

Abstract
4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK, 1) and its metabolite, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL, 2) are both potent pulmonary carcinogens in rats. The metabolism of NNK to NNAL is stereoselective and reversible, with (S)-NNAL being the major enantiomer formed from NNK. In rats, (R)-NNAL undergoes facile glucuronidation and is rapidly excreted in urine, while (S)-NNAL is preferentially retained in tissues and converted to NNK. We hypothesized that the lung carcinogenicity of NNK in the rat is due in part to preferential retention of (S)-NNAL in lung, reconversion to NNK, then metabolic activation of NNK to pyridyloxobutyl(POB)-DNA adducts. We tested this hypothesis by treating male F344 rats with 10 ppm of NNK, (R)-NNAL, or (S)-NNAL in the drinking water. After 1, 2, 5, 10, 16, or 20 weeks of treatment, POB-DNA adducts in liver and lung DNA were quantified by HPLC-ESI-MS/MS. At each time point, total adduct levels were higher in lung than liver. O2-[4-(3-pyridyl)-4-oxobut-1-yl]thymidine (O2-POB-dThd, 13) was the major adduct detected. Total adduct levels in the rats treated with (S)-NNAL were 0.6–1.3 times as great as those in the NNK group in lung and 0.7–1.4 times in liver, and 6–14 times higher than those in the (R)-NNAL group in lung and 11–17 times in liver. These results suggest that (S)-NNAL is stereoselectively retained in tissues. This study demonstrates for the first time the accumulation and persistence of specific POB-DNA adducts in rat lung and liver during chronic treatment with NNK, (R)-NNAL and (S)-NNAL, and supports the hypothesis that preferential retention of (S)-NNAL in the lung, followed by reconversion to NNK, then metabolic activation of NNK, is critical for lung carcinogenesis by NNK and NNAL.
Introduction
4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK, 1, Chart 1), a tobacco-specific nitrosamine found only in tobacco products and their smoke, is a well-established pulmonary carcinogen in rodents, inducing tumors independent of the route of administration (1). Its lung tumorigenicity is particularly great in the rat, in which it also induces tumors of the liver, pancreas, and nasal cavity (2). NNK and the related tobacco-specific nitrosamine, N′-nitrosonornicotine (NNN), are considered by the International Agency for Research on Cancer to be human carcinogens (3).
Chart 1.
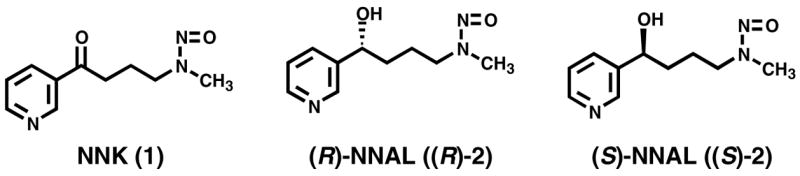
Structures of NNK, (R)-NNAL and (S)-NNAL
NNK requires metabolism to express its mutagenicity and presumably its carcinogenicity (1). In this study, we investigate the hypothesis that metabolism of NNK to (S)-4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol [(S)-NNAL, (S)-2], followed by reoxidation to NNK, is an important step leading to pulmonary DNA adduct formation in the rat. DNA adducts can cause permanent mutations and are critical in cancer induction by genotoxic carcinogens such as NNK (4). Cytochrome P450-catalyzed α-hydroxylation is the major bioactivation pathway by which NNK forms DNA adducts and expresses its carcinogenicity. α-Methyl hydroxylation of NNK (Scheme 1) results in the formation of a reactive electrophile 9, which pyridyloxobutylates DNA to produce pyridyloxobutyl (POB)-DNA adducts. The formation of POB-DNA adducts is believed to be an important mechanism of NNK carcinogenesis in rodents (5–9), and likely in smokers (10–11). The structures of four POB-DNA adducts - O6-[4-(3-pyridyl)-4-oxobut-1-yl]-2′-deoxyguanosine (O6-POB-dGuo, 12), O2-[4-(3-pyridyl)-4-oxobut-1-yl]thymidine (O2-POB-dThd, 13), 7-[4-(3-pyridyl)-4-oxobut-1-yl]-2′-deoxyguanosine (7-POB-dGuo, 14), and O2-[4-(3-pyridyl)-4-oxobut-1-yl]-2′-deoxycytidine (O2-POB-dCyd, 15) - have been characterized (Chart 2) (12–14). A sensitive high performance liquid chromatography (HPLC)-electrospray ionization (ESI)-tandem mass spectrometry (MS/MS) method has been developed for quantitative analysis of four stable POB-DNA adducts: O6-POB-dGuo, O2-POB-dThd, 7-[4-(3-pyridyl)-4-oxobut-1-yl]guanine (7-POB-Gua, 16) and O2-[4-(3-pyridyl)-4-oxobut-1-yl]cytosine (O2-POB-Cyt, 17) (Chart 2) (15). α-Hydroxylation of NNK at the methylene carbon adjacent to the N-nitroso group generates methanediazohydroxide (10), which methylates DNA producing methyl-DNA adducts including O6-methylGua (18), 7-methylGua (19) and O4-methylThy (20) (Scheme 1) (1).
Scheme 1.

Overview of NNK and NNAL metabolism and DNA adduct formation. NTH: neutral thermal hydrolysis.
Chart 2.

Structures of POB-DNA adducts 12–17. dR: 2′-deoxyribosyl.
A common pathway of NNK metabolism in virtually all biological systems examined is conversion to NNAL, catalyzed by carbonyl reductases, 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1), and the aldo-keto reductases AKR1C1–4 (16). NNAL, like NNK, is a potent pulmonary carcinogen in rats (1). NNAL can also be bioactivated through cytochrome P450-catalyzed α-hydroxylation to form both methyl-DNA adducts (18–20) and pyridylhydroxybutyl (PHB)-DNA adducts (Scheme 1) (1, 14, 17).
Carbonyl reduction of NNK to NNAL is stereoselective and reversible. Several lines of evidence indicate that (S)-NNAL is important in lung tumorigenesis by NNK. (S)-NNAL is the predominant enantiomer formed from NNK in liver and lung microsomes and cytosol (16,18 ). Both (S)-NNAL and (R)-NNAL can be enzymatically reoxidized to NNK, but the reductive pathway predominates (18–21). In rat liver microsomes, reoxidation of (S)-NNAL to NNK was significantly faster than of (R)-NNAL (18). In experiments carried out in vivo in rats, (S)-NNAL was stereoselectively retained in lung and was more extensively reconverted to NNK than was (R)-NNAL, which was excreted extensively as glucuronides (20, 21). Twenty-four hours after administration of racemic [5-3H]NNAL to rats, NNAL comprised an average of 75% of total radioactivity in the lung with an (S):(R) ratio of >20 (20). When [5-3H]NNAL enantiomers were administrated to rats, less than 5% of the (R)-NNAL dose was excreted as NNK or NNK metabolites, compared to almost 20% of (S)-NNAL. In A/J mice, (S)-NNAL, with activity similar to NNK, was significantly more potent than (R)-NNAL or racemic NNAL (19). Finally, the ratios of (S)/(R)-NNAL and their glucuronides increased over time in the urine of humans who stopped using tobacco products, indicating the preferential retention of (S)-NNAL (22).
Based on these and related data, we hypothesize that the following sequence of events is important for lung tumorigenesis by NNK: metabolism to (S)-NNAL; preferential retention of (S)-NNAL in the lung; reoxidation to NNK; and metabolic activation of NNK to POB-DNA adducts (Scheme 2). In this study, we further explored this mechanism by investigating the formation of POB-DNA adducts in rats treated with NNK, (R)-NNAL and (S)-NNAL. The formation of POB-DNA adducts is an indication of the conversion of NNAL to NNK, since they can be formed from NNK, but not NNAL. If our hypothesis were correct, then similar POB-DNA adduct levels would be expected in rats treated with (S)-NNAL and NNK, while lower levels of adducts would be expected upon (R)-NNAL treatment. DNA pyridyloxobutylation of hepatic DNA in rats treated with a single dose of racemic NNAL was previously demonstrated by analysis of 4-hydroxy-1-(3-pyridyl)-1-butanone (HPB, 8)-releasing DNA adducts by gas chromatography-mass spectrometry (23). In this study, F344 rats were given 10 ppm of NNK, (R)-NNAL or (S)-NNAL in the drinking water for 20 weeks, and the amounts of individual POB-DNA adducts were quantified in liver and lung DNA by HPLC-ESI-MS/MS analysis.
