

Abstract
Appropriate cell-cell signaling is crucial for proper tissue homeostasis. Protein sorting of cell surface receptors at the early endosome is important for both the delivery of the signal and the inactivation of the receptor, and its alteration can cause malignancies including cancer. In a genetic screen for suppressors of the pro-apoptotic gene hid in Drosophila, we identified two alleles of vps25, a component of the ESCRT machinery required for protein sorting at the early endosome. Paradoxically, although vps25 mosaics were identified as suppressors of hid-induced apoptosis, vps25 mutant cells die. However, we provide evidence that a non-autonomous increase of Diap1 protein levels, an inhibitor of apoptosis, accounts for the suppression of hid. Furthermore, before they die, vps25 mutant clones trigger non-autonomous proliferation through a failure to downregulate Notch signaling, which activates the mitogenic JAK/STAT pathway. Hid and JNK contribute to apoptosis of vps25 mutant cells. Inhibition of cell death in vps25 clones causes dramatic overgrowth phenotypes. In addition, Hippo signaling is increased in vps25 clones, and hippo mutants block apoptosis in vps25 clones. In summary, the phenotypic analysis of vps25 mutants highlights the importance of receptor downregulation by endosomal protein sorting for appropriate tissue homeostasis, and may serve as a model for human cancer.
Keywords: Vps25, ESCRT, Protein sorting, MVB, Notch, Cell proliferation, Cell survival, Apoptosis
INTRODUCTION
The regulation of tissue homeostasis involves the concerted action of several signaling pathways. An imbalance in this fine-tuned signaling network leads to overgrowth or apoptosis, and patterning defects in developing organisms. Hence, it is very important for the organism to hold excessive signaling in check for the proper regulation of tissue homeostasis.
One way to regulate signaling is by endocytosis of ligand-bound receptors. The signal for inducing endocytosis is provided by mono-ubiquitylation mediated by specific ubiquitin ligases (Haglund and Dikic, 2005; Hicke and Dunn, 2003). Endocytosis is either required for efficient signaling by bringing the activated receptor into close proximity of intracellular signaling components (Gonzalez-Gaitan, 2003; Le Roy and Wrana, 2005; Seto and Bellen, 2004), or it is needed to turn off signaling by sorting the ubiquitylated cargo at the early endosome into vesicles that pinch off from the limiting membrane into the lumen of emerging multi-vesicular bodies (MVB) (Babst, 2005; Felder et al., 1990; Gruenberg and Stenmark, 2004; Katzmann et al., 2002; Odorizzi et al., 1998; Raiborg et al., 2003). MVBs fuse with lysosomes for degradation of the internalized proteins.
The sorting process of ubiquitylated proteins at the early endosome into MVBs requires class E Vps (Vacuolar Protein Sorting) proteins, first identified in Saccharomyces cerevisiae (Raymond et al., 1992). Mutants of class E vps genes in yeast cause the accumulation of ubiquitylated proteins on the limiting membrane of early endosomes (Katzmann et al., 2002). Eleven class E Vps proteins participate in the formation of four protein complexes: Hrs/Stam and three ESCRT (Endosomal Sorting Complex Required for Transport) protein complexes (reviewed by Babst, 2005). Hrs binds ubiquitylated receptors in early endosomes and delivers them to the ESCRT complexes, which catalyze the internalization of the ubiquitylated cargo into MVBs (Babst, 2005). This process separates the intracellular domain of activated signaling receptors from the cytosolic environment and, thus, inactivates them. vps mutants disrupt this process, causing aberrant endosomal structures (Raymond et al., 1992) in which activated receptors may continue to signal.
Because of the high conservation of class E Vps proteins, it is not surprising that these proteins have a similar function for protein sorting at the endosome in mammals (Babst, 2005). Additional functions of class E Vps proteins in mammals may include exosome secretion, virus budding, transcriptional control, cell cycle progression and apoptosis (de Gassart et al., 2004; Demirov and Freed, 2004; Kamura et al., 2001; Krempler et al., 2002; Pornillos et al., 2002; Schmidt et al., 1999; Wagner et al., 2003), indicating a broad range of Vps action for controlling tissue homeostasis. In addition, mutations of human TSG101 (Vps23p) have been linked to a number of tumors, including cervical, breast, prostate and gastrointestinal cancers (Li and Cohen, 1996; Li et al., 1997; Lin et al., 1998; Sun et al., 1997).
In Drosophila, loss of the class E vps genes hrs, erupted (the vps23 homolog encoding a component of ESCRT-I) and vps25 (a component of ESCRT-II) leads to accumulation of the cell surface receptors Notch (N), Delta (Dl), Thickveins and Egfr, consistent with a conserved role of these genes for endosomal protein sorting (Jekely and Rorth, 2003; Lloyd et al., 2002; Moberg et al., 2005; Thompson et al., 2005; Vaccari and Bilder, 2005). In the case of erupted and vps25, N accumulation stimulates the JAK/STAT pathway, which is known to induce cell proliferation in the eye disc (Chao et al., 2004; Reynolds-Kenneally and Mlodzik, 2005; Tsai and Sun, 2004), and gives rise to overgrowth phenotypes (Moberg et al., 2005; Thompson et al., 2005; Vaccari and Bilder, 2005).
