 4 after injury (BrdU-CCSP double positive/BrdU positive cells = 80.1 ± 16.1 and 71 ± 14.9, respectively). The increase in S-phase Clara cells was maintained in RB ablated lungs on day 14 after injury (BrdU-CCSP double positive/BrdU positive = 81.4 ± 6.6). The overall labeling index (BrdU positive/total epithelial cells) in control lungs at day 14 after injury was 1.6 ± 1.5 which is comparable to baseline levels before injury. Quantitative data represents average ± SD. Percent positive cells was calculated counting >250 BrdU positive epithelial cells representing 3–5 animals per time point. br=bronchiole; Original magnification: 1000×
Aberrant cell cycle progression induced by RB loss can result in cell cycle arrest. For example, RB deficient myocytes are unable to maintain G0 arrest upon restimulation with serum and eventually arrest in the S and G2 phases of the cell cycle (29). RB null cells in the brains of chimeric embryos show increased G2 fractions without progression into mitosis suggesting that RB null neuronal cells arrest in late S or G2 phase of the cell cycle (30). Finally, RB is essential for G1/S phase arrest after DNA damage in mouse embryonic fibroblasts (MEFs) in culture; however RB null MEFs accumulate in G2/M phase providing evidence that the G2/M phase checkpoint remains intact in the absence of RB (31). RB deficient lung epithelial cells progressed into mitosis as evidenced by increased expression of phosphorylated histone H3 (PH3) compared to controls on day 14 after injury (Fig. 2). While these data cannot exclude an arrest in mitosis, they indicate that RB deficient lung epithelial cells progress through S and G2 phases of the cell cycle. Thus, RB is essential for cell cycle exit during epithelial repair after injury, and aberrantly proliferating RB deficient epithelial cells progress into mitosis.
Non-ciliated Clara cells constitute the vast majority of aberrantly proliferating cells in the remodeling RB deficient lung
The airways are lined by diverse and specialized cell types required for normal lung function. Non-ciliated Clara cells function as progenitor cells in the conducting airways (25). Since studies in the hematopoietic system indicate that RB is essential for regulating progenitor cells (32, 33), double immunolabeling for the Clara cell marker, Clara cell secretory protein (CCSP), and BrdU was done to determine whether Clara cells constitute the aberrantly proliferating population. Clara cells accounted for 81% ± 7% of the S-phase population in RB deficient lungs on day 14 after injury (Fig. 2). This was comparable to the proportion of S-phase Clara cells in RB ablated and control lungs on day 4 after injury (80% ± 16% and 71% ± 15%, respectively) (Fig. 2). Thus, Clara cells represent the vast majority of aberrantly cycling cells in the repairing Rb deficient lung epithelium.
Aberrant cell cycle progression in RB deficient lungs is sustained for at least 9 months after a single episode of injury
Epithelial proliferation was significantly increased in RB deficient lungs 9 months after injury (Fig. 3). Moreover, the percentage of epithelial cells in S- and M-phases remained elevated providing evidence that aberrant cell cycle progression was still occurring 9 months after the initiating event (Fig. 3). A trend towards decreased overall proliferation was noted in RB ablated lungs at 9 months as compared to day 14 after injury; however this decrease reached statistical significance only for BrdU labeled cells (7.9 ± 1.3 (day 14) vs. 2.9 ± 1.0 (9 months) p=0.009) (Fig. 3). Epithelial cell cycle abnormalities were separately assessed in proximal and distal conducting airways at 9 months and day 14 after injury to determine if a regional difference existed. Although, there was a trend toward increased proliferation in the distal airways, this difference was not consistently statistically significant among the assessed cell cycle markers. While it is possible that distal airway epithelial cells are slightly more sensitive to RB loss, the trend toward increased cell cycle abnormalities in the distal airway epithelium may simply reflect the higher proportion of Clara cells in distal versus proximal conducting airways.
Figure 3. Aberrant cell cycle progression is sustained in RB ablated lungs 9 months after a single episode of injury and is associated with increased apoptosis.

