

Abstract
We have identified three new human tumor necrosis factor-α (TNF-α) promoter polymorphisms with single nucleotide (nt) substitutions at −862, −856, and −574 nt relative to the TNF-α transcription start site. The −862 and −856 nt TNF-α promoter polymorphisms occur with high frequency in Caucasian and Cambodian individuals and are each non-randomly associated with three extended HLA haplotypes. This study, in which 61 independent TNF-α promoters were analyzed spanning from −977 to +93 nt relative to the TNF-α mRNA cap site, establishes a new canonical TNF-α promoter sequence. Furthermore, we show that none of the three novel polymorphisms at −862, −856 and −574 nt or polymorphisms previously described at positions −238. −308 and +70 have an effect upon TNF-α gene expression in activated lymphocytes. Thus, these TNF-α promoter polymorphisms likely serve as markers for neighboring genes encoding HLA or other undefined molecules in the MHC that may influence disease susceptibility.
Keywords: Cambodians, Caucasians, disease susceptibility, evolution, HLA, MHC, TNF-α gene regulation, TNF-α promoter polymorphisms
The cytokine tumor necrosis factor (TNF)-α plays a critical role in lymphocyte biology as well as in a variety of infectious and autoimmune diseases (reviewed in (1)). In the case of certain infectious diseases, too much or too little TNF-α is associated with variable patterns of disease pathogenesis (for example, see (2–4); reviewed in (5)). Thus, the identification of polymorphisms that influence TNF-α gene expression and protein production could provide insight into patterns of disease pathogenesis associated with different amounts of TNF-α gene expression and subsequent protein production.
To date, five TNF-α promoter polymorphisms, located at −238, −244, −308, −376, and +70 nucleotides (nt) relative to the TNF-α transcription start site, have been identified (6–11). The −238, −244, −308, and −376 nt polymorphisms represent base variants at the respective positions (6–9, 11), whereas the +70 polymorphism is an added nucleotide (10). None of these polymorphisms occur within TNF-α promoter regions previously shown to be involved in regulation of the gene (12–16).
We sequenced TNF-α promoters from a total of 61 different chromosomes spanning from −977 to +93 nt relative to the TNF-α transcription start site and found three novel polymorphisms located at −574, −856, and −862 nt. We could not detect any effect of the three novel TNF-α polymorphisms identified in this study or of the previously identified −308, −238, and +70 polymorphisms upon transcriptional regulation of the TNF-α gene in activated T or B cells. However, the two high frequency polymorphisms located at −856 and −862 are each associated with three extended HLA haplotypes consistent with the high degree of linkage disequilibrium within the human major histocompatibility complex (MHC) (see (17) for review).
Material and methods
Determination of TNF-α promoter polymorphism
Genomic DNA was obtained from bone marrow biopsies from Caucasian patients in remission from multiple myeloma (MM), non-Hodgkin’s lymphoma (NHL) as previously described (18), or from peripheral blood mononuclear cells derived from normal Cambodian individuals from Svay Rieng, Cambodia (19), and from normal Caucasian individuals from Boston, Massachusetts, by a quick isolation method (2%).
Polymerase chain reaction (PCR) was performed using primers to amplify the sequences between −977 and +93 nt relative to the TNF-α transcription start site using a primer at −977: 5′ CCG GAT CCT ATG GCC ACA TGT AGC GGCT 3′ with an artificial Bam-H1 end, and a primer at +93: 5′ GCT CTA GAC GTC TGA GGG TTG TTT TCAG 3′ with an artificial Xba-I end Samples were first denatured for 5 min at 95°C, followed by 35 cycles of a 45 s denaturing step at 92°C, a 45 s annealing step at 68°C, and a 45 s extension step at 74°C. A final extension step was performed for 10 min at 72°C. The PCR products were separated by agarose gel electrophoresis, excised from the ethidium bromide-stained gel, and purified using a Qiagen gel extraction kit. A negative control (minus DNA) was included in each PCR reaction. The fragments were then either sequenced directly using an automated sequencer (attaining readable sequence between −950 and +70 nt relative to the TNF-α transcription start site) or digested with Bam-HI and Xba-I and subcloned into the Bam-HI and Xba-I sites of the POCAT reporter plasmid as previously described (14) and sequenced by the dideoxy method (determining sequences between −977 and +93 nt). Each mutation was verified by an independent PCR reaction from the original sample and subsequent sequencing. We PCR amplified a total of 61 TNF-α promoter fragments derived from 31 unrelated Caucasians and from 10 unrelated Cambodians, After PCR amplification, both DNA strands were sequenced in 12 of the Caucasian and in all 10 of the Cambodian individuals. Of the remaining 19 Caucasian individuals, a TNF-α promoter fragment from a single DNA strand was cloned and sequenced.