Cell death in Drosophila is under the control of the pro-apoptotic genes reaper, head involution defective (hid; W – FlyBase) and grim (Cashio et al., 2005). The activation of these genes results in caspase activation, most notably Dronc (Nc – FlyBase), the Caspase-9 homolog. In living cells, Dronc is kept inactive by binding to Diap1 (Drosophila inhibitor of apoptosis protein 1; Thread – FlyBase) to prevent cell death (Bergmann et al., 2003; Meier et al., 2000). Reaper, Hid and Grim induce cell death through the binding to and stimulation of proteolytic degradation of Diap1 (Holley et al., 2002; Ryoo et al., 2002; Yoo et al., 2002). Dronc is released from Diap1 inhibition and, with the scaffolding protein Ark, forms the active apoptosome, which activates Drice (Ice – FlyBase) and Dcp-1, caspase-3-like proteins, inducing cell death.
Here, we extend the phenotypic characterization of Drosophila vps25, a component of the ESCRT-II complex. Paradoxically, although vps25 mutants were recovered as recessive suppressors of GMR-hid-induced apoptosis, vps25 mutant cells nevertheless die. Consistent with previous reports, before they die, they stimulate non-autonomous proliferation. However, non-autonomous proliferation does not account for the suppression of GMR-hid. Instead, vps25 clones appear to enhance the apoptotic resistance of adjacent tissues by increasing Diap1 protein levels in a JAK/STAT-independent manner. Furthermore, vps25 clones die through the activation of Hid and JNK, the inhibition of which causes dramatic overgrowth phenotypes in vps25 mosaics. In addition, we detect inappropriate Hippo signaling in vps25 clones, and hippo mutants block apoptosis in vps25 clones. In conclusion, our studies present a mechanistic model by which the impairment of ESCRT function induces overgrowth, and may explain tumorous phenotypes, such as those caused by mutations of TSG101 in humans.
MATERIALS AND METHODS
Isolation of vps25, ark and hippo alleles
For GheF screening (Xu et al., 2005), isogenized FRT42D P[y+] males were starved for 12 hours and then incubated with 25 mM EMS in 5% sucrose solution for 24 hours (Lewis and Bacher, 1968). After 3 hours of recovery, the males were mated to GheF; FRT42D P[w+] females and incubated at 25°C. F1 animals (32,000) were screened for suppression of GMR-hid. Two vps25, 37 ark and five hippo alleles were recovered.
Stocks
vps25K2 and vps25N55 are described in the Results. arkH16 carries a premature stop codon at residue 42, arkG8 changes Cys346 to Trp (Srivastava et al., 2006). Both alleles are strong loss-of-function alleles of ark. hippo3D has a premature stop codon at residue 185 (M.L. and A.B., unpublished). UAS-NDN, UAS-puc, UAS-upd and E(spl)m8 2.61-lacZ were provided by Georg Halder and Jennifer Childress (MD Anderson Cancer Centre, Houston), UAS-dMyc by David Stein (University of Texas at Austin), FRT42D arm-lacZ M(2) by Graeme Mardon (Baylor College of Medicine, Houston) and the MARCM stock by Hugo Bellen (Baylor College of Medicine, Houston). The GMR-hidw− transgene was isolated by mobilizing GMR-hid using Δ2-3 transposase. This transgene has lost the w+ marker, but maintains the hid ORF.
Mosaic analysis
Clones of genetically marked homozygous vps25 mutant cells were obtained by FLP/FRT mitotic recombination (Xu and Rubin, 1993), using ey-FLP or hs-FLP. In each experiment, multiple clones of 10-20 eye imaginal discs were analyzed, unless otherwise noted. The MARCM technique (Lee and Luo, 2001) was used to induce UAS-based transgenes (UAS-NDN, UAS-diap1, UAS-dMyc, UAS-puc) in vps25 clones.
Immunohistochemistry
Eye-antennal and wing imaginal discs from third instar larvae were dissected and labeled using standard procedures with antibodies against the following antigens: Hid and Diap1 (gift of Hermann Steller and Hyung-Don Ryoo, Rockefeller University, New York), Expanded (gift of Georg Halder, MD Anderson Cancer Centre, Houston), pSTAT (gift of Richard Sorrentino, MD Anderson Cancer Centre, Houston), Ubiquitin (Sigma), Notch and Delta (Developmental Studies Hybridoma Bank), BrdU (Becton Dickinson Biosciences), anti-cleaved Caspase-3 (Caspase-3*, Cell Signaling Technology), pJNK and β-Gal (Promega). Secondary antibodies were from Jackson ImmunoResearch. The in situ cell death detection kit was from Roche. All images were taken with a Zeiss AxioImager equipped with ApoTome technology.
DNA sequencing and transgenic rescue
To identify the mutations in the vps25 alleles, PCR products of genomic DNA encompassing CG14750 were sequenced. For transgenic rescue, genomic DNA containing the CG14750 transcription unit, including the flanking regions up to the neighboring genes, was cloned into the transformation vectors pCaSpeR-hs and pUAST. For each vector, two independently obtained transgenic lines rescued the phenotypes of vps25 mutants.