Quantitative data for cell cycle markers and TUNEL analysis on RB ablated and control adult lungs from mice treated with doxycycline throughout gestation. BrdU incorporation and PH3 and Ki67 expression were assessed by immunohistochemistry 9 months after injury. Quantified data is presented as average percent positive cells + SEM (A). RB ablated lungs showed a statistically significant increase in S-phase cells (BrdU), mitotic cells (PH3) and overall proliferation (Ki67) as compared to controls (*p=0.04, *p=0.01 and *p=0.02, respectively). Apoptosis was assessed by TUNEL assay on lung sections before injury (day 0) and on day 4, day 14 and 9 months after injury. Representative results from the 9 month time point are shown for RB ablated (B) and control (C) lungs. Percent TUNEL positive cells (arrow) were quantified and data represented as average percent positive cells + SEM (D). Apoptosis was increased in RB ablated lungs as compared to controls at all time points analyzed with the increase reaching statistical significance on day 14 (*p=2.9×10−5) and 9 months (*p=0.006) after injury. Data is representative of 5–11 animals per time point. br=bronchiole; Original magnification: 1000×
Tumor incidence was not increased in RB ablated lungs at 9 months after injury despite the prolonged period of sustained proliferation. Gross and microscopic examination of RB deficient lungs from mice at 8–16 months of age showed no increase in tumor incidence irrespective of naphthalene treatment (Table 1). A possible explanation for lack of tumor formation despite sustained proliferation is that RB ablated cells are selectively lost after injury resulting in a RB proficient epithelium. While this explanation is highly unlikely given the relatively uniform RB ablation throughout the conducting airway (15), Cre mediated recombination was directly assessed at the cellular level by performing in situ β-galactosidase assays on RB deficient lungs from mice harboring the ROSA26 reporter locus. LacZ is expressed in this reporter strain only in cells expressing functional Cre recombinase and their descendants (34). The vast majority of epithelial cells in RB deficient lungs at 9 months after injury were β-galactosidase positive (data not shown). Therefore, RB ablated cells are not selectively eliminated during epithelial regeneration after injury.
Table 1.
Tumor Incidence
| Naphthalene
|
No Naphthalene
|
|||
|---|---|---|---|---|
| RB Ablated | Controls | RB Ablated | Controls | |
| Prenatal* | 1/16 | 0/15 | 0/28 | 0/10 |
| Postnatal** | 0/9 | 0/7 | 0/11 | 0/5 |
Animals analyzed at 8–16 months of age.
RB ablated mice treated with doxycycline during gestation.
RB ablated mice treated with doxycycline as adults or not treated with doxycycline.
Increased cell death could account for lack of tumor formation in Rb deficient lungs. Indeed, apoptosis was detected in RB ablated lungs at baseline before injury and on day 14 and 9 months after injury. The increase in TUNEL positive epithelial cells in RB ablated lungs was not statistically significant over controls prior to injury, but did reach statistical significance on day 14 and 9 months after injury (Fig. 3). Additionally, apoptosis in RB ablated lungs at day 14 and 9 months after injury was significantly elevated over RB ablated lungs prior to injury (p=0.003 and p=0.008, respectively) (Fig. 3). Finally, RB ablated lungs could be blindly identified based upon morphologic features of apoptosis when assessed by a pathologist (KAWB). Thus, sustained cell cycle progression in RB deficient lungs was accompanied by apoptotic cell death. The 6–7 fold increase in apoptosis in RB ablated injured lungs as compared to controls corresponds to a 4–8 fold increase in proliferation (Fig. 1). Taken together, these data provide evidence that increased cell death accounts, at least in part, for the absence of tumor formation in RB ablated lungs despite long term aberrant cell cycle progression.
Postnatal and prenatal RB ablation result in similar phenotypic outcomes
To determine whether timing of RB ablation significantly impacts the phenotypic outcomes in the lung epithelium in vivo, RB recombination was induced postnatally rather than during development by treating adult mice with doxycycline. RB recombination was consistently detected in the lungs of double transgenic mice treated with doxycycline but not in controls lacking Cre recombinase (Fig. 4 and data not shown). Surprisingly, RB recombination was also detected in double transgenic adult lungs in the absence of doxycycline treatment (Fig. 4). Despite the finding that postnatal Rb ablation occurs independently of doxycycline treatment, this mouse model remains valuable for assessing the effects of developmental versus postnatal RB ablation since Rb ablation during development is strictly dependent upon doxycycline induction (15).
Figure 4. Prenatal and postnatal RB ablation occurs throughout the lung epithelium and is present in both Clara and ciliated cells.

