

1. Introduction
Specific enzymatic mechanisms in mammalian cells generate two free-radical species: nitric oxide (NO) via NO synthases (NOS) and superoxide (O2−) via NADPH oxidases (NOX). NO, generated by NOSs that are expressed constitutively in most cell types, regulates a wide range of physiological signals, acting substantially through S-nitrosylation of active site and allosteric cysteine residues [1]. NOXs as well are expressed constitutively in many cell types, and their activity is coupled to diverse physiological stimuli [2–4]. In addition, reactive oxygen species (ROS) generated from O2−, in particular hydrogen peroxide (H2O2), can oxidatively modify cysteine thiols and thereby modulate protein function [5].
The extent and mechanisms of interaction between redox-based cellular signals conveyed by NO and ROS remain major unresolved issues, and it has been assumed that NO and O2− are mutually eliminative in vivo [6,7]. We report here that the small GTPase Rac, a component of at least some major forms of NOX [2–4,8], also interacts with and regulates the activity of the constitutively-expressed NOSs (endothelial NOS, eNOS, and neuronal NOS, nNOS), and that NO and O2− production are altered in parallel, and not in opposition, by inhibition or activation of Rac. Furthermore, we demonstrate a role for Rac in transduction that utilizes NO, ROS or both.
2. Materials and methods
Additional materials and methods can be found on-line as SUPLEMENTARY MATERIAL.
2.1. Reagents for manipulation of Rac and Nox activity
Mutant human Rac 1 was overexpressed in cultured cells as either Rac 1 G12V (Rac-CA), in which turnover of bound GTP is greatly reduced and which is therefore constitutively active (Rac-CA), or as Rac 1 D17N, in which GDP is preferentially bound and which therefore functions as a dominant negative suppressor of endogenous, Rac-dependent mechanisms when overexpressed (Rac-DN). Since their use was introduced in the context of analyzing the role of Rac in cytoskeletal regulation [9], Rac-CA and Rac-DN have been employed extensively in a broad range of studies examining many effects of Rac activity in cultured cells [e.g., 10].
For acute, pharmacological inhibition of Rac, we employed a specific small-molecule inhibitor of Rac activation, NSC23766 (NSC). NSC, which was identified by structure-based virtual screening, acts as a competitive inhibitor to prevent binding to and activation of Rac by Rac-specific GEFs including Trio and Tiam1 [11].
For specific inhibition of Nox activity, we employed a peptide inhibitor of NOX activation (gp91 ds-tat) and a control peptide (scram-tat). The gp91 ds (docking sequence)-tat peptide comprises a segment of the region within the catalytic flavocytochrome subunit of NOX2 (gp91phox) to which p47phox binds, fused to the HIV tat sequence to facilitate entry into cells, and gp91 ds-tat blocks the activating interaction between p47phox and gp91phox (and/or their close homologues) [12]. The gp91 ds sequence was scrambled in the scram-tat control peptide. The peptide sequence of gp91 ds-tat is: RKKRRQRRR (tat portion) CSTRIRRQL-NH2 (gp91 ds portion). In the scram-tat peptide, the gp91 ds sequence is replaced with: CLRITRQSR-NH2.
In addition, we employed a well-characterized, stable, cell-permeant superoxide dismutase (SOD) mimetic, manganese(III)5,10,15,20-tetrakis(N-ethylpyridinium-2-yl)porphyrin (MnTE-2-PyP5+), to accelerate the removal of O2− (2O2−→ H2O2) in cultured cells [13].
2.2. Cell culture, adenoviral infection and pharmacologic Rac inhibition
Primary human aortic endothelial cells (HAEC; Clonetics) were cultured in serum-rich endothelial growth medium (EGM-2; Clonetics) and used at passages 4–6. Cells were infected at ~75% confluency by exposure for ~12 hr to myc-tagged adenoviral constructs of Rac-DN or Rac-CA and maintained in fresh medium for 36 hr, followed by incubation for an additional 12 hr with or without serum deprivation. Under these conditions, infection efficiency was greater than 75% as assessed by immunohistofluorescence of myc. human embryonic kidney (HEK) cells stably transfected with eNOS or nNOS were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% serum, infected by exposure for 12 hr to Rac-CA or Rac-DN adenoviral constructs in medium containing 2% serum, and maintained in medium containing 10% serum for an additional 24 hr before analysis. Primary cultures of rat hippocampal neurons were prepared as described from the CA1 region of day-old pups [14] and maintained in supplemented Neurobasal medium (Sigma) for 14 d before NOS activity assay. When employed in measurement of NO/O2− levels or of actin content/distribution, MnTE-2-PyP5+ (50μM) and the NOS inhibitor L-NMMA (3mM) were applied 48 hr before assay. For specific inhibition of Rac activation with NSC, HAEC were incubated with 1 mM NSC for 36 hr before NOS activity assays, ring segments of rabbit thoracic aorta were incubated with 100 μM NSC for 1 hr before measurement of cGMP or bioassay, and hippocampal neurons were incubated with 100 μM NSC for either 1 hr or 16 hr before NOS activity assay.