Screening for −308, −856, and −862 variants in B-cell malignancy samples
DNA was extracted from samples from the Dana-Farber Cancer Institute bone marrow bank from individuals with established diagnoses of MM, NHL, chronic lymphocytic leukemia (CLL), and acute lymphoblastic leukemis (ALL) as previously described (18, 20) and was screened for the −308, −862, and −856 TNF-α promoter variants. The −308 TNF-α variants were determined by restriction fragment length polymorphism analysis using primers designed to incorporate the polymorphic site (the nucleotide A versus G) at position −307 nt relative to the TNF-α transcription start site. The −308 variant (A at −308 nt) creates a Nco-I restriction site and can be differentiated by size (107 nt for the non-variant form and 87 nt for the variant −308 form) by agarose gel electrophoresis (6). The −856 and −862 TNF-α promoter variants were determined by direct sequencing of a PCR product of a TNF-α promoter fragment between −945 and −760 nt. The primers used were: −977: 5′ CCG GAT CCT ATG GCC ATG GCC ATG TAG CCC CT 3′, and −739: 5′ TGA AGC TCT CAC TTC TCA GG 3′. The PCR conditions and the processing of the PCR fragments were as described above.
Screening homozygous B-cell lines for the −856 and −862 alleles
Homozygous cell lines from the Tenth International Histocompatability Workshop (PF97387, L0541265, DBB, HHKB, BM9, WT49, WT51, KT12, MADURA, YAR, WDV, SAVC, DKB, SL005, SPACH, BOLETH, MT14B, EJ32B, JO528239, EHM, MOU, PITOUT L0081785, BSM, DUCAF, DEU, WIN, MZ070782, TEM, HID, HOR, JHAF, E4181324) (21, 22) were screened for the −856 and −862 TNF-α promoter polymorphisms by PCR-single strand oligonucleotide polymorphism (SSOP) methodology (23). PCR amplification of the −945 to −760 region of the TNF-α promoter was performed as described above. Gel electrophoresis was carried out with a 5 μl aliquot of the 50 μl PCR-amplified product to confirm amplification. Replicate membranes were prepared by direct spotting of the PCR product onto Hybond N+ membranes (Amersham, Arlington Heights, Illinois). Membranes were allowed to dry at room temperature for 30 min followed by denaturing in 0.4 M NaOH for 5 min neutralizing in 5× standard saline citrate (SSC) for 5 min and by crosslinking by UV light as previously described (23, 24).
Prehybridization was carried out for 1 h followed by hybridization overnight at 42°C in a buffer containing 5× SSC, 0.1% sodium lauryl sarcosinate, 0.02% sodium dodecyl sulfate (SDS) and 1% slocking reagent (Boehringer Mannheim, Indianapolis, Indiana), employing a non-radioactive method. Detection of the TNF-α promoter polymorphisms was performed using four probes (two for −856 and two for −862), which create a single base pair mismatch. The sequences of each pair of the probes are: −856: 5′ CTTAA(C/T)GAAGACAGGCCAT 3′; −862: 5′ GTATGGGGACCCCC(C/A)CTTAA 3′. The probes were labeled at the 3′ end with digoxigenin (Genius 6 Kit, Boehringer Mannheim). After hybridization, the niernbranes were washed twice with 2× SSC at room temperature for 10 min, with tetramethylammonium chloride (TMAC) buffer (50 mM Tris-HCL, 3 M TMAC, 2 mM EDTA, 0.1% SDS) and then washed twice with TMAC buffer for 15 min at 61°C for the −862 variant and at 55°C for the −856 variant. Allele assignment was established using the chemiluminescent detection method.
Cell culture, activation, and transfection
The murine B-cell lymphoma cell line A20 and the antigen-specific murine T-cell clone Ar-5 were grown and transfected as previously described (14, 16), Twenty-four hours after transfection, cells were activated with phorbol myristate acetate (PMA) (Calbiochem, 200 nM) plus ionomycin (1 μM), harvested approximately 18 h later and chloramphenicol acetyltransferase (CAT) assays were performed as previously described (8, 10). Human Raji B cells were transfected by the lipofectamine method, stimulated approximately 18 h later with PMA (Calbiochem, 200 nM) and CAT assays were performed as previously described (13).
Plasmids
We cloned the wild type (WT) and polymorphic TNF-α promoters (−862, −856/+70, −574, −307), all spanning from −977 to +93 nt relative to the TNF-α mRNA cap site, into the Bam HI and Xba I sites of POCAT as previously described (14). We PCR amplified the TNF-α promoter bearing the −237 variant from the homozygous cell line DUCAF. All constructs were confirmed by dideoxy sequencing and are isogenic except for the allelic variant bases noted.
Results
Identification of novel TNF-α promoter polymorphisms at −574, −854, and −862 nt
We used PCR to amplify 61 TNF-α promoter fragments spanning a region from −977 to +93 nt relative to the TNF-α transcription start site derived from unrelated Caucasian and Cambodian individuals. This analysis revealed three previously unidentified promoter polymorphisms at −574 (G to A), −856 (C to T), and −862 (C to A) nt relative to the TNF-α transcription start site (Fig. 1).
Fig. 1. The human TNF-α gene promoter from−977 to +93 nt relative to the TNF-α transcription start site.

The three new polymorphisms detected at −862, −856, and −574 nt; the previously detected Polymorphisms, marked by an asterisk (*), at −237, −243, −307, and −375 nt; and the +70 polymorphic addition are noted in bold. The more uncommon polymorphic allele is shown below. The start site of transcription is at +1. Regulatory motifs in the human TNF-α promoter which have been shown to have a functional role in TNF-α gene regulation (12–16) are underlined.