PCR analysis on lung DNA from postnatal (PN) day 1 or adult double transgenic mice treated with doxycycline (Dox) during gestation (Prenatal Dox), for 3 weeks at 2–3 months of age (Postnatal Dox) or not treated with doxycycline (No Dox) (A). Control lanes show migration of the recombined (RBRec), wild type (RBWT), and floxed (RBLoxP) RB alleles. Adult mice were homozygous for RBLoxP, and day 1 pups were homozygous or heterozygous for RBLoxP. RBRec was detected in the PN day 1 lungs only after doxycycline treatment. In contrast, RBRec was detected in adult lungs in the absence and presence of doxycycline treatment. Thyroid stimulating hormone beta subunit (TSHbeta) was amplified in each sample as an endogenous internal control to verify equivalent template DNA quality and quantity. Data is representative of 5 No Dox and 36 Prenatal Dox (15 homozygous and 21 heterozygous for RBLox) PN day 1 lungs, and ≥ 3 adult lungs for each treatment group. Varied levels of recombination among samples is due, at least in part, to differences in the relative proportion of conducting airway epithelium represented in the lung tissue used to isolate DNA. Enzymatic staining for β-galactosidase performed on lung sections from adult double transgenic mice haRBoring the ROSA26 reporter locus after prenatal doxycycline treatment (B), postnatal doxycycline treatment (C) or no doxycycline treatment (D). Representative low (top panels) and high (bottom panels) power images are shown. β-galactosidase staining (blue) was epithelial specific and present throughout the conducting airways in all groups indicating active Cre recombinase and thus RB ablation. Epithelial staining in the conducting airways was less uniform after postnatal treatment and no treatment as compared to prenatal treatment (compare B to C and D). Ciliated (arrows) and Clara cells (arrowheads) were stained in all three treatment groups. Data is representative of at least 3 animals for each treatment group. br=bronchiole; Original magnification: 200× (top panels) 1000× (bottom panels)
The conducting airways in the mature mouse lung are lined predominantly by ciliated cells and non-ciliated Clara cells. To determine the extent of postnatal RB ablation in these distinct cell types, in situ β-galactosidase analysis was performed on lungs from double transgenic mice containing the ROSA26 locus. As expected, β-galactosidase positive cells were restricted to the epithelium (Fig. 4). The majority of epithelial cells in the conducting airways were β-galactosidase positive and included both ciliated and Clara cells. Doxycycline administration during gestation induces Cre recombinase expression in early progenitor cells within the lung epithelium (35). Accordingly, the overall β-galactosidase staining was more uniform after doxycycline treatment during development as compared to postnatal treatment (Fig. 4 and (15)). A similar pattern of β-galactosidase staining was seen in the conducting airways of double transgenic adult mice not treated with doxycycline (Fig. 4). Scattered β-galactosidase positive alveolar cells, consistent with the location of Type II cells, were detected and appeared to be more frequent in the presence of doxycycline treatment. Thus, both prenatal and postnatal RB ablation is epithelial specific, occurs in the majority of conducting airway epithelial cells, and is present in both Clara and ciliated cells.
Postnatal RB ablation resulted in similar phenotypic outcomes as that seen after RB ablation during development. Epithelial quiescence was maintained in RB deficient lungs after doxycycline administration to adult mice at 2–3 months of age (Fig. 5). Similar to RB ablation during development, postnatal RB loss resulted in aberrant cell cycle progression during epithelial repair after injury (Fig. 5). Non-quiescent Ki67 positive cells were increased in RB ablated lungs on day 14 after injury as compared to controls. Moreover, cells progressed into S-phase and mitosis. Despite the aberrant cell cycle progression, lung tumors were not detected in mice analyzed at 8–16 months of age either in the presence or absence of naphthalene induced injury (Table 1). Taken together, the data further demonstrate that RB function is critical during epithelial repair after injury and provide evidence that prenatal versus postnatal timing of RB ablation does not significantly affect phenotypic outcome.
Figure 5. Postnatal RB ablation results in aberrant cell cycle progression after injury.