2.3. Measurement of intracellular NO and O2−
Cells were washed in PBS and incubated for 30 min at 37° C in Hank’s balanced salt solution supplemented with 200mM HEPES and 5mM glucose, and containing either 5uM 4,5-diaminofluorescein diacetate (DAF-FM; Molecular Probes) for detection of NO or 10μM dihydroethidium (DHE; Molecular Probes) for detection of O2−. Cells loaded with DAF-FM were washed and incubated for 15–30 min, and DHE-loaded cells were washed without further incubation, before analysis by laser-scanning confocal microscopy.
3. Results
3.1. Rac1 co-regulates NO production by NOS and O2− production by NOX
On the assumption of mutual elimination, we were surprised to find that, in primary human aortic endothelial cells (HAEC, which contain at least one isoform of both NOS and NOX, including NOS3 and NOX2), expression (via an adenoviral vector) of a dominant-negative (GDP-bound) form of Rac (Rac1 D17N, Rac-DN), which prevented production of O2− as assessed by DHE fluorescence (Figure 1A), did not increase but rather substantially reduced basal levels of NO as assessed by DAF-FM fluorescence (Figure 1A). Conversely, expression of a constitutively-active (GTP-bound) form of Rac (Rac1 G12V, Rac-CA) resulted in large increases in both NO and O2− levels (Figure 1A).
Fig. 1.
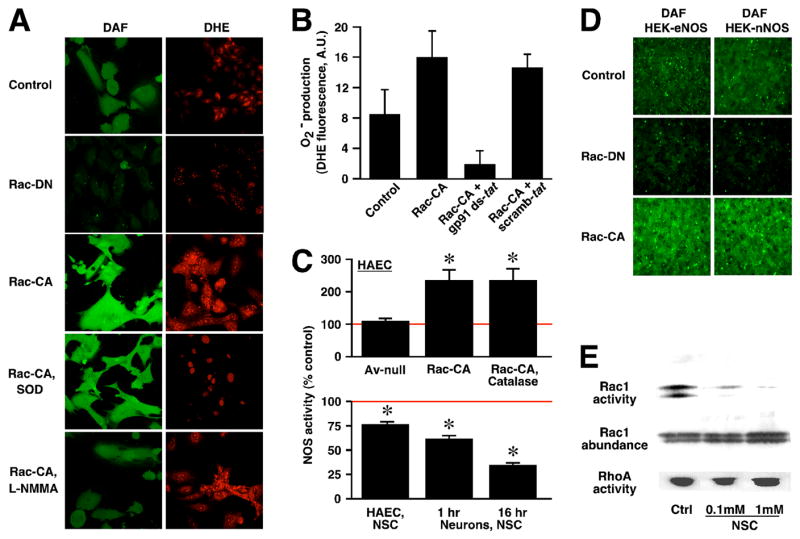
Rac co-regulates NO and O2− production. (A) In HAEC, NO (DAF fluorescence) and O2− (DHE fluorescence) are increased by expression of Rac-CA and decreased by expression of Rac- DN. Reducing levels of NO by inhibiting NOS (L-NMMA) or of O2− with MnTE-2-PyP5+ (SOD mimetic) has little effect on levels of the other species. (B) Rac1 regulates endothelial O2−production by a p47phox–dependent NADPH oxidase (NOX). HAEC expressing Rac-CA were treated with a peptide inhibitor of NOX activation (gp91 ds-tat), or with a control peptide (scramb-tat). Expression of Rac-CA resulted in large increases in O2− production as assessed by DHE fluorescence, which were eliminated by treatment with gp91 ds-tat. Note that treatment with gp91 ds-tat also suppressed basal O2− production. For each condition, average DHE fluorescence/cell (mean ± S.D.) was quantified for 1–5 cells in each of 20–24 randomly selected microscopic fields of view, obtained from 3 experiments in which all conditions were examined. (C) Upper histogram: expression of Rac-CA enhances endogenous eNOS enzymatic activity in HAEC as assessed by measuring the conversion of 3H-arginine to 3H-citrulline, and enhancement is not influenced significantly by eliminating intracellular H2O2 with PEG-catalase (p < 0.05; n=3). Asterisk indicates significant re. uninfected control. Av-null indicates adenoviral vector control. Lower histogram: treatment with a small-molecule inhibitor of Rac activation (NSC) significantly inhibits endogenous NOS activity of HAEC (NSC: 1 mM for 16 hr) and of cultured rat primary hippocampal neurons (NSC: 100 μM for 1 hr or 16 hr) (p < 0.05; n=4 for all groups). Asterisk indicates significant re. untreated control cultures. (D) Levels of NO are increased by Rac-CA expression and decreased by Rac-DN expression in HEK cells stably transfected with either eNOS or nNOS. (E) Basal activity of Rac in HAEC and specific inhibition of Rac by NSC. Cultured HAEC were exposed to NSC overnight and Rac and RhoA activities were assessed by substrate-binding activation assays. NSC eliminates basal (Ctrl) Rac activity but has no effect on RhoA activity. Rows depict representative Western blots for Rac or RhoA following substrate binding and pulldown (top and bottom rows), or of starting material (cell lysate; middle row).