The −862 and −856 variants were detected respectively in 11 and six of the 43 TNF-α promoters derived from Caucasians, and the −862 and −856 variants were found respectively in six and two of the 20 TNF-α promoters derived from Cambodians. One Caucasian individual bearing the −856 polymorphism also carried the +70 nt variant. In contrast, the new −574 polymorphism was observed in only one of the 61 TNF-α promoters, derived from a Caucasian individual. Of the 61 TNF-α promoters examined, we did not identify any individuals bearing the −238, −244, or −375 alleles. The −308 nt variant was found in 12 of the 61 promoters (nine Caucasians and three Cambodians), consistent with its prevalence in other sample sets tested (9, 25).
In Genbank, there are nine independent human TNF-α promoter sequences which variably span from −1056 to +127 relative to the TNF-α transcription start site and, when these sequences are compared, there are variations in each of the reported sequences. Our analysis of 61 TNF-α promoters derived from two distinct populations produced a unique consensus TNF-α promoter sequence. We have determined the most frequent allelic form of each promoter residue spanning from −977 to +93 relative to the TNF-α mRNA cap site (Fig. 1). This data revealed that the previously reported −238, −244, −308, and −376 TNF-α promoter polymorphisms actually occur at positions −237, −243, −307, and −375 respectively when counting 5′ from the transcription start site (the ‘A’ noted at position +1 (Fig. 1}). Throughout the remainder of the paper, these mutations are referred to by their correct position relative to the TNF-α mRNA cap site.
The −862, −856, −574, −307, −237, and +70 polymorphisms do not affect TNF-α transcription
To determine whether the newly identified TNF-α promoter polymorphisms had an effect upon TNF-α gene expression, we cloned a WT TNF-α promoter and TNF-α promoters bearing the −862, −856/+70, and the −574 variants and fused them to the CAT reporter gene (-862M, −856/+70M, −574M). We also constructed TNF-α promoter CAT reporter genes bearing the −307 and −237 polymorphisms to test the effect of the −307 and −237 TNF-α promoter variants upon TNF-α gene expression. These constructs were transfected into the antigen-specific murine T-cell clone Ar-5 or the murine B-cell lymphoma line A20, and either mock stimulated or stimulated the cells with phorbol ester (PMA) and ionomycin. The −862, −856/+70, −574, −307, and −237 TNF-α promoter variants did not affect constitutive or PMA/ionomycin-stimulated TNF-α gene expression in Ar-5 T or A20 B cells (Fig. 2).
Fig. 2. Testing the functional effect of the−862, −856/+70, −574, −307, and −237 polymorphisms upon TNF-α gene expression in A20 B cells (A) and Ar-5 T cells (B).
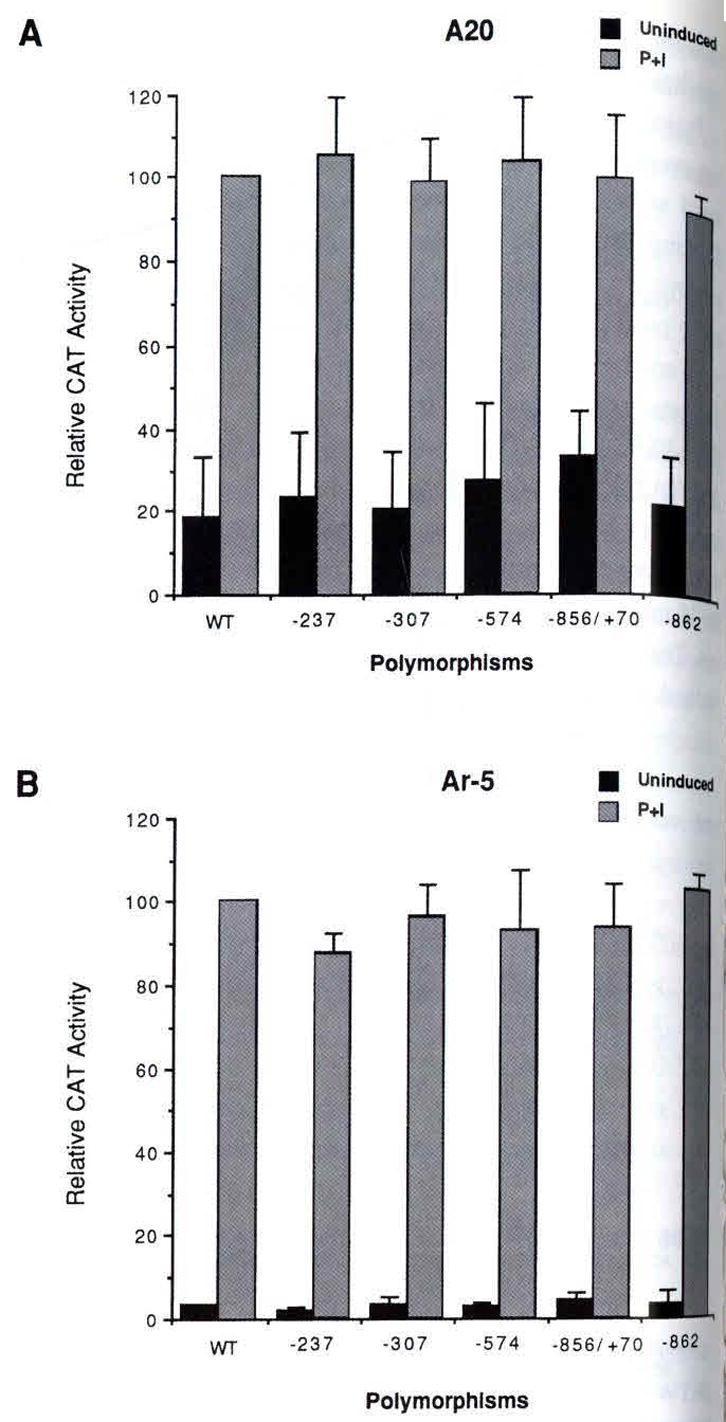
Cells were transfected with the wild type (′977 TNF-α promoter) CAT (WT) and polymorphic TNF-α promoter CAT plasmids tarrying the variants noted in the figure. Twenty-four hours later the cells were mock stimulated (Uninduced) or induced with PMA plus ionomycin (P+I) as descibed in Material and methods. Quantification of the conversion of 14C-chloramphenicol to its acetylated forms was obtained using a Betagen Betascope (Waltham, MA) and in A and B three independent transfections were averaged.