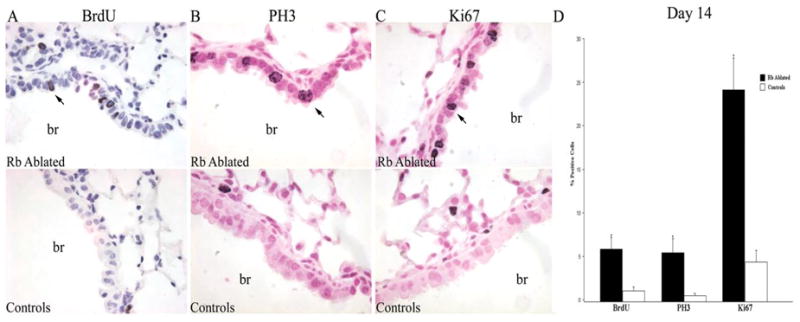
Immunohistochemical analysis for BrdU incorporation (A), and PH3 (B) and Ki67 (C) expression on day 14 after injury in RB ablated and control adult lungs from mice treated with doxycycline as adults. Percent positive cells (arrows) were quantified and data represented as average percent positive cells + SEM (D). RB ablated lungs showed a statistically significant increase in S-phase cells (BrdU), mitotic cells (PH3) and overall proliferation (Ki67) as compared to controls (*p=0.03, *p=0.04 and *p=0.004, respectively). Data is representative of 5–6 animals per time point. br=bronchiole; Original magnification: 1000×
DISCUSSION
Quiescence is established and maintained in adult RB deficient lung epithelium providing evidence that RB is not required for these processes during lung homeostasis. In contrast, RB function is essential for establishing cellular quiescence in the context of epithelial remodeling after injury. Furthermore, the aberrant epithelial cell cycle progression in RB ablated lungs was noted 9 months after a single episode of injury providing evidence that epithelial cells do not compensate for RB loss during epithelial remodeling after injury. This is in stark contrast to the compensation that occurs in the postnatal lung after RB ablation in the absence of injury. These studies provide evidence that RB plays a critical and unique role in epithelial remodeling after injury.
The essential role for RB in lung regeneration after injury differs from skeletal muscle wherein RB is essential for muscle development but is not required for muscle regeneration after cardiotoxin induced injury (36, 37). Interestingly, there is a unique requirement for RB in stress erythropoiesis (33). Although RB is not critical for steady state hematopoiesis, RB loss is associated with increased erythroblasts that fail to undergo terminal maturation under stress conditions. These studies suggest that RB loss confers a growth advantage on progenitor cells. Consistent with this concept, Walkley et al. showed that RB is dispensable in hematopoietic stem cells and raised the hypothesis that the requirement for RB in self-renewal is lineage dependent, with progenitor cells having a greater dependence on RB for their division than stem cells (32). The current studies identify RB as a critical regulator of progenitor cells in the repairing lung epithelium. Thus, our data support the notion that RB has unique and essential functions during cellular regeneration after injury, and that progenitor cells are critically dependent on RB for control of cell division.
RB is required in specific cellular contexts and time periods
RB ablation targeted to the lung epithelium resulted in aberrant cellular proliferation and apoptosis in specific cellular contexts, namely in the newborn lung and during regeneration after injury (current data and (15)). These findings are consistent with RB being essential during limited time periods wherein cells transition into quiescence. Cells withdraw from the cell cycle and enter G0 in response to mitogen deprivation (for review, (38)). This process is dependent upon p27 accumulation and reduction in cyclin D activity. In contrast, cell cycle arrest in response to DNA damage requires p21 and not p27. Interestingly, RB has previously been reported to transcriptionally regulate p21 specifically in epithelial cells, and to increase p27 stability by targeting Skp2, a component of the Skp1-Cullin-F-box protein (SCF) E3 ubiquitin ligase complex, for degradation (39, 40). Additionally, p27 was shown to be required for RB mediated senescence in cells in culture (41). Thus, deregulated p21 and/or p27 activity pose a potential link between loss of RB function and inability to enter quiescence.
RB is classically viewed as an essential regulator of cell cycle; however, RB function is also important in regulating cell survival, chromatin remodeling, genomic stability and cellular ploidy (1, 2, 42). Many of these functions depend upon RB mediated gene regulation resulting from Rb interactions with the E2F family of transcription factors. Transcriptional control of DNA replication genes by the E2F/RB pathway is important for maintaining proper cell cycle control and regulating additional cellular processes including ploidy (42). Apoptosis in RB deficient cells also results, at least in part, from deregulation of E2F/RB target genes. Although the response to RB loss is cell type specific, apoptosis in RB null cells frequently occurs in a p53 dependent manner. Interestingly, p53 and its proapoptotic target gene Bax are induced in the lung epithelium after hyperoxia and bleomycin induced injury (43, 44). However, no change in p53, Bax, or activated p53 expression was detected in RB ablated lungs compared to controls, and protein expression was not induced after naphthalene injury (Supplemental Figure).