RESULTS
Isolation and characterization of recessive suppressors of GMR-hid
Overexpression of the pro-apoptotic gene hid under the control of the eye-specific Glass-Multimer-Reporter (GMR-hid) causes a strong eye-ablation phenotype as a result of the induction of apoptosis (Fig. 1A) (Grether et al., 1995). Using the recently described GheF (GMR-hid ey-FLP) method (Xu et al., 2005), we conducted an EMS-mutagenesis screen on chromosome arm 2R, aimed at identifying recessive suppressors of the GMR-hid eye-ablation phenotype. The GheF method takes advantage of the ey-FLP/FRT system, which induces homozygous mutant clones specifically in the eye by mitotic recombination (Newsome et al., 2000). Mutants that suppress the GMR-hid eye-ablation phenotype were selected for further analysis.
Fig. 1. Isolation and characterization of K2 and N55 alleles.
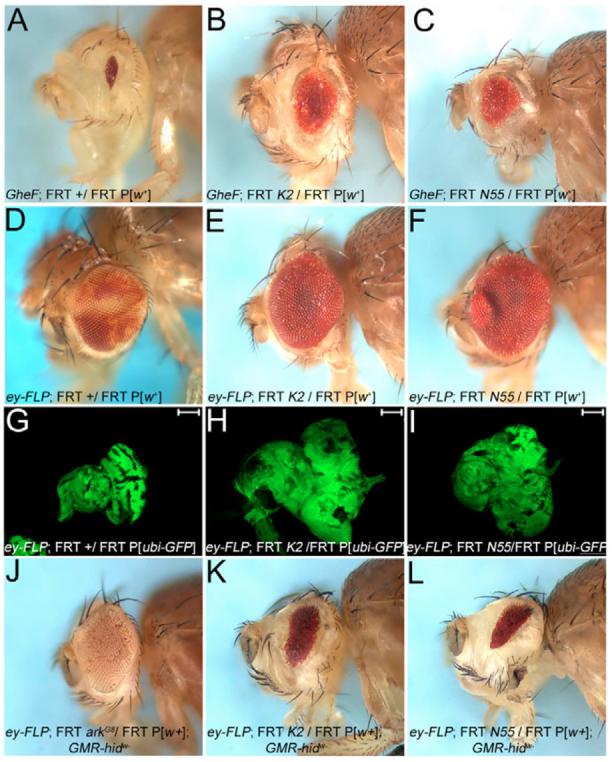
Genotypes are indicated in each panel. FRT, FRT42D. (A) The unmodified GMR-hid ey-FLP (GheF) eye-ablation phenotype. (B,C) K2 (B) and N55 (C) alleles are moderate suppressors of the GheF-eye phenotype. (D-F) Adult eyes of ey-FLP-induced mosaics of wild-type (D), and K2 (E) and N55 (F). K2 and N55 mosaic eyes lack mutant clones (marked by the absence of red pigment) and are overgrown. (G-I) Eye-antennal discs from third instar larvae of K2 (H) and N55 mosaics (I) are overgrown and disorganized compared with wild type (G). Scale bar: 100 μm. (J-L) Non-autonomous suppression of GMR-hidw− in K2 and N55 mosaics. (J) Control: in ark mosaics, largely ark mutant clones (marked by an absence of eye pigment) suppress GMR-hidw−. (K,L) In K2 (K) and N55 (L) mosaics, the suppression of GMR-hidw− is mediated by wild-type tissue (red pigment). K2 and N55 clones do not contribute.
In the GheF screen, two mutants, su(GMR-hid)K2 and su(GMR-hid)N55 (referred to as K2 and N55) were independently recovered as moderately strong recessive suppressors of the GMR-hid eye ablation phenotype (Fig. 1B,C). These mutants are homozygous lethal in trans to each other, and thus affect the same genetic function. To characterize these alleles, we induced K2 and N55 mosaics via the ey-FLP/FRT system in the eye (without simultaneous expression of GMR-hid). Surprisingly, homozygous mutant clones (marked by absence of red eye pigment; compare with Fig. 1D) were not recovered, although the twin-spots were (Fig. 1E,F), suggesting that the mosaic eyes are composed of wild-type and heterozygous tissue (marked by red eye pigment). Even more surprisingly, K2 and N55 mosaic eyes were larger than wild type (Fig. 1D-F). Thus, the mutant clones do not contribute to the adult eye tissue, but appear to be able to induce overgrowth in wild-type tissue. Analysis of third instar eye-antennal K2 and N55 mosaic discs confirms these conclusions. These discs are overgrown and disorganized when compared with wild type (Fig. 1G-I). At this stage, mutant clones are detectable (marked by absence of GFP labeling in Fig. 1G,H). However, as shown below, they are eliminated by apoptosis.
These results pose an apparent paradox. Although we have identified K2 and N55 as being recessive suppressors of GMR-hid, the mutant clones do not survive. We determined whether K2 and N55 mutant clones contribute to the suppression of GMR-hid by clonal analysis. However, the GMR-hid transgene used for GheF screening is marked with w+, and does not allow a clonal analysis based on red/white pigment selection in eyes. Thus, we generated a GMR-hid transgene that has lost w+, termed GMR-hidw−, allowing the determination of the genetic identity of the rescued tissue of GMR-hidw− in K2 and N55 mosaics. As a positive control, the strong suppression of GMR-hidw− by a mutant of ark, an essential component of the cell death pathway, is largely mediated by ark mutant tissue (marked by absence of eye pigment in Fig. 1J). By contrast, in K2 and N55 mosaics, the rescued tissue of GMR-hidw− is genetically wild type or heterozygous (marked by red pigment in Fig. 1K,L), suggesting that K2 and N55 mutant tissue does not contribute to the suppression of GMR-hidw−. Therefore, because the surviving wild-type tissue is exposed to GMR-driven hid expression, this tissue may have an increased apoptotic resistance induced by K2 and N55 mutant clones. Thus, these are unprecedented phenotypes, which merit further analysis.