We also tested the effect of the −;307 polymorphism upon TNF-α gene expression in the human Burkitt’s lymphoma cell line, Raji. Similar to results obtained in Ar-5 and A20 cells, we did not detect an effect of the −307 polymorphism, nor did we detect an effect of the −862, −856/+70, and −237 TNF-α promoter variants upon upon constitutive or PMA-inducible TNF-α gene expression in Raji cells (Fig. 3). We conclude that the −862, −856/+70, −574, −307, and - 237 TNF-α promoter polymorphisms do not have a detectable effect upon TNF-α gene expression in the T and B-cell model systems tested.
Fig. 3. Testing the functional effect of the−862, −856/+70, −307, and −237 polymorphisms upon TNF-α gene expression in Raji B cells. An autoradiogram of a CAT assay of Raji cells transfected with the wild type (−977 TNF-α promoter) CAT (WT) and polymorphic TNF-α promoter CAT plasmids carrying the variants noted in the figure.
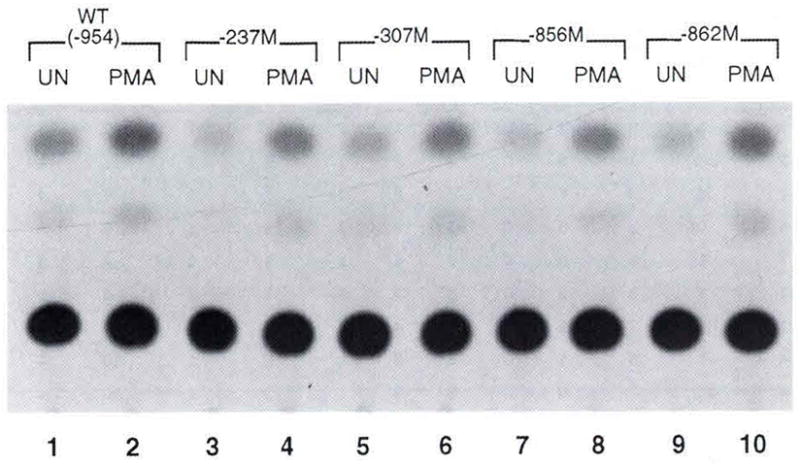
Cells were transfected using lipofectamine as described in the methods section Twenty-four hours later the cells were mock stimulated (UN) or induced with PMA as descibed in Material and methods. The figure shows a representative experiment. Raji cells were co-transfected with a CMV-βgal plasmid (a gift from D. Thanos) and, prior to performing CAT assays, extracts were normalized to β-gal activity.
The −856 and −862 TNF-α promoter variants are associated with extended HLA haplotypes
Markers in the HLA class III region, including complement and HSP 70 alleles, TNF microsatellite markers and the −307 TNF-α promoter variants are in linkage disequilibrium with HLA-DR and HLA-B genes (17, 24, 26). To determine whether the high frequency −862 and −856 TNF-α promoter polymorphisms were in linkage disequilibrium with extended HLA haplotypes, we screened homozygous human B-cell lines of known HLA-DR and HLA-B types (21), TNF microsatellite (24), and complotype markers (27) using a PCR-SSOP method.
As shown in Table 1, this analysis revealed that the −862 and −856 TNF-α promoter variants are in linkage disequilibrium with extended HLA haplotypes (21, 24, 27). The −856 variant is associated with three extended haplotypes, including HLA-DR2, B18 haplotype, which is a marker for C2 deficiency (HLA-DR2, C2*0, C4A*4, BF*F1, C4B*2; see (24)). The −856 variant is also associated with the extended haplotype defined by HLA-DR4. B38, which is associated with pemphigus vulgaris and clozapine-induced agranulocytosis, found in Ashkenazi Jewish populations (24), and includes the TNF microsatellite variant marker TNFa10 (HLA-DR4, C2*C, C4A*2, BF*S, C4B*1, HLA-B38) (24). The extended haplotype associated with 21-hydroxylase deficiency, which is defined by HLA-DR7, B47 (28, 29), is also associated with the −856 variant. The −862 variant is associated with the extended haplotypes HLA-DR1, B14; HLA-DR1, B35; and HLA-DR4. B62 (30).