Scheme 2.

Proposed mechanism for centrality of (S)-NNAL in NNK lung tumorigenesis in rodents.
Materials and methods
Caution
NNK and NNAL are carcinogenic They should be handled in a well-ventilated hood with extreme care, and with personal protective equipment.
Chemicals
NNK was purchased from Toronto Research Chemicals (Toronto, Canada). Purity was greater than 98%. (R)- and (S)-NNAL were synthesized, as described previously (24). Chemical purities were at least 99% based on normal phase HPLC analysis (24). Enantiomeric purities were greater than 99% by chiral stationary phase -gas chromatography- nitrosamine selective detection analysis (25). POB-DNA adducts were synthesized (15,26 ). Micrococcal nuclease (LS004797, 15 kU) and phosphodiesterase II (LS003603, 10 U) were obtained from Worthington Biochemical Corporation (Lakewood, NJ). Alkaline phosphatase (567752, 30 U/μL) was purchased from Roche Molecular Biochemicals (Indianapolis, IN). The reagents and enzymes for DNA isolation were purchased from Gentra Systems (Minneapolis, MN). All other chemicals and solvents were acquired from Sigma-Aldrich Chemical Co. (Milwaukee, WI) or Fisher Scientific (Fairlawn, NJ).
Animal experiment
The study was approved by the University of Minnesota Institutional Animal Care and Use Committee. A total of 216 male F344 rats, 7 weeks old, were purchased from Charles River Laboratories (Kingston, NY). They were housed two animals per micro-isolator cage with corn cob bedding in the Research Animal Resources facility of the University of Minnesota under the following conditions: temperature 20–24 °C; relative humidity 50 ± 10%; 12 h light/dark cycle. They were given NIH-07 diet (Harlan, Madison, WI) and tap water ad libitum. The animals were allowed one week for acclimatization to the facility prior to carcinogen treatment. The rats were randomly divided into four groups of 54 rats: (1) control; (2) NNK; (3) (R)-NNAL; (4) (S)-NNAL. Mean body weights at the beginning of the experiment were: 170 ± 12 g for control group; 174 ± 8 g for NNK group; 181 ± 7 g for (R)-NNAL group; 179 ± 11 g for (S)-NNAL group. Body weights were recorded once a month. The rats in the treatment groups received 10 ppm of the appropriate carcinogen in the drinking water, and the control rats were given tap water. Aqueous solutions of carcinogens were prepared weekly and stored at 4°C, conditions under which they are known to be stable. These solutions were placed in the plastic water bottles of the rat cages twice weekly and water consumption was recorded. Nine rats per group were sacrificed by CO2 overdose at 1, 2, 5, 10, 16, and 20 weeks. Tissues were harvested and stored at −80°C until DNA isolation.
Quantitation of POB-DNA adducts by HPLC-ESI-MS/MS-SRM
DNA was isolated from liver and lung of three rats per group, by following the modified Puregene® DNA isolation protocol (Gentra Systems), as previously described except that NaBH3CN was not used (27). DNA was analyzed for POB-DNA adducts by HPLC-ESI-MS/MS-selected reaction monitoring (SRM), as described previously (15). The characteristics of the method including limits of detection, accuracy, precision, and recoveries have been presented (15). In brief, 0.5–2 mg of each DNA sample, with addition of four deuterated internal standards, was subjected to neutral thermal hydrolysis (100 °C, 30 min), followed by enzymatic hydrolysis with micrococcal nuclease, phosphodiesterase II and alkaline phosphatase. The hydrolysate was partially purified by solid phase extraction (Strata-X cartridge, Phenomenex, CA). Then the sample was delivered by an Agilent 1100 capillary HPLC system (Agilent Technologies, Inc. Wilmington, DE) into a Finnigan TSQ Quantum Discovery Max triple quadrupole mass spectrometer (Thermo Electron, San Jose, CA). The ESI source was operated in the positive mode. POB-DNA adducts and corresponding internal standards were detected by monitoring the formation of the POB ion from the parent ion. The ion transitions are listed in Table 1. Contents of dGuo, dCyd and dThd were determined by HPLC (15). The amount of DNA was calculated from dGuo content by considering that 1 mg of DNA contains 3 μmol nucleotides (28), while dGuo accounts for 22% of total nucleotides in rat DNA, as previously determined in our laboratory. Adduct levels were expressed as fmol adduct/mg DNA. Adduct levels at the 20-week time point were also expressed as fmol per μmol appropriate nucleoside, i.e. fmol O2-POB-dThd/μmol dThd and fmol O2-POB-Cyt/μmol dCyd, as listed in Table 3 in the Supporting Information. Levels of POB-DNA adducts were expressed as the mean values ± S.D. of single analyses of three DNA samples per group.
Table 1.
Ion transitions for SRM of POB-DNA adducts and corresponding internal standards
| Ion transitions | ||
|---|---|---|
| Adducts | Parent ion ([M + 1]+, m/z) | Daughter ion (m/z) |
| O6-POB-dGuo (12) | 415.1 | 148.1 [POB]+ |
| [pyridine-D4]O6-POB-dGuo | 419.1 | 152.1 ([pyridine-D4]POB])+ and [Gua+H]+ |
| O2-POB-dThd (13) | 390.1 | 148.1 [POB]+ |
| [pyridine-D4]O2-POB-dThd | 394.1 | 152.1 ([pyridine-D4]POB])+ |
| 7-POB-Gua (16) | 299.1 | 148.1 [POB]+ |
| [pyridine-D4]7-POB-Gua | 303.1 | 152.1 ([pyridine-D4]POB])+ and [Gua+H]+ |
| O2-POB-Cyt (17) | 259.1 | 148.1 [POB]+ |
| [pyridine-D4]O2-POB-Cyt | 263.1 | 152.1 ([pyridine-D4]POB])+ |
Statistical analyses
The method of analysis for water consumption was a repeated measures approach using a generalized linear mixed model, which includes both fixed and random effects. The groups and time intervals are considered fixed effects and the rate of change over time for each cage is treated as a random effect. The variance/covariance structure for the data was estimated using restricted maximum likelihood (REML). Two-way analysis of variance (ANOVA) was used for comparisons of levels of total and individual POB-DNA adducts between groups (NNK, (R)-NNAL and (S)-NNAL) and time (1, 2, 5, 10, 16, 20 weeks). Since the individual adducts were obtained from the same tissue sample, a repeated measures ANOVA was chosen to compare these POB-DNA adducts within each group. Time was treated as a fixed effect and, again, REML was used to estimate the variance/covariance structure. Due to a highly skewed distribution, the POB-DNA adducts were analyzed in the natural log scale. Adjustments for multiple comparisons between groups or between POB-DNA adducts were made according to the Tukey method. A p-value < 0.05 was considered to be statistically significant, except for comparisons of individual POB-DNA adducts within each group at each time point, for which a more restrictive p-value cutoff of less than 0.01 was used to judge statistical significance.
Results
Animal Experiment
The design was based on a carcinogenicity study of NNK and NNAL, in which male F344 rats were given 5 ppm of NNK or NNAL in the drinking water for 128 weeks (2). Twenty-seven of 30 NNK-treated rats and 26 of 30 NNAL-treated rats had lung tumors compared to 6/80 controls. In a preliminary study for this project, POB-DNA adducts were readily detected in liver DNA isolated from rats treated with 2 ppm of NNK in the drinking water for 8 weeks (data not shown). Another study demonstrated that less than 5% of (R)-NNAL and about 20% of (S)-NNAL was converted to NNK in rats (21). To ensure the detection of POB-DNA adducts at each time point in rats treated with the enantiomers of NNAL, the 10 ppm doses of (R)-NNAL and (S)-NNAL were chosen for the current experiment. The same dose of NNK was used for comparison.
Overall, the rats treated with (R)-NNAL consumed more water than those in the control group, the NNK group and the (S)-NNAL group. The differences were significant (P < 0.05). There was a small but significant decrease in water consumption over time in each group (P < 0.05) (Figure 1A-D). There were no effects of carcinogen treatment on body weights (Figure 1E). All rats survived the 20-week experiment.
Figure 1.