The timing of RB dependent phenotypes in the lung epithelium has striking parallels with RB function in developing retina and skeletal muscle. RB is critical for cell cycle exit in retinal transitional cells during a limited time window during development (for review (45)). RB ablation prior to this critical time period results in abnormal retinal development, whereas RB loss at later time points results in no abnormalities. This appears to be true in skeletal muscle as well. RB function is required in myoblasts whereas RB loss has no effect in mature myocytes (36, 37). The critical requirement for RB in limited time periods is likely relevant to RB mediated tumor suppression since retinoblastoma is almost exclusively a disease of childhood occurring within the temporal window wherein retinoblasts undergo final maturation (46).
Postnatal and prenatal RB ablation result in similar phenotypic outcomes
Germline versus acute loss of RB function results in different phenotypic outcomes in cells in culture. Quiescent MEFs undergo cell cycle reentry after acute, but not germline, RB loss (16). In addition, acute RB ablation in senescent MEFs leads to reversal of the senescence associated phenotype whereas MEFs with germline RB loss undergo and maintain senescence similar to wild type cells. Keratinocytes undergoing acute loss of RB in culture are completely refractory to growth arrest when induced to differentiate (14). In contrast, RB null keratinocytes derived after conditional RB ablation in mice in vivo undergo growth arrest similar to wild type cells, albeit with a 24 hour delay. These findings suggest that precise timing of RB ablation affects phenotypic outcomes, and that RB is critical for maintaining cellular quiescence and senescence. Additionally, these studies raise the possibility that phenotypes in adult mouse models could reflect secondary affects of developmental compensation after RB loss during embryogenesis.
RB ablation in the postnatal lung did not lead to aberrant cell cycle control under homeostatic conditions providing evidence that RB is not required to maintain lung epithelial quiescence. RB, however, was essential for establishing lung epithelial quiescence after injury regardless of whether RB was ablated during development or in the postnatal lung. These results directly demonstrate that prenatal versus postnatal timing of RB ablation does not significantly affect the injury induced phenotype. Furthermore, the studies provide direct evidence that the injury induced phenotype is not simply secondary to epithelial alterations occurring during development.
Relationship to human carcinogenesis
Carcinogenesis occurs through sequential steps including tumor initiation and promotion. Initiation depends upon somatic mutations. Promotion mechanisms are less well defined but involve epigenetic factors such as inflammation and substances that trigger cell death and proliferation. The current studies demonstrate that RB and cytotoxic damage cooperate to transform the normally quiescent lung epithelium into an organ with sustained epithelial cell death and proliferation. Cytotoxic damage could thereby function as a tumor promoter by creating a cellular context wherein RB function is particularly critical.
Loss of RB function by somatic mutation or deregulation of other proteins in the RB pathway is known to be critical, if not essential, for lung carcinogensis. However, mechanisms underlying tumor promotion are poorly understood. In the liver, many tumor promoters are cytotoxic and therefore indirectly trigger hepatocyte proliferation by causing cell death (for review, (47)). Cytotoxic damage and compensatory proliferation induced by mitogen production are also important components of the tumor promoting microenvironment linking inflammation and carcinogenesis (48). Since the liver is comprised of quiescent differentiated cells, induction of hepatocyte proliferation is a prerequisite for transformation. In corollary, the adult lung epithelium is quiescent and therefore transition of RB null epithelial cells from a quiescent to a constitutively replicating state after cytotoxic damage may set the stage for tumorigenesis.
Lung carcinogenesis is associated with tobacco smoking in 80–85% of cases. Increased cancer incidence in tobacco users has traditionally been attributed solely to the mutagenic affects of cigarette smoke. However, reevaluation of the data suggests that cytotoxic damage and chronic epithelial remodeling induced by tobacco smoke act as critical promoters of carcinogenesis by selecting rather than simply inducing tumorigenic mutations (49, 50). Repetitive smoking may therefore promote carcinogenesis by creating a microenvironment that facilitates preferential expansion of cells with mutations that confer a proliferative advantage or resistance to cytotoxic damage.