Table 1.
Association of the −856 and −862 TNF-α promoter variants with extended HLA haplotypes
| Complotype
|
Markers in the TNF locus
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HLA-DR | C4B | C4A | BF | C2 | e | d | −307 | −856 | −862 | a | b | HLA-B | No. haplotypes studied |
| 3 | 1 | 0 | S | C | 3 | 1 | 2 | 1 | 1 | 2 | 3 | 8 | 10 |
| 2 | 1 | 3 | S | C | 3 | 3 | 1 | 1 | 1 | 11 | 4 | 7 | 6 |
| 7 | 1 | 3 | F | C | 3 | 3 | 1 | 1 | 1 | 7, 8 | 4 | 44 | 4 |
| 4 | 0 | 3 | S | C | 3 | 3 | 1 | 1 | 1 | 6, 7 | 5 | 44 | 4 |
| 7 | 1 | 6 | S | C | 3 | 4 | 1 | 1 | 1 | 2 | 5 | 57 | 6 |
| 11 | 1 | 3 | S | C | 3 | 3 | 1 | 1 | 1 | 5 | 5 | 35 | 6 |
| 1 | 1, 2 | 2 | S | C | 1 | 4 | 1 | 1 | 2 | 2 | 1 | 14 | 6 |
| 4 | 1 | 3 | S | C | 3 | 3 | 1 | 2 | 1 | 10 | 4 | 35 | 4 |
| 4 | 1 | 2 | S | C | 3 | 3 | 1 | 2 | 1 | 10 | 4 | 38 | 6 |
| 1 | 0 | 3, 2 | F | C | 1 | 4 | 1 | 1 | 2 | 5 | 5 | 35 | 6 |
| 4 | 3 | 3 | S | C | 1 | 4 | 1 | 1 | 2 | 2 | 1 | 62 | 4 |
| 2 | 2 | 4 | S | 0 | 3 | 3 | 1 | 2 | 1 | 10 | 4 | 18 | 4 |
| 3 | 0 | 3 | F1 | C | 3 | 4 | 1 | 1 | 1 | 1 | 5 | 18 | 8 |
The −856, −862, and −307 TNF-α promoter variants are shown with reference to extended HLA haplotypes. Cell fines of known homozygous extended haplotypes (21, 22), including known complotype, TNFmicrosatellite markers, and −307 variants (24), end selected families with known extended haplotypes were screened for expression of the −856 and −862 TNF-α promoter variants as described in Material and methods. The number under each column refers to the particular allele variant of the TNF-α promoter that is associated with the HLA-DR and HLA-B specificities as noted. The TNF-α −856/C allele is noted as 1 and the rarer −856/T allele is noted as 2, The −862/C TNF-α. allele is noted as 1 and the rarer −856/T allele is noted as 2. The complotype, TNF microsatellite. and −307 variant data included in the table is from the study by Turbay et al. (24): the −307/A allele is noted as 1 and the rarer −307/G allele is noted as 2.
Interestingly, none of the TNF-α promoter variants found at −856, −862, and −307 occur simultaneously in the homozygous cell lines (Table 1). Furthermore, none of the TNF-α promoter variants studied (−862, −856, −307) are present in the three extended haplotypes marked by HLA-DR2. B7; HLA-DR4, B44; HLA-DR7, B44 (30). We also found no evidence of these variants in six HLA-DR11, B35 haplotypes derived from one homozygous cell line (J0528239; see (22)) and four independent examples from four families with heterozygous individuals (Table 1, line 6).
To ascertain the frequency of occurrence of the −856 and −862 TNF-α promoter polymorphisms relative to the −307 TNF-α promoter polymorphism, an additional 105 promoters were examined from Caucasian individuals with B-cell malignancies and specifically screened for these polymorphisms. Interestingly, the −862 variant occurs with approximately the same frequency as the −307 variant in Caucasians, 17.4% versus 18.6% respectively, and was twice as frequent in the Cambodian population (Table 2). The −856 variant occurred in 13.6% of the Caucasian and 10% of the Cambodian promoters tested (Table 2). Furthermore, we did not detect a significant increase of the −862, −856, or −307 variants in patients carrying the diagnosis of NHL or MM versus normal control individuals (Table 2).
Table 2.