Rac and other constituents including p47phox were identified as obligate components of active multicomponent NOX (NOX2) in phagocytic cells [2], and considerable evidence indicates that at least some of these components, including Rac, are required for NOX activity in other cell types [2–4, 8]. Nonetheless, effects of Rac on O2− production that are independent of NOX are possible in principle. We found that treatment of HAEC with a peptide shown to specifically inhibit the binding of p47phox to the membrane-associated catalytic flavocytochrome subunit of NOX2 (gp91phox) [12] abrogated the increase in O2− production induced by Rac-CA expression and suppressed basal O2− production, as assessed by DHE fluorescence (Figure 1B). These results demonstrate directly that Rac1 governs O2− generation in endothelial cells by regulating the activity of a NOX, which is dependent upon p47phox or a close homologue, consistent with and extending previous results [2–4, 15].
Rac-dependent changes in levels of NO might be accounted for by an effect on NOS activity or abundance, or on NO metabolism. Assay of NOS function in intact cells by measuring the conversion of 3H-arginine to 3H-citrulline, a specific indicator of enzymatic activity, demonstrated directly that expression of Rac-CA potentiated NO production by NOS in HAEC (Figure 1C), in the absence of changes in endothelial NOS (eNOS) expression as assessed by western blotting or effects on cellular uptake of 3H-arginine (not shown). Further, the Rac-dependent increase in NOS activity was unaffected by eliminating hydrogen peroxide with cell-permeable polyethylene glycol (PEG)-linked catalase (as assessed by 2′,7′-dichlorodihydrofluorescein diacetate fluorescence, not shown), and was thus independent of increased production of the major reactive oxygen species resulting from NOX activity (Figure 1B). More generally, the finding that altering Rac activity concomitantly increased or decreased levels of both NO and O2− is explained by conjoint regulation by Rac of NOS and NOX, rather than by any action of NO or O2− on production or metabolism of the other species (or any lack of specificity in the fluorescent indicators employed [16]), because greatly reducing O2− with the SOD mimetic, MnTE-2-PyP5+, or NO with the NOS inhibitor, NG-monomethyl-L-arginine (L-NMMA), did not substantially affect levels of the other species in cells expressing Rac-CA (Figure 1A). In addition, protein nitrotyrosine content, a measure of peroxynitrite formation from NO/O2− combination, was indistinguishable in control and Rac-CA-expressing cells as assessed by western blotting (not shown).
The predominant form of NOS in endothelial cells (including HAEC) is eNOS. The generality of our results was demonstrated by the finding that NO production was potentiated by Rac-CA and inhibited by Rac-DN in HEK cells stably transfected with either eNOS or nNOS (Figure 1D). Thus, Rac can regulate basal NO production by the major, constitutively-expressed forms of NOS. Note that, as for nNOS, NOXs are distributed broadly within the central nervous system [17].
Because mutant, overexpressed Rac might not replicate the actions of endogenous Rac and might suppress other G-proteins by effectively depleting exchange factors that are not exclusive to Rac, we verified and extended our findings by employing a specific small-molecule inhibitor of Rac activation, NSC. NSC was reported to have no effect on other closely-related members of the Rho family of G-proteins (Cdc42, RhoA) [11]. Treatment overnight (16 hr) with NSC substantially decreased basal eNOS activity of HAEC, as well as basal nNOS activity of cultured rodent primary hippocampal neurons, and significant decreases in nNOS activity of cultured neurons were evident following even brief exposure to NSC (1 hr) (Figure 1C). In addition, we verified that Rac exhibited basal activity in HAEC (consistent with suppression of NOS activity, and of resting NO and O2− production, by inhibition of Rac) and that treatment with NSC, at concentrations (0.1–1 mM) and over intervals (1–16 hr) employed in evaluating effects on NOS activity, greatly inhibited Rac without effects on RhoA, as assessed by substrate-binding activation assays (Figure 1E).
3.2. Rac activation mediates ligand-induced stimulation of NOS
Basal and stimulus-induced NOS activity can be regulated separately [18]. To determine whether Rac governs stimulus-induced and physiologically relevant NOS activity, we examined the effects of expressing Rac-DN in endothelial cells of intact aorta, and of exposure to NSC, on the acetylcholine (ACh)-induced increase in smooth muscle cyclic guanosine monophosphate (cGMP) and decrease in smooth muscle tone, which together constitute the prototypic example of the cellular role of NO (vasodilation).