If smoking promotes carcinogensis by causing chronic injury, one would predict that pulmonary diseases that arise in response to chronic lung injury would be associated with an increased risk of lung cancer. Indeed, patients with chronic interstitial lung disease are at increased risk for developing lung cancer independent of smoking history (for review (20, 21)). Although the pathogenetic mechanisms are unknown, an association between chronic injury and lung cancer provides correlative evidence that cytotoxic damage and epithelial remodeling promote lung carcinogenesis. The current studies identify RB as a critical regulator in the context of lung epithelial repair after cytotoxic damage, and suggest RB as a potential molecular link between chronic lung injury and carcinogenesis.
Supplementary Material
Acknowledgments
Grant support: National Heart Lung and Blood Institute R01 HL079193 (KAWB). We thank J.A. Whitsett, S.I. Wells, J.C. Rhodes and D.S. Askew for critical review of the manuscript.
References
- 1.Cobrinik D. Pocket proteins and cell cycle control. Oncogene. 2005;24:2796–2809. doi: 10.1038/sj.onc.1208619. [DOI] [PubMed] [Google Scholar]
- 2.Dimova DK, Dyson NJ. The E2F transcriptional network: old acquaintances with new faces. Oncogene. 2005;24:2810–26. doi: 10.1038/sj.onc.1208612. [DOI] [PubMed] [Google Scholar]
- 3.Clarke AR, Maandag ER, van Roon M, et al. Requirement for a functional Rb-1 gene in murine development. Nature. 1992;359:328–30. doi: 10.1038/359328a0. [DOI] [PubMed] [Google Scholar]
- 4.Lee EY, Chang CY, Hu N, et al. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–94. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- 5.Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- 6.Wu L, de Bruin A, Saavedra HI, et al. Extra-embryonic function of Rb is essential for embryonic development and viability. Nature. 2003;421:942–7. doi: 10.1038/nature01417. [DOI] [PubMed] [Google Scholar]
- 7.Iavarone A, King ER, Dai XM, Leone G, Stanley ER, Lasorella A. Retinoblastoma promotes definitive erythropoiesis by repressing Id2 in fetal liver macrophages. Nature. 2004;432:1040–5. doi: 10.1038/nature03068. [DOI] [PubMed] [Google Scholar]
- 8.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–12. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 9.Mayhew CN, Bosco EE, Fox SR, et al. Liver-specific pRB loss results in ectopic cell cycle entry and aberrant ploidy. Cancer Res. 2005;65:4568–77. doi: 10.1158/0008-5472.CAN-04-4221. [DOI] [PubMed] [Google Scholar]
- 10.Kucherlapati MH, Nguyen AA, Bronson RT, Kucherlapati RS. Inactivation of conditional Rb by Villin-Cre leads to aggressive tumors outside the gastrointestinal tract. Cancer Res. 2006;66:3576–83. doi: 10.1158/0008-5472.CAN-05-2699. [DOI] [PubMed] [Google Scholar]
- 11.Haigis K, Sage J, Glickman J, Shafer S, Jacks T. The related retinoblastoma (pRb) and p130 proteins cooperate to regulate homeostasis in the intestinal epithelium. J Biol Chem. 2006;281:638–47. doi: 10.1074/jbc.M509053200. [DOI] [PubMed] [Google Scholar]
- 12.Maddison LA, Sutherland BW, Barrios RJ, Greenberg NM. Conditional deletion of Rb causes early stage prostate cancer. Cancer Res. 2004;64:6018–25. doi: 10.1158/0008-5472.CAN-03-2509. [DOI] [PubMed] [Google Scholar]
- 13.Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell. 2003;4:181–9. doi: 10.1016/s1535-6108(03)00220-4. [DOI] [PubMed] [Google Scholar]
- 14.Ruiz S, Santos M, Segrelles C, et al. Unique and overlapping functions of pRb and p107 in the control of proliferation and differentiation in epidermis. Development. 2004;131:2737–48. doi: 10.1242/dev.01148. [DOI] [PubMed] [Google Scholar]
- 15.Wikenheiser-Brokamp KA. Rb family proteins differentially regulate distinct cell lineages during epithelial development. Development. 2004;131:4299–4310. doi: 10.1242/dev.01232. [DOI] [PubMed] [Google Scholar]