K2 and N55 mutant clones die, but induce non-autonomous proliferation
To examine the K2 and N55 mutant phenotypes in more detail, we performed TUNEL and BrdU labeling as assays for apoptosis and cell proliferation, respectively, in imaginal discs of third instar larvae. TUNEL labeling was increased in N55 mutant clones (Fig. 2A-C), consistent with the observed lack of mutant clones in the adult eye (Fig. 1E,F). N55 clones can grow to a fairly large size, suggesting that they are not growth impaired (Fig. 2A-C). However, these clones are completely removed by apoptosis during pupal stages (data not shown). Similar data were obtained for K2 (data not shown).
Fig. 2. TUNEL and BrdU analysis of N55 mosaics.
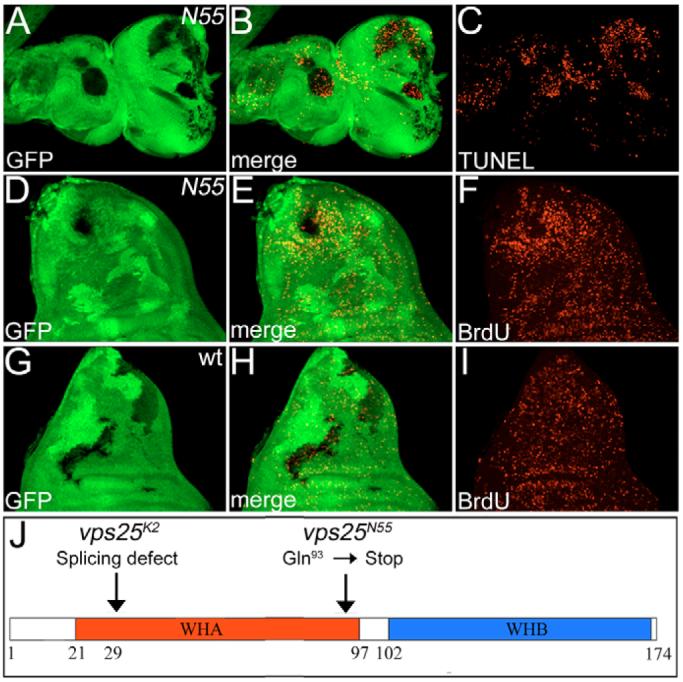
(A-C) Increased TUNEL-positive cell death in N55 mutant clones (genotype: ey-FLP; FRT42D N55/FRT42D P[ubi-GFP]) of eye-antennal imaginal discs from third instar larvae. N55 clones are marked by the absence of GFP. (D-F) BrdU labeling of N55 mosaic wing imaginal discs. Note the increased proliferation in tissue adjacent to N55 clones (hs-FLP; FRT42D N55/FRT42D P[ubi-GFP]). (G-I) BrdU labeling of wild-type (wt) clones (hs-FLP; FRT42D +/FRT42D P[ubi-GFP]) in wing imaginal discs serves as a control to D-F. BrdU incorporation is homogeneous inside and outside the clones. (J) Schematic outline of the Vps25 protein and the molecular lesions in Vps25K2 and Vps25N55. WHA and WHB are winged-helix domains A and B.
Interestingly, cell proliferation in N55 mosaics, as demonstrated by BrdU incorporation, is significantly increased in tissue adjacent to the mutant clones. This non-autonomous cell proliferation is best visible in wing imaginal discs, where N55 clones appear to be the origin for the increased proliferation of adjacent tissue (Fig. 2D-F); wing discs with wild-type clones show a homogenous distribution of proliferating cells both within and outside of the clones (Fig. G-I). Similar data were obtained in eye-antennal imaginal discs (data not shown).
In addition to the apoptotic and proliferation phenotypes, N55 mutant clones fail to differentiate. Elav (a neuronal differentiation marker) labeling demonstrates that N55 cells are unable to differentiate (data not shown).
In summary, these analyses reveal that the wild-type function of K2 and N55 is required for the appropriate control of apoptosis, cell proliferation and cell differentiation. The overgrowth phenotype in K2 and N55 mosaics (Fig. 1H,I) can most likely be explained by emission of signaling molecules from the mutant cells initiating non-autonomous proliferation in the adjacent wild-type tissue.
K2 and N55 are mutants of the Drosophila vps25 homolog
To understand the molecular cause of these phenotypes, we identified the mutant gene in K2 and N55. By P-element (Zhai et al., 2003) and deficiency mapping, K2/N55 was located to cytological region 44D4-44D5 on the polytene map. Both alleles failed to complement the lethality of a P-element-induced mutation (l(2)44Dbk08904) which is inserted in the gene CG14750. DNA sequencing of CG14750 of K2 revealed a transversion from T to A in the second base of the only intron, presumably causing a splicing defect and, subsequently, premature termination of translation by an in-frame stop codon in the intron. CG14750 of N55 carries a premature termination codon at amino acid 93 (Fig. 2J). Genomic constructs of CG14750 rescue the phenotypes associated with K2 and N55 mutants (data not shown), suggesting that K2 and N55 affect CG14750.