The frequencies of the novel and previously reported TNF-α promoter polymorphisms located at +70, −163, −237, −243, −307, −574, −856, and −862 in unrelated Caucasaian and Cambodian individuals
| Number of TNF-α promoters examined | + 70(+C) | −237(A) | −243(A) | −307(A) | −574(A) | −856(T) | − 862(A) | |
|---|---|---|---|---|---|---|---|---|
| Caucasians | ||||||||
| Individuals with B-cell malignancies | ||||||||
| Non-Hodgkin’s lymphoma | 60(7) | – | – | – | 9 | 1 | 12 | 12 |
| Multiple myeloma | 54 (10) | 1 | – | – | 16 | – | 6 | 10 |
| CLL | 4(2) | – | – | – | 1 | – | – | 3 |
| ALL | 1 | – | – | – | – | – | – | – |
| Normal controls | (24) | – | – | – | 5 | – | 5 | 4 |
| Cambodians | (20) | – | – | – | 3 | – | 2 | 6 |
|
| ||||||||
| Total | 166 (61) | 1 | – | – | 34 | 1 | 25 | 35 |
DNA was derived from the bone marrow of Caucasian individuals with the diagnosis of non-Hodgkin’s lymphoma (30 individuals), multiple myeloma (27 individuate), chronic lymphocytic leukemia (CLL) (4 individuals), acute lymphoblastic leukemia (ALL) (1 individual), or DNA derived from peripheral blood lymphocytes from normal Caucasians (12 individuals) or Cambodians (10 individuals) without a history of malignancy. The parentheses indicate the number of TNF-α promoters that were sequenced from −977 to +93 relative to the TNF-α mRNA cap site. TNF-α promoter sequences were amplified by PCR and sequenced and screened for polymorphisms as described in Material and methods.
Discussion
The TNF-α gene occurs in the class III region of the major histocompatibility locus on human chromosome 6 (see (31) for review). Here, we have described three new TNF-α single nt promoter polymorphisms, two of which are in linkage disequilibrium with extended HLA haplotypes. The finding that these polymorphisms occur in genetically separated groups of Caucasian and Asian descent and in independently derived homozygous cell lines indicates that these mutations are ancestral and did not arise recently. Given our sample size of 61 promoters, there is greater than a 95% chance that we would have detected any single nt polymorphism occurring in the human TNF-α promoter between −977 and +89 if the hetero-zygosity of that polymorphism were 0.048 or greater.
Previous studies have shown that the major regulatory elements necessary for the inducible regulation of the TNF-α gene via multiple stimuli in a variety of cell types are contained within a relatively compact promoter region contained within −200 base pairs relative to the TNF-α mRNA cap site (12–16). Consistent with this observation, the −862, −856, and −574 polymorphisms did not have an effect upon the transcriptional regulation of the gene in T and B lymphocytes.
Our results also demonstrate conclusively that the −307 variant does not influence TNF-α gene expression in activated lymphocytes, in agreement with two studies (32, 33) and at variance with two other studies (34, 35). Others have speculated that the −237 and −243 TNF-α variants would have an effect upon TNF-α gene regulation because they occur within a stretch of DNA that bears a resemblance to a ‘Y-box’ (CCAAT) enhancer motif (7). Here, we have formally shown that the −237 variant also has no effect upon TNF-α gene expression in activated T or B cells using promoter-reporter fusion genes. This data is consistent with functional studies, using the mouse TNF-α promoter, which have demonstrated that mutations in the murine equivalent of these sequences had no effect upon the transcriptional activity of the TNF-α promoter in lipopolysaccharide-stimulated monocytes (36).
Using homozygous cell lines (21, 22), we have shown that the −856 and −862 TNF-α promoter variants are associated with extended HLA haplotypes. Previous studies have shown an association between the −307, −237, and −243 variants and extended HLA haplotypes (7, 8, 19, 24, 25). Thus, the human TNF-α promoter polymorphisms located at positions −237, −243, −307, −862, and −856 relative to the TNF-α transciption start site are all non-randomly associated with other neighboring genes on chromosome 6 and all serve as markers for extended HLA haplotypes.
Interestingly, different patterns of TNF-α secretion have previously been associated with particular extended HLA haplotypes (37, 38). Our data argue that differential patterns of TNF-α secretion are not due to upregulation of TNF-α gene transcription secondary to the TNF-α promoter polymorphisms tested. Thus, the increased TNF-α bioactivity observed in association with extended HLA haplotypes must involve a different aspect of regulation of TNF-α.
As would be anticipated from the critical role that TNF-α plays in inflammation and lymphocyte biology, the regions of the promoter involved in the regulation of the gene are highly conserved. In fact, our analysis revealed no nt variability near any of the TNF-α upstream promoter elements previously shown to be involved in the regulation of the gene. It is interesting to speculate that TNF-α promoter variants that are in linkage disequilibrium with certain HLA genes are tolerated only in regions of the TNF-α promoter that are functionally silent.
Acknowledgments
This work was supported by grants from the NIH (CA58735) and an Established Investigator Award from the American Heart Association to A.E.G, from the NIH to E.J.Y. (HL59838, HL29583, AI 35630, MH47029), and by a NARSAD Fellowship to J.C.D. We are grateful to Brigitta Brinkman, Pedro Flores-Villanueva, and CorVerweij for critical comments on the manscript.