- 16.Sage J, Miller AL, Perez-Mancera PA, Wysocki JM, Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature. 2003;424:223–8. doi: 10.1038/nature01764. [DOI] [PubMed] [Google Scholar]
- 17.Minna JD, Roth JA, Gazdar AF. Focus on lung cancer. Cancer Cell. 2002;1:49–52. doi: 10.1016/s1535-6108(02)00027-2. [DOI] [PubMed] [Google Scholar]
- 18.Strong LC, Herson J, Haas C. Cancer mortality in relatives of retinoblastoma patients. J Natl Cancer Inst. 1984;73:303–11. doi: 10.1093/jnci/73.2.303. [DOI] [PubMed] [Google Scholar]
- 19.Kleinerman RA, Tarone RE, Abramson DH, Seddon JM, Li FP, Tucker MA. Hereditary retinoblastoma and risk of lung cancer. J Natl Cancer Inst. 2000;92:2037–9. doi: 10.1093/jnci/92.24.2037. [DOI] [PubMed] [Google Scholar]
- 20.Daniels CE, Jett JR. Does interstitial lung disease predispose to lung cancer? Curr Opin Pulm Med. 2005;11:431–7. doi: 10.1097/01.mcp.0000170521.71497.ba. [DOI] [PubMed] [Google Scholar]
- 21.Artinian V, Kvale PA. Cancer and interstitial lung disease. Curr Opin Pulm Med. 2004;10:425–34. doi: 10.1097/00063198-200409000-00017. [DOI] [PubMed] [Google Scholar]
- 22.Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182:311–22. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 23.Buckpitt A, Chang AM, Weir A, et al. Relationship of cytochrome P450 activity to Clara cell cytotoxicity. IV Metabolism of naphthalene and naphthalene oxide in microdissected airways from mice, rats, and hamsters. Mol Pharmacol. 1995;47:74–81. [PubMed] [Google Scholar]
- 24.Park KS, Wells JM, Zorn AM, et al. Transdifferentiation of ciliated cells during repair of the respiratory epithelium. Am J Respir Cell Mol Biol. 2006;34:151–7. doi: 10.1165/rcmb.2005-0332OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Winkle LS, Buckpitt AR, Nishio SJ, Isaac JM, Plopper CG. Cellular response in naphthalene-induced Clara cell injury and bronchiolar epithelial repair in mice. Am J Physiol. 1995;269:L800–18. doi: 10.1152/ajplung.1995.269.6.L800. [DOI] [PubMed] [Google Scholar]
- 26.Plopper CG, Suverkropp C, Morin D, Nishio S, Buckpitt A. Relationship of cytochrome P-450 activity to Clara cell cytotoxicity. I Histopathologic comparison of the respiratory tract of mice, rats and hamsters after parenteral administration of naphthalene. J Pharmacol Exp Ther. 1992;261:353–63. [PubMed] [Google Scholar]
- 27.Herrera RE, Sah VP, Williams BO, Makela TP, Weinberg RA, Jacks T. Altered cell cycle kinetics, gene expression, and G1 restriction point regulation in Rb-deficient fibroblasts. Mol Cell Biol. 1996;16:2402–07. doi: 10.1128/mcb.16.5.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Classon M, Salama S, Gorka C, Mulloy R, Braun P, Harlow E. Combinatorial roles for pRB, p107, and p130 in E2F-mediated cell cycle control. Proc Natl Acad Sci U S A. 2000;97:10820–5. doi: 10.1073/pnas.190343497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Novitch BG, Mulligan GJ, Jacks T, Lassar AB. Skeletal muscle cells lacking the retinoblastoma protein display defects in muscle gene expression and accumulate in S and G2 phases of the cell cycle. J Cell Biol. 1996;135:441–56. doi: 10.1083/jcb.135.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lipinski MM, Macleod KF, Williams BO, Mullaney TL, Crowley D, Jacks T. Cell-autonomous and non-cell-autonomous functions of the Rb tumor suppressor in developing central nervous system. Embo J. 2001;20:3402–13. doi: 10.1093/emboj/20.13.3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harrington EA, Bruce JL, Harlow E, Dyson N. pRB plays an essential role in cell cycle arrest induced by DNA damage. Proc Natl Acad Sci U S A. 1998;95:11945–50. doi: 10.1073/pnas.95.20.11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell. 2007;129:1081–95. doi: 10.1016/j.cell.2007.