A BLAST search identified CG14750 as the Drosophila homolog of vps25 in S. cerevisiae. It is a member of the class E Vps proteins (Raymond et al., 1992), and a component of ESCRT-II, which functions to catalyze the feeding of ubiquitylated transmembrane receptors into intraluminal vesicles of MVBs, targeting them for degradation in lysosomes. From now on, we refer to K2 and N55 as vps25K2 and vps25N55, respectively. Drosophila vps25 encodes a protein of 174 amino acids and contains two winged helix (WH) domains, WHA and WHB (Fig. 2J). Because both WHA and WHB are essential for ESCRT-II function (Hierro et al., 2004; Teo et al., 2004), and because of the molecular lesions of vps25K2 and vps25N55 (Fig. 2J), these alleles are likely to be very strong hypomorphic alleles, if not null alleles. Recently, two papers reported the isolation of vps25 mutants in entirely different genetic screens (Thompson et al., 2005; Vaccari and Bilder, 2005). Our phenotypic characterization of vps25 is largely consistent with these studies.
Increased Notch and JAK/STAT signaling in vps25 mosaics
In yeast, vps25 mutants cause aberrant endosomal structures in which ubiquitylated proteins accumulate (Katzmann et al., 2002). In Drosophila vps25 mutant clones, similar abnormal endosomal structures have been observed and ubiquitin immunoreactivity is strongly increased (Fig. 3A-C) (Thompson et al., 2005; Vaccari and Bilder, 2005) (note that vps25 clones are positively marked by GFP using the MARCM technique). This analysis suggests that in vps25 mutant cells, ubiquitylated proteins accumulate, and are presumably not degraded.
Fig. 3. Accumulation of ubiquitin, N and Dl in vps25 clones, and increased pSTAT immunoreactivity adjacent to vps25 clones.
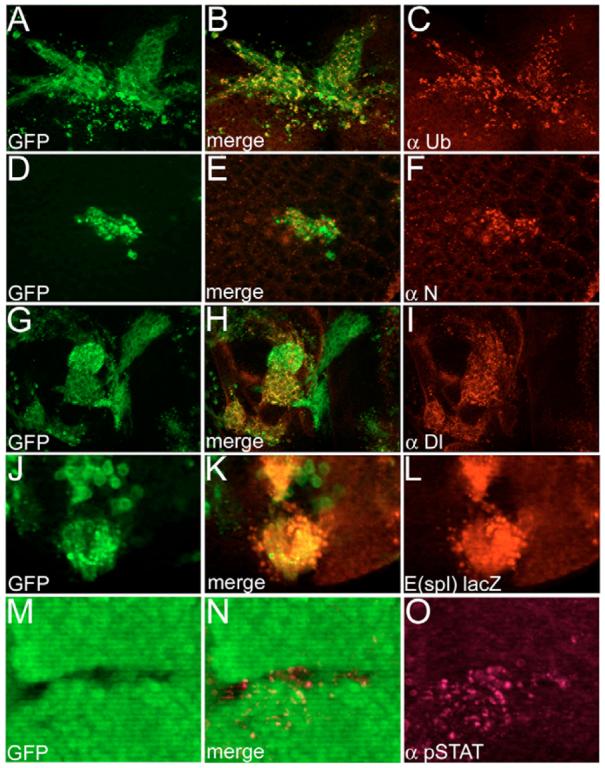
(A-C) Accumulation of ubiquitin in vps25 clones (genotype: hs-FLP UAS-GFP; FRT42D vps25N55/FRT42D tub-Gal80; tub-Gal4). Note vps25 clones are positively marked with GFP using the MARCM technique (Lee and Luo, 2001). (D-F) Accumulation of N in GFP-marked vps25 clones (genotype as in A-C). (G-I) Accumulation of Dl in GFP-marked vps25 clones (genotype as in A-C). (J-L) Increased N activity in GFP-marked vps25 clones (hs-FLP UAS-GFP; E[spl]m8 2.61-lacZ FRT42D vps25N55/FRT42D tub-Gal80; tub-Gal4). (M-O) Increased STAT activity inside and outside of vps25 clones (ey-FLP; FRT42D vps25N55/FRT42D P[ubi-GFP]) by anti-pSTAT labeling. Clones are marked by the absence of GFP.
Cell surface receptors are able to signal after endocytosis as long as they are not incorporated into MVBs (Gonzalez-Gaitan, 2003; Seto and Bellen, 2004), because their intracellular domains are still exposed to the cytosol in the early endosome. Because vps25 mutants are likely to impair MVB function, these receptors may still continue to signal. Thus, the proliferation phenotype of vps25 mosaics may be explained by continued signaling activity. Consistently, N protein and its ligand Dl accumulate in vps25 clones (Fig. 3D-I), resulting in increased N activity as shown by the N reporter E(spl)m8 2.61-lacZ (Fig. 3J-L) (Thompson et al., 2005; Vaccari and Bilder, 2005). However, not all known target genes of N are upregulated in vps25 clones. The expression of cut, another N target, is unaltered in vps25 mutant clones in wing discs (data not shown). N is able to promote global growth in the eye by inducing unpaired (upd; os – FlyBase) expression (Chao et al., 2004; Reynolds-Kenneally and Mlodzik, 2005; Tsai and Sun, 2004). Upd is a ligand of Domeless, the receptor of the JAK/STAT signaling pathway (Brown et al., 2001). Consistently, non-autonomous STAT activity is stimulated outside of vps25 mutant clones in eye discs (Fig. 3M-O). In summary, these data link N activation with the mitogenic activity of the JAK/STAT pathway, and this may be the cause of non-autonomous proliferation in vps25 mosaics.