References
- 1.Aggarwal BB, Puri RK, editors. Human cytokines: their role in disease and therapy. Cambridge: Blackwell Science; 1995. [Google Scholar]
- 2.Garcia I, Mlyazaki Y, Araki K, et al. Transgenic mice expressing high levels of soluble TNF-R1 fusion protein are protected from lethal septic shock and cerebral malaria, and are highly sensitive to Listeria monocytogenes and Leishmania major infections. Eur J Immunol. 1995;25:2401–7. doi: 10.1002/eji.1830250841. [DOI] [PubMed] [Google Scholar]
- 3.Flynn JL, Goldstein MM, Chan J, et al. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity. 1995;2:561–72. doi: 10.1016/1074-7613(95)90001-2. [DOI] [PubMed] [Google Scholar]
- 4.Lucas R, Juilliard P, Redard M, et al. Lack of TNF receptor 2 but not of TNFR1 protects mice from experimental murine cerebral malaria. Eur Cytokine Netw. 1996;7:247. [Google Scholar]
- 5.Goldfeld A, Tsai EY. TNF-alpha and genetic susceptibility to parasitic disease. Exp Parasitol. 1996;84:300–3. doi: 10.1006/expr.1996.0117. [DOI] [PubMed] [Google Scholar]
- 6.Wilson AG, di Giovine FS, Blakemore AIF, Duff GW. Single base polymorphism in the human tumor necrosis factor alpha gene detectable by Nco1 restriction of PCR product. Hum Mol Genet. 1992;1:353. doi: 10.1093/hmg/1.5.353. [DOI] [PubMed] [Google Scholar]
- 7.D’Alfonso S, Richiardi PM. A polymorphic variation in a putative regulation box of the TNFA promoter region. Immunogenetics. 1994;39:150–4. doi: 10.1007/BF00188619. [DOI] [PubMed] [Google Scholar]
- 8.Zimmerman PA, Guderian RH, Nutman TB. A new TNFA promoter allele identified in South American Blacks. Immunogenetics. 1996;44:485–6. [PubMed] [Google Scholar]
- 9.Brinkman BMN, Giphart MJ, Verhoef A, et al. Tumor necrosis factor alpha −308 gene variants in relation to major histocompatibility complex alleles and Felty’s syndrome. Hum Immunol. 1994;41:259–66. doi: 10.1016/0198-8859(94)90044-2. [DOI] [PubMed] [Google Scholar]
- 10.Brinkman BMN, Kaijzel EL, Huizinga TWJ, Giphart MJ, Breedveld FC, Verweij CL. Detection of a novel c-insertion polymorphism within the human tumor necrosis factor alpha gene. Hum Genetics. 1995;96:493. doi: 10.1007/BF00191815. [DOI] [PubMed] [Google Scholar]
- 11.Hamann A, Mantzoros C, Vidal-Puig A, Flier JS. Genetic variability in the TNF alpha promoter is not associated with type II diabetes mellitus. Biochem Biophys Res Common. 1995;211:833–9. doi: 10.1006/bbrc.1995.1887. [DOI] [PubMed] [Google Scholar]
- 12.Goldfeld AE, Doyle C, Maniatis T. Human tumor necrosis factor-α gene regulation by-virus and lipopolysaccharide. Proc Natl Acad Sci U S A. 1990;87:9769–73. doi: 10.1073/pnas.87.24.9769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldfeld AE, Strominger JL, Doyle C. Human tumor necrosis factor-α gene regulation in phorbol ester stimulated B and T cell lines. J Exp Med. 1991;174:73–81. doi: 10.1084/jem.174.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldfeld AE, McCaffrey PG, Strominger JL, Rao A. The human tumor necrosis factor-α gene is regulated by a novel cyclosporin-sensitive promoter element in activated T cells. J Exp Med. 1993;178:1365–79. doi: 10.1084/jem.178.4.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsai EY, Jain J, Pesavento PA, Rao A, Goldfeld AE. TNF-α gene regulation in activated T cells involves ATF-2/Jun and NFATp. Mol Cell Biol. 1996;16:459–67. doi: 10.1128/mcb.16.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsai EY, Yie J, Thanos D, Goldfeld AE. Cell-type specific regulation of the human tumor necrosis factor-α gene in B cells by NFATp and ATF-2/Jun. Mol Cell Biol. 1996;16:5232–44. doi: 10.1128/mcb.16.10.5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Granja CB, Salazar M, Yunis EJ. Population genetics and human leukocyte antigen polymorphism. In: Tilney NL, Strom TB, Paul LC, editors. Transplantation biology. Philadelphia (PA): Lippincott-Raven Publishers; 1996. pp. 311–25. [Google Scholar]
- 18.Gribben JG, Freedman AS, Newberg D, et al. Immunologic purging of marrow assessed by PCR before autologous bone marrow transplantation for B cell lymphoma. N Eng J Med. 1991;325:1525–33. doi: 10.1056/NEJM199111283252201. [DOI] [PubMed] [Google Scholar]
- 19.Goldfeld AE, Delgado JC, Sok T, et al. Association of an HLA-DQ allele with clinical tuberculosis. J Am Med Assoc. 1998;279:226–8. doi: 10.1001/jama.279.3.226. [DOI] [PubMed] [Google Scholar]