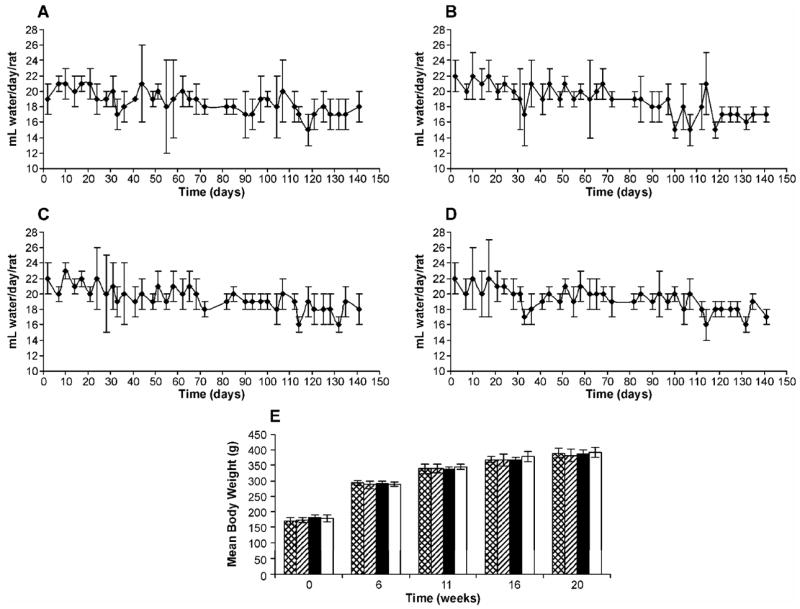
Water consumption by (A) control rats; (B) NNK-treated rats; (C) (R)- NNAL-treated rats; (D) (S)-NNAL-treated rats; and (E) body weights of rats in control, NNK, (R)-NNAL and (S)-NNAL groups during the 20-week experiment. Values for water consumption and body weights are the mean ± S.D. of measurements of all rats in each group. Symbol designations are: ▩, control; ▨, NNK; ■. (R)-NNAL; □, (S)-NNAL.
POB-DNA adducts in NNK-treated rats
POB-DNA adducts were not detected in liver and lung DNA isolated from control rats, but were readily detected in liver and lung DNA from NNK-treated rats (Figure 2A–D). The amounts of POB-DNA adducts in NNK-treated rats are summarized in Figure 3A–C (also see Tables 1A and 2A in the Supporting Information). After the first week of treatment, levels of O2-POB-dThd were the highest among four POB-DNA adducts in both liver and lung DNA: 650 fmol/mg DNA in liver and 1080 fmol/mg DNA in lung. Those of 7-POB-Gua were 490 fmol/mg DNA in liver and 750 fmol/mg DNA in lung, while those of O2-POB-Cyt were 170 fmol/mg DNA in liver and 240 fmol/mg DNA in lung. O6-POB-dGuo was not detected in liver DNA, and its level in lung DNA was 45 fmol/mg DNA. At each sacrifice interval, the relative adduct levels in lung and liver DNA were in the same order: O2-POB-dThd > 7-POB-Gua > O2-POB-Cyt > O6-POB-dGuo (Figure 3A–C). In liver, there was a significant difference between levels of each individual adduct (P < 0.01), except for levels of 7-POB-Gua and O2-POB-dThd at week one, and levels of 7-POB-Gua and O2-POB-Cyt at week 20. In lung, levels of individual adducts were significantly different from each other (P < 0.01), except for 7-POB-Gua and O2-POB-dThd at weeks 16 and 20. Statistical comparisons between each POB-DNA adduct within the NNK group are shown in Table 4A in the Supporting Information. O2-POB-dThd was the major adduct detected. It accounted for 49.6–70.0% of total adducts in liver DNA and 51.1–73.4% in lung DNA. The percentage of O2-POB-dThd of the adducts measured increased throughout the course of NNK treatment (Tables 1A and 2A in the Supporting Information). The percentage of 7-POB-Gua of total POB adduct levels measured decreased over time, accounting for 18.7–39.1% in liver DNA and 15.0–35.5% in lung DNA. Those of O2-POB-Cyt were 6.2–13.0% in liver DNA and 6.5–13.3% in lung DNA. O6-POB-dGuo contributed to 0.3–2.1% of total adduct levels in lung DNA. The levels of O6-POB-dGuo were 24–250 times lower than those of O2-POB-dThd.
Figure 2.
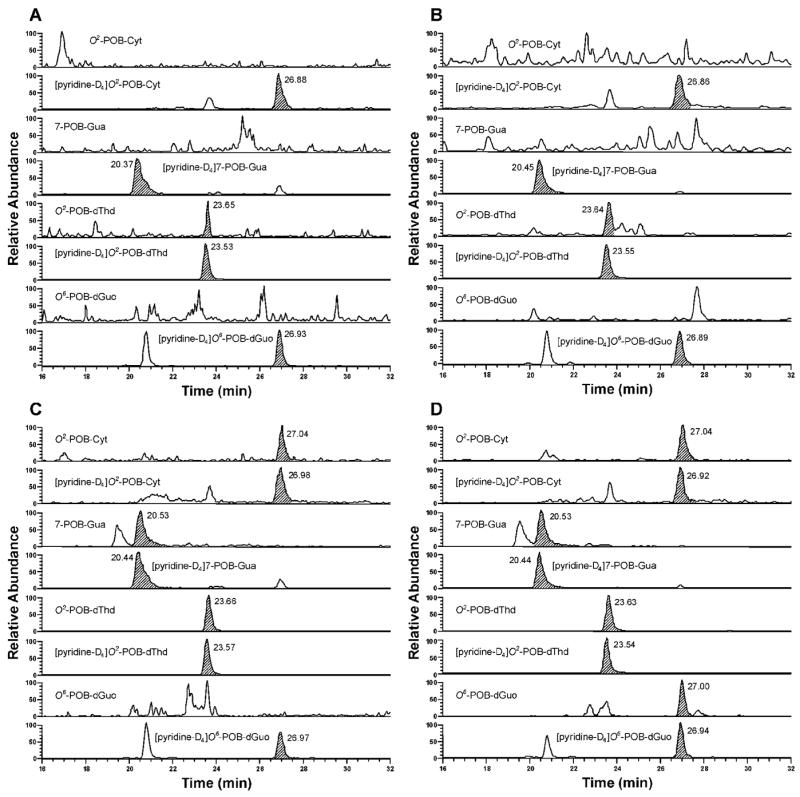
Typical SRM chromatograms obtained upon analysis of (A) liver DNA from control rats; (B) lung DNA from control rats; (C) liver DNA from NNK-treated rats; and (D) lung DNA from NNK-treated rats after 16 weeks of treatment. Individual POB-DNA adducts or internal standards were monitored as indicated on each channel. In Figure 2A and 2B, the peaks eluting at the position of O2-POB-dThd, the areas of which are 0.2 % of [pyridine-D4]O2-POB-dThd, originate from the internal standard.
Figure 3.