N is required for non-autonomous proliferation in vps25 mosaics
To determine a genetic requirement of N signaling for non-autonomous proliferation in vps25 mosaics, we expressed a dominant-negative N (NDN) transgene (Sen et al., 2003) in vps25 clones (referred to as vps25/NDN) using MARCM (Lee and Luo, 2001). STAT activity in vps25/NDN eye mosaics was strongly reduced or absent compared with in vps25 clones (Fig. 4A-C). Furthermore, BrdU-positive cell proliferation was not significantly increased in vps25/NDN mosaics (Fig. 4D-F). Consistently, eye imaginal discs obtained from vps25/NDN mosaics are normal in shape and size (data not shown). These observations suggest that the increased N activity in vps25 clones accounts for the non-autonomous proliferation phenotype of vps25 mosaics through activation of the JAK/STAT pathway. A similar conclusion was obtained by analyzing vps25 mosaics in a heterozygous Stat92E mutant background (Vaccari and Bilder, 2005). Interestingly, Dl protein does not accumulate in vps25/NDN clones (Fig. 4G-I). This observation suggests that N controls Dl protein levels in vps25 clones.
Fig. 4. N is required for non-autonomous proliferation of vps25 mosaics, but not for apoptosis.
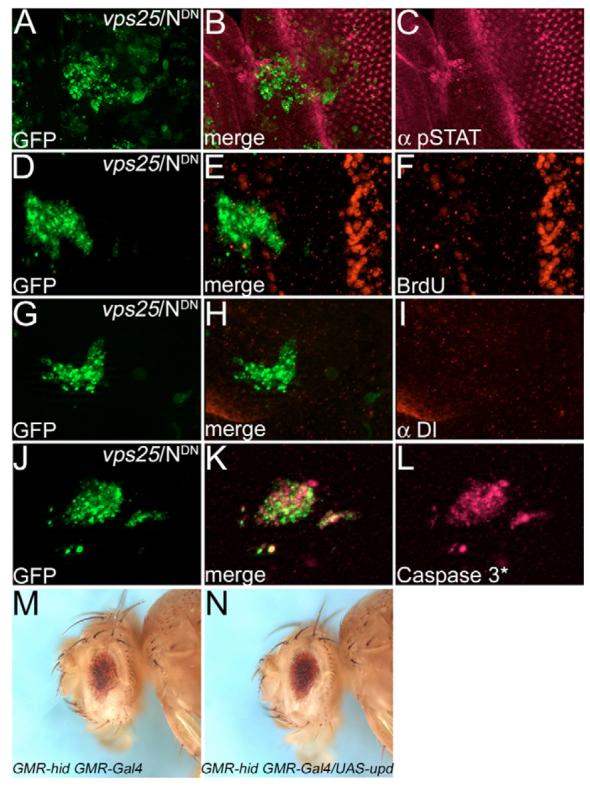
All vps25 clones expressing NDN (vps25/NDN) were labeled with GFP using MARCM. (A-I) vps25/NDN clones have reduced levels of pSTAT (A-C), non-autonomous cell proliferation (BrdU labeling in D-F), and Dl (G-I). The strong BrdU signal in E,F is due to the second mitotic wave. (J-L) Caspase-3*-positive apoptosis is unchanged in vps25/NDN clones. Genotype in A-L: hs-FLP UAS-GFP; FRT42D vps25N55 UAS-NDN/FRT42D tub-Gal80; tub-Gal4. (M) The unmodified GMR-hid GMR-Gal4 eye-ablation phenotype. (N) Overexpression of upd is not sufficient to suppress GMR-hid GMR-Gal4. Genotype: GMR-hid GMR-Gal4/UAS-upd.
We determined whether non-autonomous proliferation mediated by Upd and JAK/STAT signaling is sufficient for the suppression of GMR-hid, as observed for vps25 mosaics (Fig. 1B,C). However, although overexpression of upd in the fly eye gives rise to enlarged eyes (Muller et al., 2005), it is not sufficient for suppression of GMR-hid (Fig. 4M,N). Thus, the suppression of GMR-hid in vps25 mosaics is not caused by non-autonomous proliferation through Upd signaling. Another mechanism may account for the observed suppression (see below).
N signaling has also been implicated in inducing cell death in eye imaginal discs (Miller and Cagan, 1998). Thus, we tested whether increased N signaling accounts for the cell death phenotype of vps25 clones. However, vps25/NDN clones labeled with activated caspase-3 (Caspase-3*) antibody were indistinguishable from vps25 clones (Fig. 4J-L). Similar results were obtained by TUNEL labeling (data not shown). Thus, although N induces non-autonomous proliferation in vps25 mosaics, it is not responsible for the apoptotic phenotype of vps25 clones.
We also tested the possibility that the activation of cell death might activate N signaling, and thus induce compensatory proliferation. To address this issue, we blocked cell death by the expression of diap1 in vps25 mutant clones (see below). However, pSTAT activity and cell proliferation was still evident under these conditions (data not shown), establishing that the activation of the N pathway and the induction of cell death in vps25 clones are independent of each other.