- 20.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang SY, Milford E, Hammerling U, Dupont B. Description of the reference panel of B-lymphoblastoid cell lines for factors of the HLA system: the B-cell line panel designed for the Tenth International Histocompatibility Workshop. In: Dupont B, editor. Immunobiology of HLA. New York: Springer-Verlag; 1989. pp. 11–19. [Google Scholar]
- 22.Charron D, editor. Genetic diversity of HLA functional and medical implication. Paris, France: EDK; 1997. [Google Scholar]
- 23.Salazar M, Yunis JJ, Delgado MB, Bing D, Yunis EJ. HLA-DQB1 allele typing by a new PCR-RFLP method: correlation with a PCR-SSO method. Tissue Antigens. 1992;40:116–23. doi: 10.1111/j.1399-0039.1992.tb02102.x. [DOI] [PubMed] [Google Scholar]
- 24.Turbay D, Lieberman J, Alper CA, et al. Tumor necrosis factor constellation polymorphism and clozapine-induced agranulocytosis in two different ethnic groups. Blood. 1997;89:4167–74. [PubMed] [Google Scholar]
- 25.Wilson AG, de Vries N, Pociot F, di Giovine FS, van der Putte LBA, Duff GW. An allelic polymorphism within the human tumor necrosis factor alpha promoter region is strongly associated with HLA A1, B8, and DR3 alleles. J Exp Med. 1993;177:557–60. doi: 10.1084/jem.177.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Udalova IA, Nedospasov SA, Webb GC, Chaplin DD, Turetskaya RL. Highly informative typing of the human TNF locus using six adjacent polymorphic markers. Genomes. 1993;16:180–6. doi: 10.1006/geno.1993.1156. [DOI] [PubMed] [Google Scholar]
- 27.Alper CA, Awdeh ZL, Yunis EJ. Complotypes and extended haplotypes in laboratory medicine. Complement Inflamm. 1989;6:8–18. doi: 10.1159/000463067. [DOI] [PubMed] [Google Scholar]
- 28.Dupont B, Pollack MS, Levine MS, O’Neill GJ, Hawkins BR, New MI. Joint report. Congenital adrenal hyperplasia. In: Terasaki PI, editor. Histocompatibility Testing. Los Angeles: UCLA Press; 1980. pp. 693–706. [Google Scholar]
- 29.Fleishnick E, Raum D, Alosco SM, et al. Extended MHC haplotypes in 21-hydroxylase-deficiency congenital hyperplasia: shared genotypes in unrelated patients. Lancet. 1983;1:152–6. doi: 10.1016/s0140-6736(83)92757-5. [DOI] [PubMed] [Google Scholar]
- 30.Awdeh ZL, Raum D, Yunis EJ, Aper CA. Extended HLA/complement allele haplotypes: evidence for T/t-like complex in man. Proc Natl Acad Sci U S A. 1983;80:259–63. doi: 10.1073/pnas.80.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gruen JR, Weissman SM. Evolving views of the major histocompatibility complex. Blood. 1997;90:4252–65. [PubMed] [Google Scholar]
- 32.Brinkman BMN, Zuijdgeest D, Kaijzel EL, Breedveld FC, Verweij CL. Relevance of the tumor necrosis factor alpha (TNF-α) −308 promoter polymorphism in TNF-α gene regulation. J Inflamm. 1996;46:32–6. [PubMed] [Google Scholar]
- 33.Stuber F, Udalova IA, Book M, et al. −308 tumor necrosis factor (TNF) polymorphism is not associated with survival in severe sepsis and is unrelated to lipopolysaecharide inducibility of the human TNF promoter. J Inflamm. 1996;46:42–7. [PubMed] [Google Scholar]
- 34.Wilson AG, Symons JA, McDowell TL, McDevirt HO, Duff GW. Effects of a polymorphism in the human tumor necrosis factor alpha promoter on transcriptional activation. Proc Natl Acad Sci US A. 1997;94:319–9. doi: 10.1073/pnas.94.7.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kroeger KM, Carville KS, Abraham LJ. The −308 tumor necrosis factor-alpha promoter polymorphism effects transcription. Mol Immunol. 1997;4:391–9. doi: 10.1016/s0161-5890(97)00052-7. [DOI] [PubMed] [Google Scholar]
- 36.Drouet C, Shakhov AN, Jongeneel CV. Enhancers and transcription factors controlling the inducibility of the tumor necrosis factor alpha promoter in primary macrophages. J Immunol. 1991;147:1694–700. [PubMed] [Google Scholar]
- 37.Jacob CO, Fronek Z, Lewis G, Koo M, Hansen JA, McDevitt HO. Heritable histocompatibility complex class II-associated differences in production of tumor necrosis factor-α: relevance to genetic predisposition to systemic lupus erythematosus. Proc Natl Acad Sci U S A. 1990;87:1233–7. doi: 10.1073/pnas.87.3.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia-Merino A, Alper CA, Usuku K, et al. Tumor necrosis factor microsatellite haplotypes in relation to extended haplotypes, susceptibility to diseases associated with the major histocompatibility complex and TNF secretion. Hum Immunol. 1996;50:11–21. doi: 10.1016/0198-8859(96)00064-x. [DOI] [PubMed] [Google Scholar]