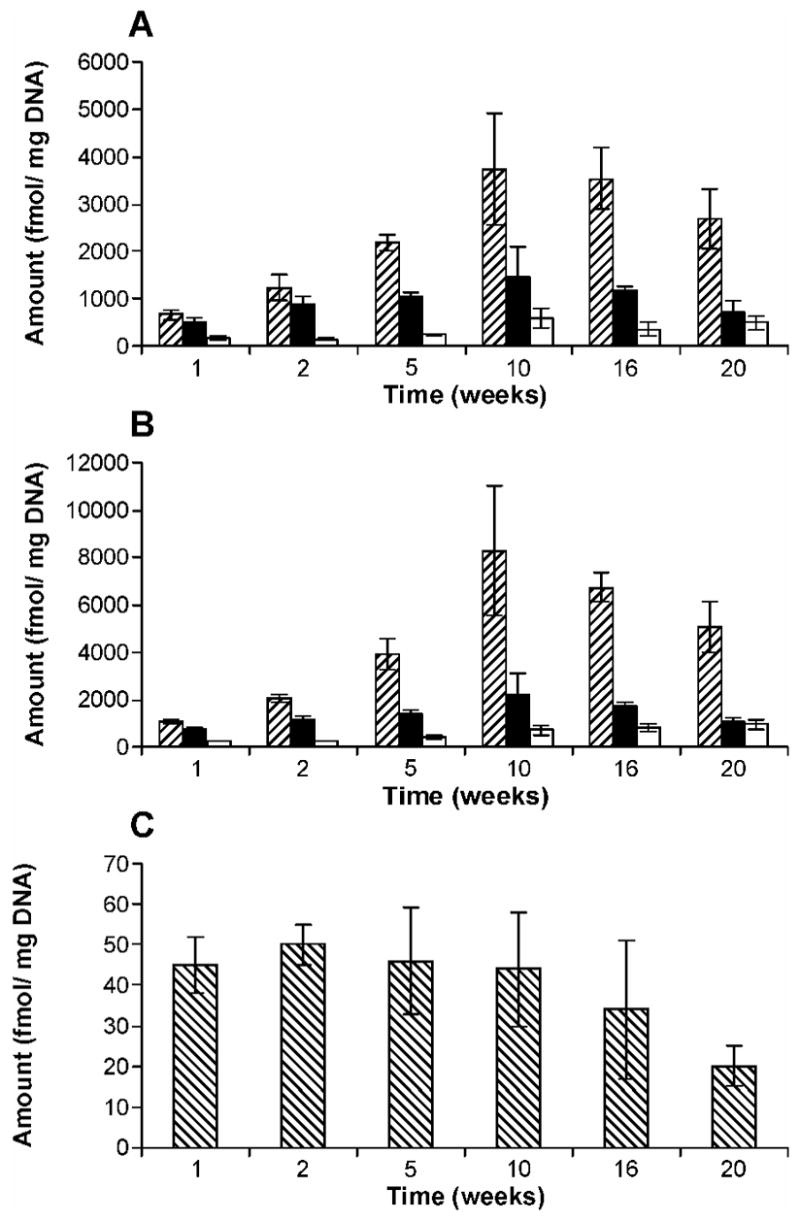
Levels of POB-DNA adducts in (A) liver and (B) lung DNA of NNK-treated rats; (C): O6-POB-dGuo in lung DNA of NNK-treated rats on a different scale. Symbol designations are: ▨, O2-POB-dThd; ■, 7-POB-Gua; □, O2-POB-Cyt; ▧, O6-POB-dGuo. Each value is the mean ± S.D. of single analyses of DNA samples from three rats per group.
In both liver and lung, total adduct levels, as well as those of O2-POB-dThd, 7-POB-Gua and O2-POB-Cyt, increased during the initial period of NNK treatment (Figures 3A, B; 4A, B). The maxima were observed at 10 weeks, except for O2-POB-Cyt in lung DNA which continuously increased over time (Figure 3B). After 10 weeks, total levels of POB-DNA adducts declined gradually. The maximum amounts of POB-DNA adducts were as follows: O2-POB-dThd, 3740 fmol/mg DNA in liver (10 weeks) and 8260 fmol/mg DNA in lung (10 weeks); 7-POB-Gua, 1460 fmol/mg DNA in liver (10 weeks) and 2220 fmol/mg DNA in lung (10 weeks); O2-POB-Cyt: 580 fmol/mg DNA in liver (10 weeks) and 940 fmol/mg DNA (20 weeks) in lung. In lung DNA, the levels of O6-POB-dGuo were steady during the first 10 weeks, and then declined slightly (Figure 3C). Total adduct levels in rat lung were 1.6–1.9 times higher than those in liver during the period of NNK treatment.
Figure 4.
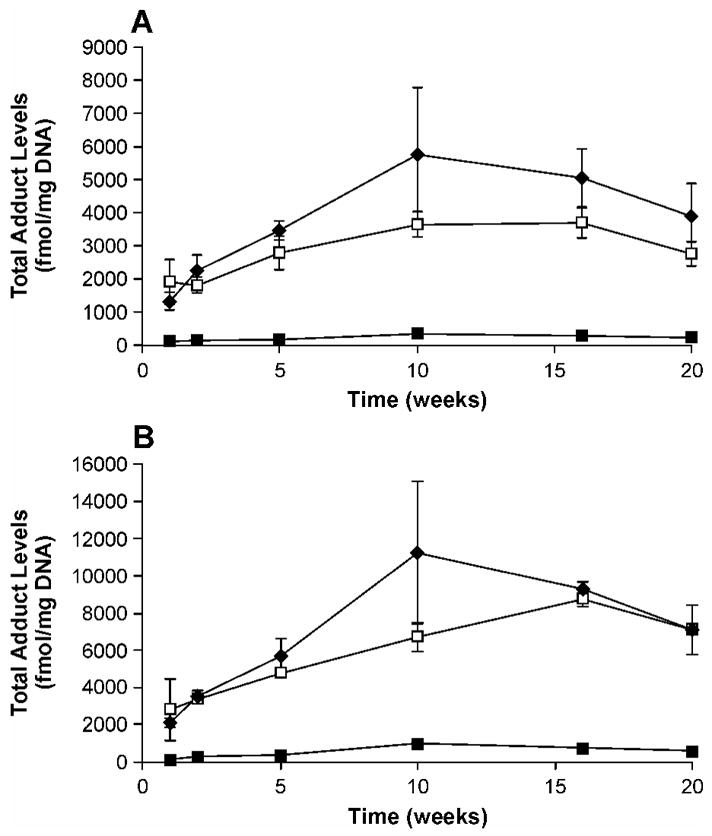
Plots of total adduct levels (fmol/mg DNA) versus time (weeks) in (A) liver and (B) lung DNA of NNK, (R)- and (S)-NNAL-treated rats. Symbol designations are: ◆, NNK, □, (S)-NNAL; ■, (R)-NNAL. Values are the sum of amounts of all POB-DNA adducts measured in lung or liver DNA at each time point ± S. D.
POB-DNA adducts in (R)- and (S)-NNAL-treated rats
POB-DNA adducts were formed in liver and lung DNA of both (R)- and (S)-NNAL–treated rats (Figure 5A-D), with total adduct levels produced from (S)-NNAL significantly higher than those from (R)-NNAL at each time point (P < 0.05) (Figure 6A-F). Averaging over the time points for each individual POB-DNA adduct, levels of adducts from (S)-NNAL treatment were also significantly greater than those from (R)-NNAL treatment in liver and lung (P < 0.05), except there were too few measurable values for O2-POB-Cyt in the (R)-NNAL group to do a comparison. The amounts of POB-DNA adducts in NNAL-treated rats are summarized in Figures 4A-B and 6A-F (also see Tables 1B, C and Tables 2B, C in the Supporting Information). In liver DNA, treatment with (S)-NNAL for one week produced 1890 fmol/mg DNA of total POB-DNA adducts while (R)-NNAL only gave 110 fmol/mg DNA. Similarly, treatment with (S)-NNAL for one week generated 2810 fmol/mg DNA of POB-DNA adducts in lung DNA while (R)-NNAL gave 190 fmol/mg DNA. At various intervals, (S)-NNAL gave 11–17 times more POB-DNA adducts than (R)-NNAL in liver DNA and 6–14 times more in lung DNA (Figure 4A, B). Consistent with the results of NNK treatment, the amounts of O2-POB-dThd were the highest among the four POB-DNA adducts in liver and lung DNA from both the (R)-NNAL and (S)-NNAL groups. In rats treated with (S)-NNAL, O2-POB-dThd accounted for 52.4–74.2% of total adduct levels in liver and 50.2–76.1% in lung. In rats treated with (R)-NNAL, O2-POB-dThd contributed 60.9–83.9% of total adduct levels in liver and 51.8–78.8% in lung. The percentages of O2-POB-dThd increased over time. Similar to the results in the rats treated with NNK, the orders of the relative adduct levels in lung and liver DNA were: O2-POB-dThd > 7-POB-Gua > O2-POB-Cyt > O6-POB-dGuo for both (R)-and (S)-NNAL (Figure 6A-F). In rats treated with (S)-NNAL, levels of individual adducts were significantly different from each other (P < 0.01), except for levels of 7-POB-Gua and O2-POB-dThd in liver at week one and levels of 7-POB-Gua and O2-POB-Cyt in lung at week 20. In rats treated with (R)-NNAL, there was a significant difference between levels of O2-POB-dThd and 7-POB-Gua in liver at weeks 5 and 20, while significant differences were observed among all adducts in lung at each time point (P < 0.01). Comparisons among individual POB-DNA adduct levels in the (R)-NNAL and (S)-NNAL groups are shown in Table 4B, C in the Supporting Information. O6-POB-dGuo was detected only in lung DNA and the levels were the lowest among the four adducts at each time point. The levels of O6-POB-dGuo were 33–263 times lower than those of O2-POB-dThd in DNA of the rats treated with (R)-NNAL and 28–320 times lower in those treated with (S)-NNAL. In the (R)-NNAL group, total adduct levels increased over time until the maximum was found at 10 weeks with total adduct levels of 340 fmol/mg DNA in liver and 1040 fmol/mg DNA in lung (Figure 4A–B). However, for (S)-NNAL, the maximum total adduct levels were reached at 16 weeks with 3680 fmol/mg DNA in liver and 8730 fmol/mg DNA in lung (Figure 4A–B). For both (R)-NNAL and (S)-NNAL, total adduct levels in lung DNA were higher than those in liver DNA. (R)-NNAL treatment generated 1.8–3.1 times more POB-DNA adducts in lung than liver, and (S)-NNAL gave 1.5–2.6 time more in lung than liver.