In ring segments of rabbit aorta, expression of Rac-DN, which was restricted to the endothelium (Figure 2A), significantly suppressed ACh-induced, NO-dependent production of cGMP in smooth muscle as determined by radioimmunoassay (Figure 2B), and smooth muscle relaxation as determined in bioassays (Figure 2C). The proportionately greater effect of Rac-DN expression on cGMP production vs. relaxation may reflect a non-linear relationship between guanylate cyclase activity and smooth muscle tone as well as a contribution of NO- and/or cGMP-independent mechanisms to the vasodilatory action of ACh. Exposure of intact aortic segments to NSC also significantly decreased ACh-induced cGMP production (Figure 2D) and vasorelaxation (Figure 2E). Inhibition of vasorelaxation was reversed rapidly following removal of NSC (Figure 2E, insert). We also determined that exposure to NSC had no effect on smooth muscle contractility (no change in the contractile response to potassium-induced depolarization) or on transduction downstream of NOS (no change in nitroglycerin-induced relaxation) (not shown). Differences between the effects of Rac-DN and of NSC on the form of the ACh dose-response curve may reflect in part inhibition by NSC of non-endothelial Rac. In addition, we found that NSC significantly inhibited stimulation of NOS in cultured HAEC by vascular endothelial growth factor (VEGF) (Figure 2F), demonstrating a role for Rac in regulating activation of NOS by ligands that trigger disparate transduction mechanisms (G-protein coupled and receptor tyrosine kinase).
Fig. 2.
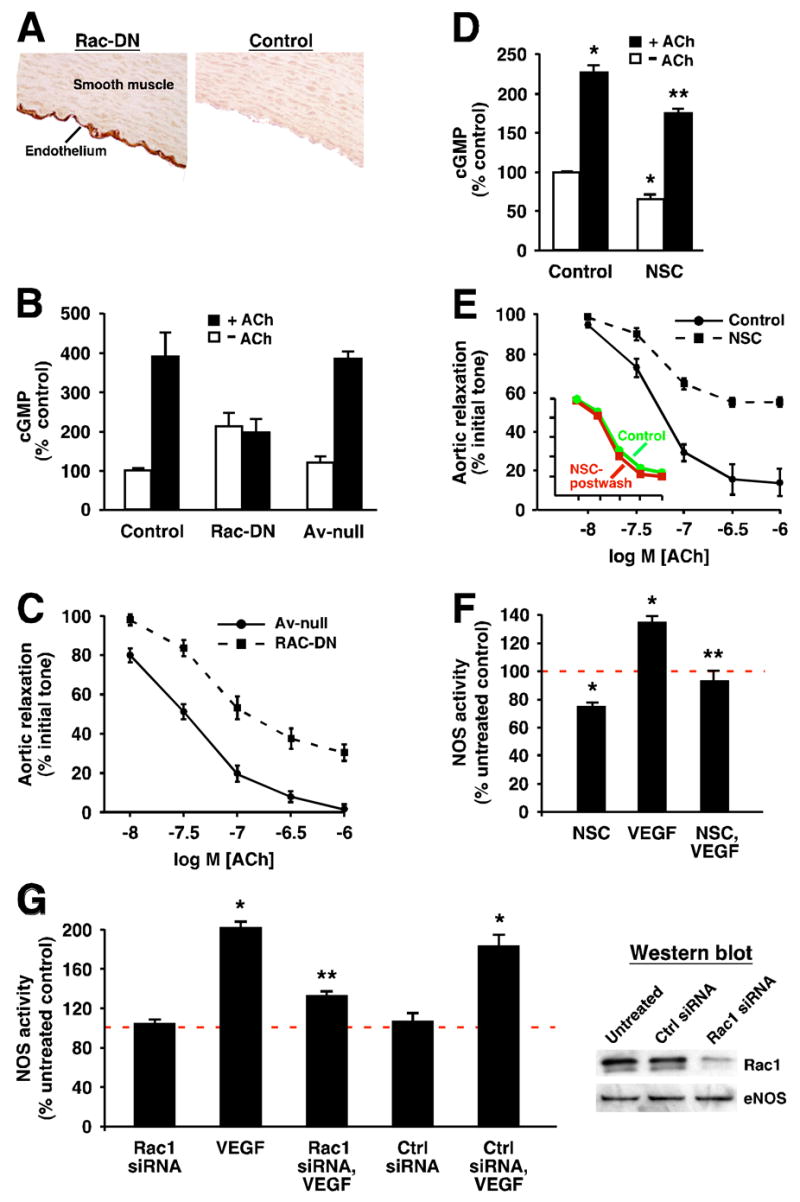
Suppressing Rac activity inhibits ligand-induced NOS activation. (A) In intact aorta, expression of myc-tagged Rac-DN is restricted to the endothelium after application of the adenoviral vector to the aortic lumen (see Supplementary Materials and Methods), as revealed by myc immunohistochemistry. (B) Radioimmunoassay shows that cGMP production induced in aortic smooth muscle by ACh (10−6 M) is inhibited significantly by endothelial expression of Rac-DN (p<0.05; n=6). In (B) and (C), Av-null indicates adenoviral vector control. (C) Expression of Rac-DN in aortic endothelium significantly inhibits ACh-induced smooth muscle relaxation (vasodilation) as assessed by bioassay of aortic ring segments (p<0.05 at all doses of ACh; n=6). (D) Treatment of intact aorta with NSC (0.1 mM for 1 hr), significantly inhibits both basal and ACh-induced cGMP production (p<0.05; n=5 for each group). Single asterisk indicates significant re. unstimulated control, and double asterisks indicate significant re. ACh-stimulated control. (E) Treatment of intact aorta with NSC as in (D) significantly inhibits ACh-induced vasodilation at all doses of ACh (p < 0.05; n=3). The effect of NSC is reversed by washing (inset). (F) Treatment of HAEC with NSC (1mM; 36 hr) significantly inhibits both basal NOS activity and NOS activation by VEGF (100ng/ml) (p < 0.05; n=4 for all groups). Single asterisk indicates significant re. untreated control (red line=100% of untreated control), and double asterisks indicate significant re. VEGF. (G) Treatment of primary bovine aortic endothelial cells with Rac1-specific siRNA, which greatly reduces levels of Rac1 (~80%–90%) but does not affect levels of eNOS (as assessed by Western blotting, shown at right), significantly inhibits NOS activation by VEGF (100 ng/ml) (p<0.05, n=3; significance indicated as in (F)). Note that NOS activity is approximately doubled by VEGF stimulation, and that basal NOS activity is not affected significantly by Rac knockdown. Ctrl siRNA= transfection with non-specific siRNA duplexes (see Supplementary Materials and Methods).