Non-autonomous survival through upregulation of Diap1 protein
Because N signaling does not induce cell death in vps25 clones, we analyzed the underlying cause of the apoptotic phenotype. vps25 clones contain increased protein levels of the cell death inducer Hid (Fig. 5A-C). Hid, as well as Reaper and Grim, induce apoptosis by stimulating ubiquitin-mediated degradation of Diap1, an inhibitor of the caspase Dronc (Holley et al., 2002; Meier et al., 2000; Ryoo et al., 2002; Yoo et al., 2002). Indeed, Diap1 protein levels were markedly reduced in vps25 mutant clones (Fig. 5D-F), suggesting that Diap1 no longer inhibits Dronc.
Fig. 5. Non-autonomous increase of Diap1 protein levels, and evidence of two cell death pathways.
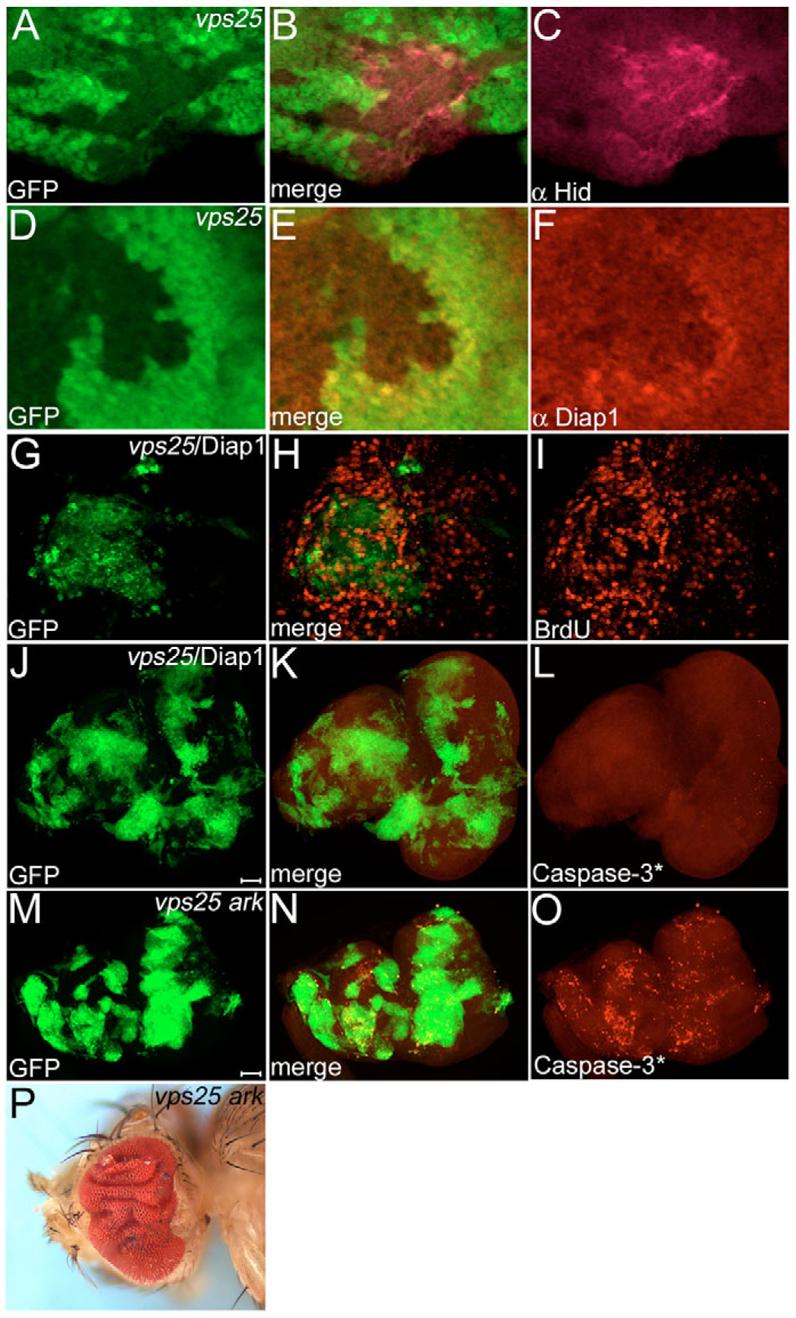
(A-C) Increased levels of Hid protein in vps25 clones (genotype: ey-FLP; FRT42D vps25N55/FRT42D P[ubi-GFP]) of third instar eye discs. Clones are marked by the absence of GFP. (D-F) Diap1 protein levels are reduced in, and increased adjacent to, vps25 clones (genotype as in A-C). (G-I) BrdU labeling of GFP-marked vps25/Diap1 mosaics (hs-FLP UAS-GFP/UAS-Diap1; FRT42D vps25N55/FRT42D tub-Gal80; tub-Gal4). Overexpression of Diap1 does not block non-autonomous proliferation. (J-L) Caspase-3*-labeling of GFP-marked vps25N55/Diap1 mosaics. Caspase-3* is completely blocked. Genotype as in G-I. Scale bar: 100 μm. (M-O) Caspase-3*-labeling of GFP-marked vps25 ark double mutant mosaics. Caspase-3*-dependent cell death is detectable in vps25 ark clones (hs-FLP UAS-GFP; FRT42D vps25N55 arkH16/FRT42D tub-Gal80; tub-Gal4). Scale bar: 100 μm. (P) Adult eyes of ey-FLP-induced mosaics of vps25 ark double mutants are severely overgrown and folded. Note that vps25 ark clones (ey-FLP; FRT42D vps25N55 arkH16/FRT42D P[w+]) are absent (marked by lack of red pigment).