Figure 5.

Typical SRM chromatograms obtained upon analysis of (A) liver DNA from (R)-NNAL-treated rats after 10 weeks of treatment; (B) lung DNA from (R)-NNAL-treated rats after 10 weeks of treatment; (C) liver DNA from (S)-NNAL-treated rats after 16 weeks of treatment; and (D) lung DNA from (S)-NNAL-treated rats after 16 weeks of treatment. Individual POB-DNA adducts or internal standards were monitored as indicated on each channel.
Figure 6.
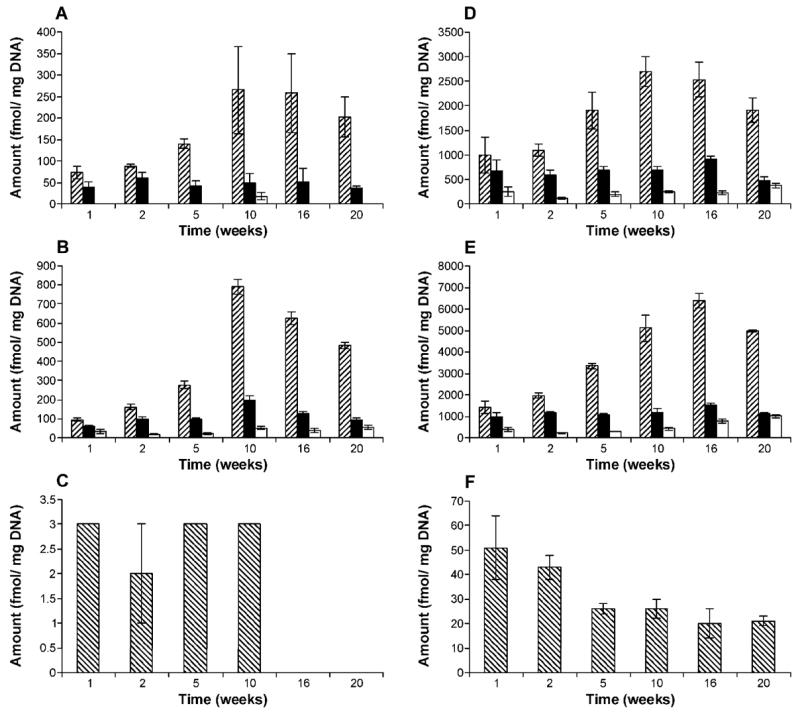
Levels of POB-DNA adducts in (A) liver and (B) lung DNA of (R)-NNAL-treated rats; (D) liver and (E) lung DNA of (S)-NNAL-treated rats; panel (C): O6-POB-dGuo in lung DNA of (R)-NNAL-treated rats on a different scale; panel (F): O6-POB-dGuo in lung DNA of (S)-NNAL-treated rats on a different scale. Symbol designations are: ▨, O2-POB-dThd; ■, 7-POB-Gua; □, O2-POB-Cyt; ▧, O6-POB-dGuo. Each value is the mean ± S.D. of single analyses of DNA samples from three rats per group.
Comparison of POB-DNA adduct levels in NNK, (R)-NNAL and (S)-NNAL-treated rats
In both liver and lung, there was no significant difference between the total amounts of POB-DNA adducts produced from (S)-NNAL and those from NNK, and total adduct levels of both NNK and (S)-NNAL were significantly higher than those of (R)-NNAL (P < 0.05) (Figure 4A, B). Except for 7-POB-Gua and O2-POB-Cyt in liver and O6-POB-dGuo in lung, the amounts of individual POB-DNA adducts were not significantly different in rats treated with NNK and (S)-NNAL, while levels of each individual adduct in NNK and (S)-NNAL-treated rats were significantly higher than those in the (R)-NNAL group (P < 0.05). After the first week of treatment, total adduct levels in the (S)-NNAL group were 1.4 times higher than those in the NNK group in liver, and 1.3 times higher in lung (Figure 7A). The ratios of total adduct levels from (S)-NNAL to those from NNK decreased slowly with continuous treatment. At 10 weeks, the lowest ratios of 0.6 and 0.7 were found in lung and liver, respectively. Then the ratios remained steady at 1.0 for lung and 0.7 for liver during the remainder of the experiment. In contrast to (S)-NNAL, the ratios of total adduct levels from (R)-NNAL to those from NNK were relatively low: 0.05–0.08 for liver and 0.07–0.09 for lung during the experiment (Figure 7B).
Figure 7.
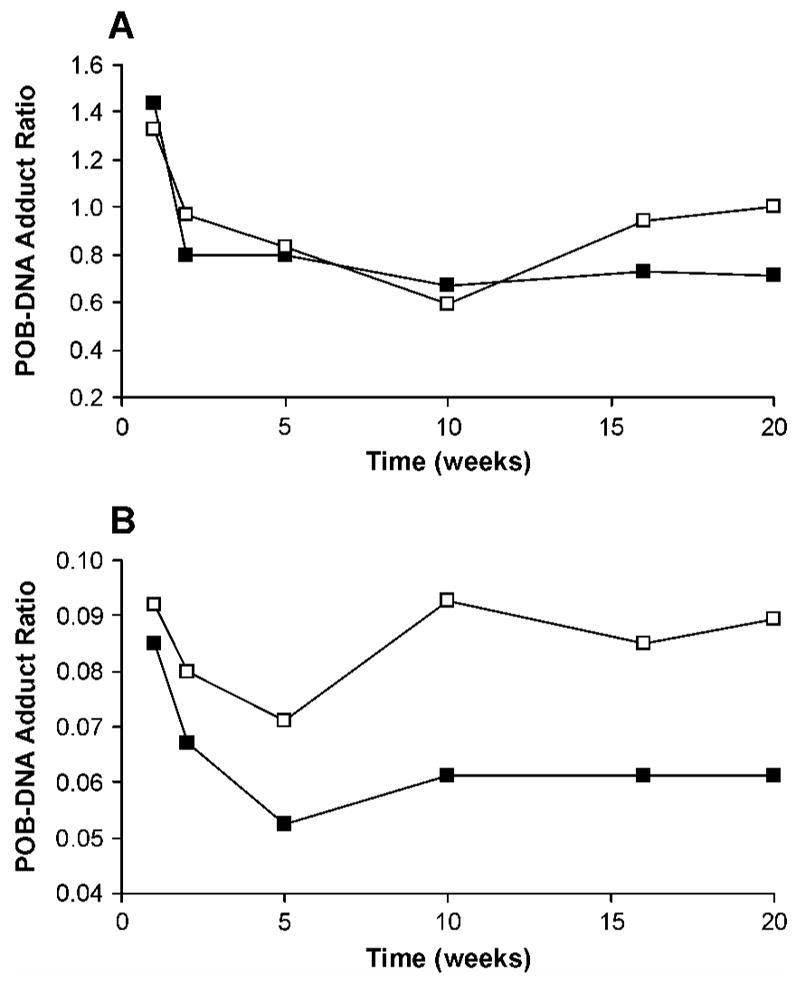
Plots of total POB-DNA adduct ratios of (A) (S)-NNAL to NNK and (B) (R)-NNAL to NNK versus time (weeks). The ratio is calculated by dividing total adduct levels from (S)-NNAL or (R)-NNAL by those from NNK. Symbol designations are: □, lung; ■. liver.