Because NSC prevents Rac activation, the suppressive effects of NSC on stimulus-induced NOS activation should be replicated by removing Rac. This prediction was confirmed fully by the finding that greatly reducing (>80%) the Rac content of primary bovine aortic endothelial cells (BAEC) with siRNA significantly suppressed NOS activation by VEGF, in the absence of any changes in eNOS abundance (Figure 2G). Note that, in contrast to the effects of Rac-DN and of sustained exposure to NSC (which would promote Rac-GDP formation), knockdown of Rac did not significantly suppress basal NOS activity (Figure 2G). Removing Rac would not in and of itself substantially alter basal NOS activity if it is the nucleotide-bound state of Rac that determines its effects on NOS (i.e., potentiation and suppression of NOS activity by, respectively, Rac-GTP and Rac-GDP).
3.3. Molecular interaction of Rac1 and eNOS
Ligand-induced increases in the activity of constitutively-expressed NOSs are often transitory. Sustained increases and decreases in basal eNOS and nNOS activity, as we observed in HEK cells following expression of Rac-CA and Rac-DN, could be provided by NOS phosphorylation [18]. However, although we do not rule out a role for NOS phosphorylation, we did not detect changes in phosphorylation of eNOS in western blots of extracts from Rac-CA-expressing HAEC using antibodies specific for phospho-Ser1177 or phospho-Ser116 (not shown), two sites where eNOS phosphorylation is altered by multiple physiological stimuli [18]. We therefore considered the possibility that Rac and constitutively-expressed NOSs form a complex in which Rac serves as an allosteric regulator of NOS activity, as suggested previously for iNOS (NOS2) [19]. Co-elution of Rac and eNOS in size-exclusion chromatography of HAEC extracts indicated co-localization in macomolecular complexes, although the large complex masses indicate the presence of other elements (Figure 3A). More directly, we found that glutathione-S-transferase (GST)-tagged Rac-CA, immobilized on glutathione-sepharose, complexed with eNOS and with nNOS respectively in extracts of eNOS- or nNOS- transfected HEK cells, and with eNOS in extracts of HAEC (Figure 3B and C). In addition, Rac-CA brought down eNOS from HAEC extracts of perinuclear and plasma membranes with comparable efficacy; thus, Rac may regulate NO production by eNOS in both compartments where it is concentrated (Supplementary Fig.). These findings are generally consistent with Kone et al. [19], who reported that Rac and iNOS interact in macrophages following induction of iNOS with bacterial lipopolysaccharide, although NOS isoforms (iNOS, eNOS, nNOS) are not known to share other allosteric regulatory proteins. In combination with their observations, our findings indicate that Rac may regulate the activity of all major forms of NOS.
Fig. 3.