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spike BT, Dirlam A, Dibling BC, et al. The Rb tumor suppressor is required for stress erythropoiesis. Embo J. 2004;23:4319–29. doi: 10.1038/sj.emboj.7600432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–1. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 35.Perl AK, Tichelaar JW, Whitsett JA. Conditional gene expression in the respiratory epithelium of the mouse. Transgenic Res. 2002;11:21–9. doi: 10.1023/a:1013986627504. [DOI] [PubMed] [Google Scholar]
- 36.Zacksenhaus E, Jiang Z, Chung D, Marth JD, Phillips RA, Gallie BL. pRb controls proliferation, differentiation, and death of skeletal muscle cells and other lineages during embryogenesis. Genes Dev. 1996;10:3051–64. doi: 10.1101/gad.10.23.3051. [DOI] [PubMed] [Google Scholar]
- 37.Huh MS, Parker MH, Scime A, Parks R, Rudnicki MA. Rb is required for progression through myogenic differentiation but not maintenance of terminal differentiation. J Cell Biol. 2004;166:865–76. doi: 10.1083/jcb.200403004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agami R, Bernards R. Convergence of mitogenic and DNA damage signaling in the G1 phase of the cell cycle. Cancer Lett. 2002;177:111–8. doi: 10.1016/s0304-3835(01)00785-6. [DOI] [PubMed] [Google Scholar]
- 39.Binne UK, Classon MK, Dick FA, et al. Retinoblastoma protein and anaphase-promoting complex physically interact and functionally cooperate during cell-cycle exit. Nat Cell Biol. 2007;9:225–32. doi: 10.1038/ncb1532. [DOI] [PubMed] [Google Scholar]
- 40.Decesse JT, Medjkane S, Datto MB, Cremisi CE. RB regulates transcription of the p21/WAF1/CIP1 gene. Oncogene. 2001;20:962–71. doi: 10.1038/sj.onc.1204169. [DOI] [PubMed] [Google Scholar]
- 41.Alexander K, Hinds PW. Requirement for p27(KIP1) in retinoblastoma protein-mediated senescence. Mol Cell Biol. 2001;21:3616–31. doi: 10.1128/MCB.21.11.3616-3631.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Srinivasan SV, Mayhew CN, Schwemberger S, Zagorski W, Knudsen ES. RB loss promotes aberrant ploidy by deregulating levels and activity of DNA replication factors. J Biol Chem. 2007;282:23867–77. doi: 10.1074/jbc.M700542200. [DOI] [PubMed] [Google Scholar]
- 43.Okudela K, Ito T, Mitsui H, et al. The role of p53 in bleomycin-induced DNA damage in the lung. A comparative study with the small intestine. Am J Pathol. 1999;155:1341–51. doi: 10.1016/S0002-9440(10)65236-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Reilly MA, Staversky RJ, Huyck HL, et al. Bcl-2 family gene expression during severe hyperoxia induced lung injury. Lab Invest. 2000;80:1845–54. doi: 10.1038/labinvest.3780195. [DOI] [PubMed] [Google Scholar]
- 45.Pacal M, Bremner R. Insights from animal models on the origins and progression of retinoblastoma. Curr Mol Med. 2006;6:759–81. doi: 10.2174/1566524010606070759. [DOI] [PubMed] [Google Scholar]
- 46.Balmer A, Zografos L, Munier F. Diagnosis and current management of retinoblastoma. Oncogene. 2006;25:5341–9. doi: 10.1038/sj.onc.1209622. [DOI] [PubMed] [Google Scholar]
- 47.Fausto N. Mouse liver tumorigenesis: models, mechanisms, and relevance to human disease. Semin Liver Dis. 1999;19:243–52. doi: 10.1055/s-2007-1007114. [DOI] [PubMed] [Google Scholar]
- 48.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–90. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 49.Rodin SN, Rodin AS. Origins and selection of p53 mutations in lung carcinogenesis. Semin Cancer Biol. 2005;15:103–12. doi: 10.1016/j.semcancer.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 50.Thilly WG. Have environmental mutagens caused oncomutations in people? Nat Genet. 2003;34:255–9. doi: 10.1038/ng1205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.