Discussion
DNA pyridyloxobutylation was previously demonstrated in NNAL-treated rats by quantitation of HPB-releasing DNA adducts, suggesting that the carcinogenicity of NNAL was due in part to reconversion to NNK (23). Numerous in vitro and in vivo metabolism studies confirmed the reoxidation of NNAL to NNK (18–21, 23, 29–30). Compared to (R)-NNAL, (S)-NNAL appeared to preferentially persist in lung and was more extensively converted to NNK in rats (20, 21). Since POB-DNA adducts are entirely generated from metabolic activation of NNK, not from NNAL, the formation of those adducts was expected to be greater from (S)-NNAL treatment than from (R)-NNAL treatment. In the current study, the formation of POB-DNA adducts was demonstrated for the first time in liver and lung DNA isolated from (R)-NNAL and (S)-NNAL-treated rats. Consistent with our hypothesis, levels of POB-DNA adducts formed in rats treated with (S)-NNAL were not significantly different from those from NNK treatment, and both were significantly higher than those from (R)-NNAL treatment (P < 0.05) (Figure 4A, B). The ratios of total adduct levels from (R)-NNAL to those from NNK ((R)-NNAL/NNK) were relatively low during the period of treatment, 0.05–0.08 in liver and 0.07–0.09 in lung, suggesting that less than 9% of (R)-NNAL was converted to NNK in vivo (Figure 7B). The (S)-NNAL/NNK DNA adduct ratio in our study was 0.6–1.4 (Figure 7A). After one-week of treatment, (S)-NNAL gave 1.3–1.4 times more adducts than NNK. Since conversion of NNAL to NNK is not as efficient as reduction of NNK to NNAL, and the conversion efficiency could not be greater than 100%, the results indicate that (S)-NNAL must be sequestered in tissues, and gradually released and converted to NNK locally. During the first week, the local concentrations of NNK, converted from sequestered (S)-NNAL, were apparently higher than those arising from NNK treatment. The ratios decreased over time reaching a minimum of 0.6 or 0.7, and then remained steady at 1.0 in lung and 0.7 in liver to the end of the experiment, suggesting that (S)-NNAL was preferentially retained in lung compared to liver. These results are consistent with those of previous studies, in which (S)-NNAL was found to be stereoselectively retained in rat tissues, preferentially in lung (20, 21). The levels of total adducts as well as individual adducts from (R)-NNAL treatment reached a maximum at 10 weeks. However, (S)-NNAL adducts reached a maximum at 16 weeks (Figure 4A, B). The delay may also be due to tissue retention of (S)-NNAL and gradual release of (S)-NNAL from binding sites. Collectively, the results of this study are consistent with a scenario in which (S)-NNAL accumulates in tissues, primarily in the lung, by binding to unknown receptors, then is gradually released from binding sites and converted to NNK, followed by further bioactivation of NNK to produce POB-DNA adducts. We propose that this mechanism is critical for the lung carcinogenicity of NNK and NNAL in rodents.
In a previous study, we determined the formation of individual POB-DNA adducts in liver and lung DNA isolated from rats treated with relatively high doses of NNK, 5.2 or 20.7 mg/kg/day for 4 days by s.c. injection (15). Total adduct levels were 4–6 times higher in liver than in lung. The results were fully consistent with previous high-dose studies which measured released HPB as an indication of POB-DNA adduct formation (6, 31–32). In the current study, the rats were chronically treated with lower doses of NNK. The dose of 10 ppm in the drinking water was comparable to 470–1200 μg/kg/day, considering the daily water consumption and body weights. Total adduct levels were 1.6–1.9 times higher in lung than in liver. Fully consistent with NNK treatment, both (R)-NNAL and (S)-NNAL administration gave more POB-DNA adducts in lung than liver. The results agree with previous studies, in which HPB-releasing DNA adducts were measured at low NNK doses. Murphy et al. found that the levels of HPB released from acid hydrolysis of lung DNA were greater than from liver DNA in rats treated by i. p. administration of 15–150 μg NNK/kg/day for four days (31). Similar results were obtained by Boysen et al in rats treated with 2 ppm of NNK in the drinking water for 8 and 16 weeks (9).
In calf thymus DNA modified with 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone (NNKOAc, 3), a chemically activated form of NNK, levels of 7-POB-Gua were the highest among four POB-DNA adducts, indicating the highest reactivity of Gua in DNA toward pyridyloxobutylating agent 9 in vitro (15). In the current study, O2-POB-dThd was the most abundant adduct in liver and lung of rats treated with NNK at each time point (Figure 3A, B). The differences were significant (P < 0.01) except that the level of O2-POB-dThd was not significantly different from that of 7-POB-Gua in liver at the first week of treatment. The percentages of O2-POB-dThd of total adducts (49.6–70.0% in liver and 51.1–73.4% in lung DNA) increased over time, while the percentages of 7-POB-Gua decreased (18.7–39.1% in liver DNA and 15.0–35.5% in lung DNA). The difference may be due to the different efficiency of in vivo cellular repair systems for O2-POB-dThd and 7-POB-dGuo. It appears that O2-POB-dThd was poorly repaired in vivo and accumulated over time, while 7-POB-dGuo was relatively efficiently removed. Furthermore, 7-POB-dGuo could also be partially lost due to spontaneous loss of HPB or depurination under physiological conditions or during DNA isolation. Levels of 7-POB-Gua were previously shown to be similar to those of O2-POB-dThd in the high dose study, which may result from the different dose regimen used (15). The short-term treatment and higher dose could result in the saturation of repair of 7-POB-dGuo resulting in its accumulation. These results support our previous conclusion that O2-POB-dThd could be a useful biomarker for tobacco carcinogenesis studies (15).
Although the miscoding properties of O2-POB-dThd are unknown, its analog, O2-ethyl-dThd, was shown to be mutagenic, causing T to A tranversions (33, 34). The highly abundant O2-POB-dThd may be important in NNK carcinogenicity and mutagenicity. An in vitro miscoding study of O2-POB-dThd would be helpful to address this question.
O6-POB-dGuo was detected only in lung. Its levels were 24–250 times lower than those of O2-POB-dThd, indicating efficient repair of O6-POB-dGuo in vivo. Levels of O6-POB-dGuo in lung were steady for the first 10 weeks, then declined slightly, indicating that the concentrations of O6-POB-dGuo reached a steady state between formation and repair in lung. The results agree with our previous study, in which O6-POB-dGuo was shown to be efficiently repaired in rat liver and lung. These results demonstrate the persistence of the mutagenic O6-POB-dGuo in rat lung and support the hypothesis that O6-POB-dGuo is important for NNK lung carcinogenesis in rats (15). In the rats treated with (R)- and (S)-NNAL, the relative adduct levels shared the same trends as in those treated with NNK: O2-POB-dThd > 7-POB-Gua > O2-POB-Cyt > O6-POB-dGuo, consistent with POB-DNA adduct formation via conversion of (R)- or (S)-NNAL to NNK.
In both liver and lung, the maximal levels of total POB-DNA adducts were found at 10 weeks for NNK and (R)-NNAL and at 16 weeks for (S)-NNAL, then decreased slowly (Figure 4A, B). During the 20-week experiment, the body weight of rats increased, while water consumptions decreased slightly. Less carcinogen intake per gram body weight of rats could slightly contribute to the decreased adduct levels at later time points, but it is not the main reason because similar results were found in a previous study, in which rats were given s.c. injections of NNK (in 0.9% saline) at a dose of 1.76 mg/kg three times a week for 4, 8, 12, 16 or 20 weeks (6). POB-DNA adducts in lung, as quantified by HPB released from DNA, doubled from 4 to 12 weeks, then declined from 12 to 20 weeks. The results of this study were also similar to those from a previous study of HPB-releasing hemoglobin adducts resulting from 2 ppm NNK administration to rats in drinking water in a 111-week course of treatment (35, 36). The maximum was reached at 16 weeks, and then adduct levels decreased during the remaining course of the experiment. A similar trend was also observed in another previous study of HPB-releasing hemoglobin adducts, in which rats were treated with 2 ppm of NNK in the drinking water for 16 weeks. The maximal amount of HPB-releasing hemoglobin adducts was found at 12 weeks and then the levels decreased between 12 and 16 weeks (9). These adduct level patterns could result from the effects of chronic NNK treatment on enzymes involved in NNK metabolic activation and detoxification (29).