Molecular interaction of Rac and NOS. (A) Rac and eNOS co-elute, in size-exclusion chromatography (Superdex-200) of HAEC extracted with 1% NP-40, in high-mass complexes (fraction 10 ≈ 650 kDa) and a population of Rac elutes independently of eNOS (fraction 21 ≈ 18 kDa, consistent with uncomplexed, monomeric Rac). The western blot shown was reacted with antibodies to Rac and eNOS simultaneously. (B) GST-Rac-CA bound to glutathione-sepharose complexes with eNOS and with nNOS in extracts of NOS-transfected HEK cells and with eNOS in extracts of HAEC. Control samples (B and C) were generated by incubating equal quantities of extract (or of purified eNOS) with GST-bound glutathione-sepharose. (C) Purified eNOS binds directly to immobilized Rac-CA and wild-type Rac. Binding of eNOS to GTP- vs. GDP-bound Rac is indistinguishable, and binding of eNOS to nucleotide-free Rac (EDTA) is substantially more efficient.
At least part of the association of Rac with NOS in cellular extracts can be ascribed to a direct interaction, because purified eNOS was bound by Rac-CA and by native (wild-type) Rac (Figure 3C). In addition, we observed consistently that binding of purified eNOS was equivalent for GDP-β-S-bound or GTP-γ-S-bound wild-type Rac, consistent with the description by Kone et al. [19] of Rac/iNOS binding, and substantially greater for nucleotide-free Rac (Fig. 3C). Taken together with our observations that NO production is potentiated by Rac-CA and suppressed by Rac-DN, NSC and Rac knockdown, equivalent binding of NOS by GDP-bound or GTP-bound Rac suggests that activation/inactivation (by nucleotide exchange) of complexed Rac, rather than complex formation, governs the function of Rac as an allosteric regulator of NOS activity. Preferential binding of NOS by the nucleotide-free form of Rac, which would be expected to exist only in the presence of an active Rac GEF, may promote the regulation of NOS by Rac in specific cellular compartments [20].
3.4. Rac-regulated NO and O2− production governs the actin-based cytoskeleton
Rac plays a prominent role in organization of the actin-based cytoskeleton [21, 22]. We evaluated the role of Rac-regulated production of NO and O2− on cytoskeletal organization. As described previously [10], expression of Rac-CA in endothelial cells increased the cellular content of filamentous actin (F-actin), as assessed by quantitation of rhodamine-phalloidin binding (Figure 4A). In addition, Rac-CA expression resulted in a dramatic redistribution of rhodamine-phalloidin-decorated actin filaments from cytosol (stress fibers) to the inner surface of the plasma membrane (cortical actin) (Figure 4B). Treatment with MnTE-2-PyP5+ substantially prevented the Rac-CA-induced increase in F-actin content, whereas L-NMMA had no significant effect by itself or in combination with MnTE-2-PyP5+ (Figure 4A). In contrast, both L-NMMA and MnTE-2-PyP5+ significantly reduced the Rac-CA-induced shift of F-actin from stress fibers to a cortical distribution, and their effects were not additive when assayed together (Figure 4B). Thus, regulation by Rac of the equilibrium between F-actin and monomeric actin (actin polymerization) requires production of O2− but not of NO, whereas the configuration of the actin-based cytoskeleton is dependent on the conjunct or concerted action of both NO and O2−, as indicated by the non-additive effects on F-actin distribution of reducing the level of each species.