α-Methylene hydroxylation of NNK and NNAL produces the same methyl-DNA adducts (Scheme 1) (1). A number of studies demonstrated that DNA methylation is important for NNK tumorigenesis in A/J mouse lung (37, 38). A study with deuterium-substituted NNK and NNAL analogues demonstrated that DNA methylation is necessary for lung tumorigenicity (37). In another study, a strong correlation between lung tumor yields and levels of O6-methylGua at 96 h after various doses of NNK indicated that O6-methylGua is a critical determinant for NNK lung tumorigenicity in A/J mice (38). Mutagenic O6-methylGua may also contribute to NNK tumorigenicity in rats since it accumulated and persisted in NNK-treated rats (7, 39). Besides reconversion of NNAL to NNK, direct α-methyl hydroxylation of NNAL, which produces PHB-DNA adducts, may also be an important mechanism for NNK and NNAL carcinogenesis in rodents. Determination of levels of methyl-DNA adducts and PHB-DNA adducts in rat liver and lung DNA used in this study would provide useful information about the relative contributions of various bioactivation pathways to NNK and NNAL carcinogenesis in rats. These studies are currently are underway in our laboratory.
In summary, chronic treatment of rats with NNK, (R)-NNAL or (S)-NNAL at low doses gave higher levels of POB-DNA adducts in lung than in liver. O2-POB-dThd was the major POB-DNA adduct found in vivo and accumulated over the course of treatment. The highly abundant O2-POB-dThd may be important for NNK and NNAL carcinogenicity. O6-POB-dGuo was found to persist in lung, supporting its important role in NNK and NNAL lung carcinogenesis in rats. This is the first study to examine the formation of NNK-derived POB-DNA adducts in (R)- and (S)-NNAL-treated rats. (S)-NNAL generated comparable levels of adducts as NNK, significantly higher than (R)-NNAL. The results support the hypothesis that preferential retention of (S)-NNAL in the lung, followed by reconversion to NNK, then metabolic activation of NNK, is critical for lung carcinogenesis by NNK and NNAL in rodents.
Supplementary Material
Supporting Information Available: Levels of POB-DNA adducts in liver and lung DNA of NNK, (R)-NNAL or (S)-NNAL-treated rats, expressed as fmol per mg DNA; levels of POB-DNA adducts in rats treated with NNK, (R)-NNAL or (S)-NNAL after 20 weeks of experiment, expressed as fmol per μmol appropriate nucleoside; statistical comparison of levels of each individual POB-DNA adduct in liver and lung of rats treated with NNK, (R)-NNAL or (S)-NNAL. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The authors acknowledge Bruce Lindgren of the University of Minnesota Cancer Center Biostatistics Core Facility for statistical analysis. Mass spectrometry was carried out in the Analytical Biochemistry Core Facility. The core facilities are supported in part by grant CA-77598 from the National Cancer Institute. We thank Steven Carmella and Adam R. Benoit for their technical assistance on CSP-GC-MS analysis of (R)- and (S)-NNAL, Dr. David Lahti for his help in animal experiments and DNA isolation, and Bob Carlson for manuscript preparation and submission. Stephen S. Hecht is an American Cancer Society Research Professor, supported in part by ACS grant RP-00-138. This work was supported by Grant CA-81301 from the National Cancer Institute.
Abbreviations
- ANOVA
analysis of variance
- HPB
4-hydroxy-1-(3-pyridyl)-1-butanone
- HPLC-ESI-MS/MS
high performance liquid chromatography-electrospray ionization-tandem mass spectrometry
- 11β-HSD1
11β-hydroxysteroid dehydrogenase type 1
- NNK
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
- NNAL
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol
- NNN
N′-nitrosonornicotine; O2-POB-dCyd, O2-[4-(3-pyridyl)-4-oxobut-1-yl]-2′-deoxycytidine; O2-POB-Cyt, O2-[4-(3-pyridyl)-4-oxobut-1-yl]cytosine
- O2-POB-dThd
O2-[4-(3-pyridyl)-4-oxobut-1-yl]thymidine
- O6-POB-dGuo
O6-[4-(3-pyridyl)-4-oxobut-1-yl]-2′-deoxyguanosine; 7-POB-dGuo, 7-[4-(3-pyridyl)-4-oxobut-1-yl]-2′-deoxyguanosine; 7-POB-Gua, 7-[4-(3-pyridyl)-4-oxobut-1-yl]guanine
- PHB
4-(3-pyridyl)-4-hydroxybut-1-yl
- POB
4-(3-pyridyl)-4-oxobut-1-yl
- REML
restricted maximum likelihood
- SRM
selected reaction monitoring
References
- 1.Hecht SS. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem Res Toxicol. 1998;11:560–603. doi: 10.1021/tx980005y. [DOI] [PubMed] [Google Scholar]
- 2.Rivenson A, Hoffmann D, Prokopczyk B, Amin S, Hecht SS. Induction of lung and exocrine pancreas tumors in F344 rats by tobacco-specific and areca-derived N-nitrosamines. Cancer Res. 1988;48:6912–6917. [PubMed] [Google Scholar]
- 3.International Agency on Research for Cancer. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Vol. 89. IARC; Lyon, France: 2006. Smokeless tobacco and tobacco-specific nitrosamines. in press. [Google Scholar]
- 4.Dragan YP. Somatic mutation theory of carcinogenesis. In: Bowden GT, Fisher SM, editors. Comprehensive Toxicology. Vol. 12. Elsevier Science; New York: 1997. pp. 31–53. [Google Scholar]
- 5.Trushin N, Rivenson A, Hecht SS. Evidence supporting the role of DNA pyridyloxobutylation in rat nasal carcinogenesis by tobacco-specific nitrosamines. Cancer Res. 1994;54:1205–1211. [PubMed] [Google Scholar]
- 6.Morse MA, Wang CX, Stoner GD, Mandal S, Conran PB, Amin SG, Hecht SS, Chung FL. Inhibition of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced DNA adduct formation and tumorigenicity in lung of F344 rats by dietary phenethyl isothiocyanate. Cancer Res. 1989;49:549–553. [PubMed] [Google Scholar]
- 7.Staretz ME, Foiles PG, Miglietta LM, Hecht SS. Evidence for an important role of DNA pyridyloxobutylation in rat lung carcinogenesis by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone: effects of dose and phenethyl isothiocyanate. Cancer Res. 1997;57:259–266. [PubMed] [Google Scholar]
- 8.Sticha KRK, Kenney PMJ, Boysen G, Liang H, Su X, Wang M, Upadhyaya P, Hecht SS. Effects of benzyl isothiocyanate and phenethyl isothiocyanate on DNA adduct formation by a mixture of benzo[a]pyrene and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in A/J mouse lung. Carcinogenesis. 2002;23:1433–1439. doi: 10.1093/carcin/23.9.1433. [DOI] [PubMed] [Google Scholar]
- 9.Boysen G, Kenney PMJ, Upadhyaya P, Wang M, Hecht SS. Effects of benzyl isothiocyanate and 2-phenethyl isothiocyanate on benzo[a]pyrene and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone metabolism in F-344 rats. Carcinogenesis. 2003;24:517–525. doi: 10.1093/carcin/24.3.517. [DOI] [PubMed] [Google Scholar]
- 10.Foiles PG, Akerkar SA, Carmella SG, Kagan M, Stoner GD, Resau JH, Hecht SS. Mass spectrometric analysis of tobacco-specific nitrosamine-DNA adducts in smokers and nonsmokers. Chem Res Toxicol. 1991;4:364–368. doi: 10.1021/tx00021a017. [DOI] [PubMed] [Google Scholar]
- 11.Schlöbe D, Hölze D, Richter E, Tricker AR. Determination of tobacco-specific nitrosamine hemoglobin and lung DNA adducts. Proc Am Assoc Canc Res. 2002;43:346. [Google Scholar]
- 12.Wang L, Spratt TE, Liu XK, Hecht SS, Pegg AE, Peterson LA. Pyridyloxobutyl adduct O6-[4-oxo-4-(3-pyridyl)butyl]guanine is present in 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone-treated DNA and is a substrate for O6-alkylhuanine-DNA alkyltransferase. Chem Res Toxicol. 1997;10:562–567. doi: 10.1021/tx9602067. [DOI] [PubMed] [Google Scholar]
- 13.Wang M, Cheng G, Sturla SJ, Shi Y, McIntee EJ, Villalta PW, Upadhyaya P, Hecht SS. Identification of adducts formed by pyridyloxobutylation of deoxyguanosine and DNA by 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone, a chemically activated form of tobacco specific carcinogens. Chem Res Toxicol. 2003;16:616–626. doi: 10.1021/tx034003b. [DOI] [PubMed] [Google Scholar]
- 14.Hecht SS, Villalta PW, Sturla SJ, Cheng G, Yu N, Upadhyaya P, Wang M. Identification of O2-substituted pyrimidine adducts formed in reactions of 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone and 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanol with DNA. Chem Res Toxicol. 2004;17:588–597. doi: 10.1021/tx034263t. [DOI] [PubMed] [Google Scholar]
- 15.Lao Y, Villalta PW, Sturla SJ, Wang M, Hecht SS. Quantitation of pyridyloxobutyl DNA adducts of tobacco-specific nitrosamines in rat tissue DNA by high performance liquid chromatography-electrospray ionization-tandem mass spectrometry. Chem Res Toxicol. 2006;19:674–682. doi: 10.1021/tx050351x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Breyer-Pfaff U, Martin HJ, Ernst M, Maser E. Enantioselectivity of carbonyl reduction of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone by tissue fractions from human and rat and by enzymes isolated from human liver. Drug Metab Dispos. 2004;32:915–922. [PubMed] [Google Scholar]
- 17.Upadhyaya P, Sturla SJ, Tretyakova N, Ziegel R, Villalta PW, Wang M, Hecht SS. Identification of adducts produced by the reaction of 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanol with deoxyguanosine and DNA. Chem Res Toxicol. 2003;16:180–190. doi: 10.1021/tx0256376. [DOI] [PubMed] [Google Scholar]
- 18.Upadhyaya P, Carmella SG, Guengerich FP, Hecht SS. Formation and metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol enantiomers in vitro in mouse, rat and human tissues. Carcinogenesis. 2000;21:1233–1238. [PubMed] [Google Scholar]
- 19.Upadhyaya P, Kenney PMJ, Hochalter JB, Wang M, Hecht SS. Tumorigenicity and metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) enantiomers and metabolites in the A/J mouse. Carcinogenesis. 1999;20:1577–1582. doi: 10.1093/carcin/20.8.1577. [DOI] [PubMed] [Google Scholar]
- 20.Wu Z, Upadhyaya P, Carmella SG, Hecht SS, Zimmerman CL. Disposition of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) in bile duct-cannulated rats: stereoselective metabolism and tissue distribution. Carcinogenesis. 2002;23:171–179. doi: 10.1093/carcin/23.1.171. [DOI] [PubMed] [Google Scholar]
- 21.Zimmerman CL, Wu Z, Upadhyaya P, Hecht SS. Stereoselective metabolism and tissue retention in rats of the individual enantiomers of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL), metabolites of the tobacco-specific nitrosamine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) Carcinogenesis. 2004;27:1237–1242. doi: 10.1093/carcin/bgh120. [DOI] [PubMed] [Google Scholar]
- 22.Hecht SS, Carmella SG, Ye M, Le K, Jensen JA, Zimmerman CL, Hatsukami DK. Quantitation of metabolites of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone after cessation of smokeless tobacco use. Cancer Res. 2002;62:129–134. [PubMed] [Google Scholar]
- 23.Hecht SS, Trushin N. DNA and hemoglobin alkylation by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and its major metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in F344 rats. Carcinogenesis. 1988;9:1665–1668. doi: 10.1093/carcin/9.9.1665. [DOI] [PubMed] [Google Scholar]
- 24.Hecht SS, Spratt TE, Trushin N. Absolute configuration of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol formed metabolically from 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis. 1997;18:1851–1854. doi: 10.1093/carcin/18.9.1851. [DOI] [PubMed] [Google Scholar]
- 25.Carmella SG, Ye M, Upadhyaya P, Hecht SS. Stereochemistry of metabolites of a tobacco-specific lung carcinogen in smokers’ urine. Cancer Res. 1999;59:3602–3605. [PubMed] [Google Scholar]
- 26.Sturla SJ, Scott J, Lao Y, Hecht SS, Villalta PW. Mass spectrometric analysis of relative levels of pyridyloxobutylation adducts formed in the reaction of DNA with a chemically activated form of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Chem Res Toxicol. 2005;18:1048–1055. doi: 10.1021/tx050028u. [DOI] [PubMed] [Google Scholar]
- 27.Wang M, Yu N, Chen L, Villalta PW, Hochalter JB, Hecht SS. Identification of an acetaldehyde adduct in human liver DNA and quantitation as N2-ethyldeoxyguanosine. Chem Res Toxicol. 2006;19:319–324. doi: 10.1021/tx0502948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gupta RC. Enhanced sensitivity of 32P-postlabeling analysis of aromatic carcinogen:DNA adducts. Cancer Res. 1985;45:5656–5662. [PubMed] [Google Scholar]
- 29.Staretz ME, Koenig LA, Hecht SS. Effects of long term dietary phenethyl isothiocyanate on the microsomal metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in F344 rats. Carcinogenesis. 1997;18:1715–1722. doi: 10.1093/carcin/18.9.1715. [DOI] [PubMed] [Google Scholar]
- 30.Staretz ME, Murphy SE, Patten CJ, Nunes MG, Koehl W, Amin S, Koenig LA, Guengerich FP, Hecht SS. Comparative metabolism of the tobacco-related carcinogens benzo[a]pyrene, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol, and N′-nitrosonornicotine in human hepatic microsomes. Drug Metab Dispos. 1997;25:154–162. [PubMed] [Google Scholar]
- 31.Murphy SE, Palomino A, Hecht SS, Hoffmann D. Dose-response study of DNA and hemoglobin adduct formation by -(methylnitrosamino)-1-(3-pyridyl)-1-butanone in F344 rats. Cancer Res. 1990;50:5446–5452. [PubMed] [Google Scholar]
- 32.Peterson LA, Mathew R, Murphy SE, Trushin N, Hecht SS. In vivo and in vitro persistence of pyridyloxobutyl DNA adducts from 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis. 1991;12:2069–2072. doi: 10.1093/carcin/12.11.2069. [DOI] [PubMed] [Google Scholar]
- 33.Grevatt PC, Solomon JJ, Bhanot OS. In vitro mispairing specificity of O2-ethylthymidine. Biochemistry. 1992;31:4181–4188. doi: 10.1021/bi00132a005. [DOI] [PubMed] [Google Scholar]
- 34.Bhanot OS, Grevatt PC, Donahue JM, Gabrielides C, Solomon JJ. In vitro DNA replication implicates O2-ethyldeoxythymidine in transversion mutagenesis by ethylating agents. Nucleic Acids Res. 1992;20:587–594. doi: 10.1093/nar/20.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hecht SS, Trushin N, Rigotty J, Carmella SG, Borukhova A, Akerkar S, Desai D, Amin S, Rivenson A. Inhibitory effects of 6-phenylhexyl isothiocyanate on 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone metabolic activation and lung tumorigenesis in rats. Carcinogenesis. 1996;17:2061–2067. doi: 10.1093/carcin/17.9.2061. [DOI] [PubMed] [Google Scholar]
- 36.Hecht SS, Trushin N, Rigotty J, Carmella SG, Borukhova A, Akerkar S, Rivenson A. Complete inhibition of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced rat lung tumorigenesis and favorable modification of biomarkers by phenethyl isothiocyanate. Cancer Epidemiol Biomarkers Prev. 1996;5:645–652. [PubMed] [Google Scholar]
- 37.Hecht SS, Jordan KG, Choi CI, Trushin N. Effects of deuterium substitution on the tumorigenicity of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in A/J mice. Carcinogenesis. 1990;11:1017–1020. doi: 10.1093/carcin/11.6.1017. [DOI] [PubMed] [Google Scholar]
- 38.Peterson LA, Hecht SS. O6-Methylguanine is a critical determinant of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone tumorigenesis in A/J mouse lung. Cancer Res. 1991;51:5557–5564. [PubMed] [Google Scholar]
- 39.Belinsky SA, White CM, Boucheron JA, Richardson FC, Swenberg JA, Anderson M. Accumulation and persistence of DNA adducts in respiratory tissues of rats following multiple administrations of the tobacco specific carcinogen 4-(N-methyl-N-nitrosamino)-1-(3-pyridyl)-1-butanone. Cancer Res. 1986;46:1280–1284. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: Levels of POB-DNA adducts in liver and lung DNA of NNK, (R)-NNAL or (S)-NNAL-treated rats, expressed as fmol per mg DNA; levels of POB-DNA adducts in rats treated with NNK, (R)-NNAL or (S)-NNAL after 20 weeks of experiment, expressed as fmol per μmol appropriate nucleoside; statistical comparison of levels of each individual POB-DNA adduct in liver and lung of rats treated with NNK, (R)-NNAL or (S)-NNAL. This material is available free of charge via the Internet at http://pubs.acs.org.