Fig. 4.
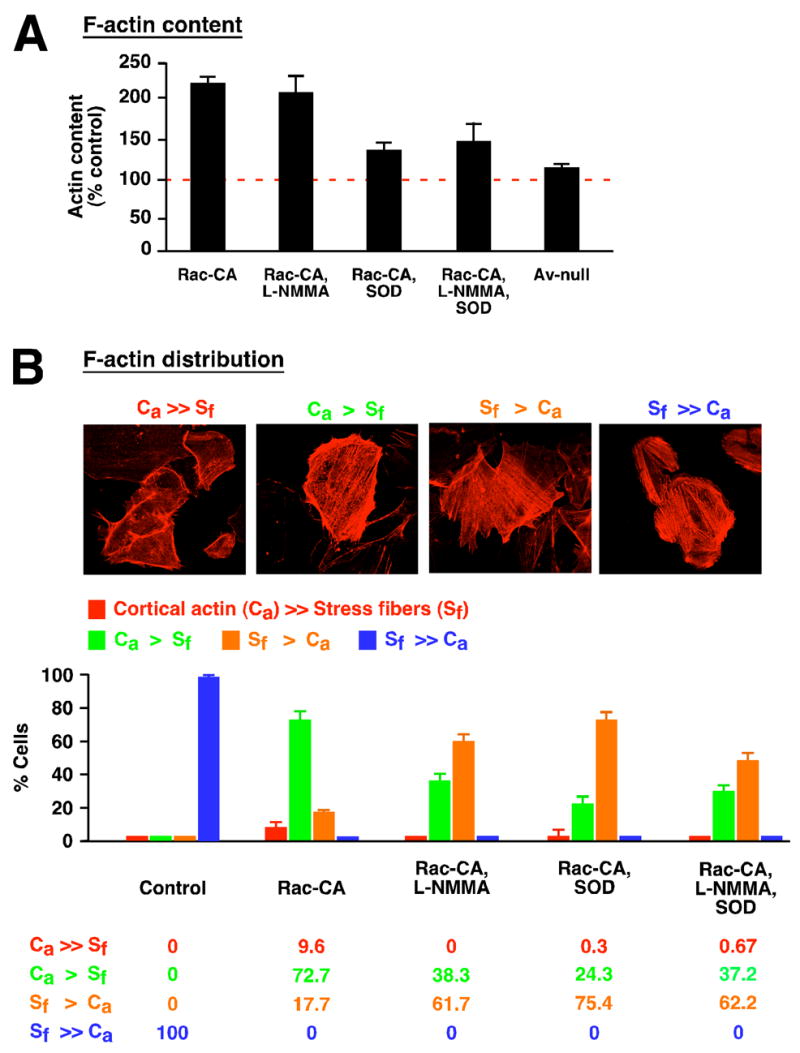
Mediation by NO and/or O2− of Rac-dependent actin polymerization and cytoskeletal organization in HAEC. (A) Expression of Rac-CA substantially increases F-actin content as assessed by quantitation of rhodamine-phalloidin binding, and this increase is inhibited significantly (n=6; p<0.005) by reducing intracellular O2− levels with MnTE-2-PyP5+. Reducing NO production with L-NMMA has little effect by itself or in combination with MnTE-2-PyP5+. Av-null indicates adenoviral vector control. (B) As revealed by the distribution of rhodamine-phalloidin fluorescence, expression of Rac-CA results in a shift in the localization of F-actin from predominantly cytosolic in the form of stress fibers (SF) to predominantly associated with the plasma membrane (cortical actin, Ca). Assignment of cells to four categories based on relative content of Sf and Ca shows that the Rac-CA-induced shift in F-actin disposition is abrogated significantly either by inhibiting NOS (L-NMMA) or by removing O2− (MnTE-2-PyP5+), and that these effects are not additive (n > 500 cells for each of the five conditions).
4. Discussion
4.1. Rac co-regulates NOS and NOX
Our findings delineate a revised perspective on redox-based cellular signal transduction, in which Rac can function as a common control element for NOS and NOX to provide for coordination of NO− and ROS-mediated signals, in contrast to the view that the interplay of NO and O2− is encompassed by their chemical interaction. Because one or more forms of NOS, NOX and Rac are present in most cell types, this role may be of very general relevance. In addition, the regulation of NOS by Rac expands the domains of cellular signals that may influence NO production and of potential roles for NO in diverse Rac-dependent cellular processes.
Coupled activation through Rac of NOX and NOS is consistent with the finding that mechanical shear stress, a principal determinant of eNOS activity, also activates Rac and increases O2− production in endothelial cells [23], and it is known that receptor ligands including VEGF, insulin and angiotensin [1,2,4], as well as leukotriene B4 (our unpublished observations), can increase production of both NO and O2−. However, we have found that other ligands including platelet-derived growth factor can stimulate O2− production in the absence of changes in levels of NO (unpublished observations).
4.2. Conjoint and independent effects of NO and ROS in redox regulation
Analysis in vitro suggests that S-nitrosothiol formation can be promoted by combinatorial NO/O2− chemistry when NO and O2− are generated simultaneously and at low levels [24], and ROS (H2O2) can potentiate S-nitrosylation in vivo [25]. Thus, conjoint effects of NO and ROS may be read out to a significant extent by enhanced S-nitrosylation of regulatory and active sites Cys residues [1]. Delineating the common and unique protein substrates of NO- and ROS-derived modifications in the context of specific transduction pathways remains a significant challenge.
Supplementary Material
Abbreviations
- NO
nitric oxide
- NOS
nitric oxide synthase
- ROS
reactive oxygen species
- NOX
NADPH oxidase
- eNOS
endothelial nitric oxide synthase
- iNOS
induced nitric oxide synthase
- nNOS
neuronal nitric oxide synthase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hess DT, Matsomoto A, Kim S-O, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 2.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 3.Hordijk PL. Regulation of NADPH oxidases: the role of Rac proteins. Circ Res. 2006;98:453–462. doi: 10.1161/01.RES.0000204727.46710.5e. [DOI] [PubMed] [Google Scholar]
- 4.Brandes RP, Kreuzer J. Vascular NADPH oxidases: molecular mechanisms of activation. Cardiovasc Res. 2005;65:16–27. doi: 10.1016/j.cardiores.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 5.Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Molec Cell. 2007;26:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 6.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 7.Gryglewski RJ, Palmer RM, Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature. 1986;320:454–456. doi: 10.1038/320454a0. [DOI] [PubMed] [Google Scholar]
- 8.Ueyama T, Geiszt M, Leo TL. Involvement of Rac1 in activation of multicomponent Nox1- and Nox3-based NADPH oxidases. Mol Cell Biol. 2006;26:2160–2174. doi: 10.1128/MCB.26.6.2160-2174.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 10.Moldovan L, Irani K, Moldovan NI, Finkel T, Goldschmidt-Clermont PJ. The actin cytoskeleton reorganization induced by Rac1 requires the production of superoxide. Antioxid Redox Signal. 1999;1:29–43. doi: 10.1089/ars.1999.1.1-29. [DOI] [PubMed] [Google Scholar]
- 11.Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci USA. 2004;101:7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O2− and systolic blood pressure in mice. Circ Res. 2001;89:408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 13.Batinic-Haberle I, Liochev SI, Spasojevic I, Fridovich I. A potent superoxide dismutase mimic: manganese β-octabromo-meso-tetrakis-(N-methylpyridinium- 4-yl) porphyrin. Arch Biochem Biophys. 1997;343:225–233. doi: 10.1006/abbi.1997.0157. [DOI] [PubMed] [Google Scholar]
- 14.Scott DB, Blanpied TA, Swanson GT, Zhang C, Ehlers MD. An NMDA receptor ER retention signal regulated by phosphorylation and aternative splicing. J Neurosci. 2001;21:3063–3072. doi: 10.1523/JNEUROSCI.21-09-03063.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Buul JD, Fernandez-Borja M, Anthony EC, Hordijk PJ. Expression and localization of NOX2 and NOX4 in primary human endothelial cells. Antioxid Redox Signal. 2005;7:308–317. doi: 10.1089/ars.2005.7.308. [DOI] [PubMed] [Google Scholar]
- 16.Jourd’heuil D. Increased nitric oxide-dependent nitrosylation of 4,5-diaminofluorescein by oxidants: implications for the measurement of intracellular nitric oxide. Free Radic Biol Med. 2002;33:676–684. doi: 10.1016/s0891-5849(02)00955-3. [DOI] [PubMed] [Google Scholar]
- 17.Infanger DW, Sharma RV, Davisson RL. NADPH oxidases of the brain: distribution, regulation, and function. Antioxid Redox Signal. 2006;8:1583–1596. doi: 10.1089/ars.2006.8.1583. [DOI] [PubMed] [Google Scholar]
- 18.Bauer PM, Fulton D, Boo YC, Sorescu GP, Kemp BE, Jo H, Sessa W. Compensatory phosphorylation and protein-protein interactions revealed by loss of function and gain of function mutants of multiple serine phosphorylation sites in endothelial nitric-oxide synthase. J Biol Chem. 2003;278:14841–14849. doi: 10.1074/jbc.M211926200. [DOI] [PubMed] [Google Scholar]
- 19.Kuncewicz T, Balakrishnan P, Snuggs MB, Kone BC. Specific association of nitric oxide synthase-2 with Rac isoforms in activated murine macrophages. Am J Physiol. 2001;281:F326–F336. doi: 10.1152/ajprenal.2001.281.2.F326. [DOI] [PubMed] [Google Scholar]
- 20.Jaffe AB, Hall A. Cell biology. Smurfing at the leading edge. Science. 2003;302:1690–1691. doi: 10.1126/science.1092874. [DOI] [PubMed] [Google Scholar]
- 21.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 22.Tzima E, Del Pozo MA, Kiosses WB, Mohamed SA, Li S, Chen S, Schwartz MA. Activation of Rac1 by shear stress in endothelial cells mediates both cytoskeletal reorganization and effects on gene expression. EMBO J. 2002;21:6791–6800. doi: 10.1093/emboj/cdf688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hwang J, Saha A, Boo YC, Sorescu GP, McNally JS, Holland SM, Dikalov S, Giddens DP, Griendling KK, Harrison DG, Jo H. Oscillatory shear stress stimulates endothelial production of O2− from p47phox-dependent NAD(P)H oxidases, leading to monocyte adhesion. J Biol Chem. 2003;278:47291–47298. doi: 10.1074/jbc.M305150200. [DOI] [PubMed] [Google Scholar]
- 24.Espey MG, Thomas DD, Miranda KM, Wink DA. Focusing of nitric oxide mediated nitrosation and oxidative nitrosylation as a consequence of reaction with superoxide. Proc Natl Acad Sci USA. 2002;99:11127–11132. doi: 10.1073/pnas.152157599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barrett DM, Black SM, Todor H, Schmidt-Ulrich RK, Dawson KS, Mikkelsen RB. Inhibition of protein-tyrosine phosphatases by mild oxidative stresses is dependent on S-nitrosylation. J Biol Chem. 2005;280:14453–14461. doi: 10.1074/jbc.M411523200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.