Strikingly, however, Diap1 immunoreactivity is increased in wild-type cells immediately abutting vps25 clones (Fig. 5D-F), suggesting that vps25 clones also promote non-autonomous cell survival. GMR-hid is sensitive to altered levels of Diap1 (Hay et al., 1995). Thus, the non-autonomous increase of Diap1 protein is likely to promote the suppression of GMR-hid in vps25 mosaics (Fig. 1B,C). This activity is independent of Upd signaling because overexpression of upd does not alter Diap1 protein levels (data not shown) and does not suppress GMR-hid (Fig. 4N). It is currently not known which signaling mechanism causes non-autonomous survival by regulating Diap1 protein levels.
Blocking cell death induces massive overgrowth of vps25 mosaics
It has recently been demonstrated that dying cells are able to induce compensatory proliferation in neighboring cells (Huh et al., 2004; Perez-Garijo et al., 2004; Ryoo et al., 2004). Thus, we tested whether compensatory proliferation contributes to non-autonomous proliferation in vps25 mosaics. If it does, then the inhibition of apoptosis either through the expression of Diap1 in vps25 clones (vps25/Diap1) or in vps25 ark double mutants (using an ark null allele, see Materials and methods) is expected to reduce proliferation and subsequently to suppress the overgrowth phenotype of vps25 mosaics. However, non-autonomous proliferation is still observed in vps25/Diap1 mosaics (Fig. 5G-I) and in vps25 ark mosaics (data not shown), suggesting that compensatory proliferation does not contribute significantly to the non-autonomous proliferation of vps25 mosaics. By contrast, eye-antennal discs of vps25/Diap1 mosaics are extremely overgrown and can be five times as large as wild-type discs (Fig. 5J). In addition, vps25/Diap1 and vps25 ark clones occupy a large fraction of the eye disc (Fig. 5J-O), suggesting that vps25 clones have no intrinsic growth disadvantage over wild-type tissue if cell death is blocked. The adult eye of vps25 ark mosaics is severely overgrown and folded (Fig. 5P). Thus, inhibiting cell death in vps25 clones gives rise to an even stronger overgrowth phenotype, as has also been observed following expression of the caspase inhibitor P35 (Thompson et al., 2005).
Hid and JNK contribute to the elimination of vps25 mutant clones
Caspase-3* labeling reveals that cell death is completely blocked in vps25/Diap1 clones (Fig. 5J-L). Surprisingly, Caspase-3* activity is still detectable in vps25 ark double mutant clones (Fig. 5M-O), suggesting that although ark, an essential component of the cell death pathway, is mutant, vps25 ark double mutant cells still die. This is also confirmed by the observation that vps25 ark clones (marked by absence of eye pigment) cannot be recovered in adult eyes of vps25 ark mosaics (Fig. 5P). Diap1 inhibits both initiator (Dronc) and Caspase-3-like caspases (Drice), whereas Ark directly only activates Dronc (Cashio et al., 2005). Thus, an alternative cell death pathway is operating in vps25 clones that can induce caspase-3-like activity independently of Ark.
We considered Jun N-terminal Kinase (JNK, encoded by basket in Drosophila), signaling as a candidate for the alternative cell death pathway. JNK activation occurs under stress conditions, and can induce apoptosis (Adachi-Yamada et al., 1999; Adachi-Yamada and O'Connor, 2002). We found increased levels of activated JNK in vps25 clones (Fig. 6A-C). It was previously shown that inactivation of Diap1 can induce JNK activation (Kuranaga et al., 2002; Ryoo et al., 2004). Thus, we tested whether this applies to vps25 clones as well. However, JNK activity is not appreciably altered in vps25/Diap1 clones (Fig. 6D-F), excluding the possibility that JNK activation occurs as a result of Diap1 inactivation. To determine a requirement of JNK for the apoptotic phenotype, we inhibited JNK in vps25 clones by overexpressing Puckered (vps25/Puc), a phosphatase that dephosphorylates JNK. However, Caspase-3* activity is still detectable in vps25/Puc clones (Fig. G-I). This caspase activity may be derived from Hid activity, as hid is expressed in vps25/Puc clones (data not shown). Thus, we expressed Puc in vps25 ark double mutant clones. In vps25 ark/Puc clones, Caspase-3* activity is mostly blocked (Fig. 6J-L), and vps25 ark/Puc mosaic discs are severely overgrown (Fig. 6J), similar to vps25/Diap1 eye discs (Fig. 5J). Taken together, these observations implicate Hid/Diap1/Dronc/Ark and JNK signaling as being contributing factors to the apoptotic phenotype of vps25 mutant clones. Interestingly, we still detect Caspase-3* activity at the clonal margin of vps25 ark/Puc clones, suggesting that a third cell death pathway may operate in vps25 clones. Alternatively, JNK inhibition by Puc may be incomplete.
Fig. 6. JNK contributes to the apoptotic phenotype of vps25 clones.
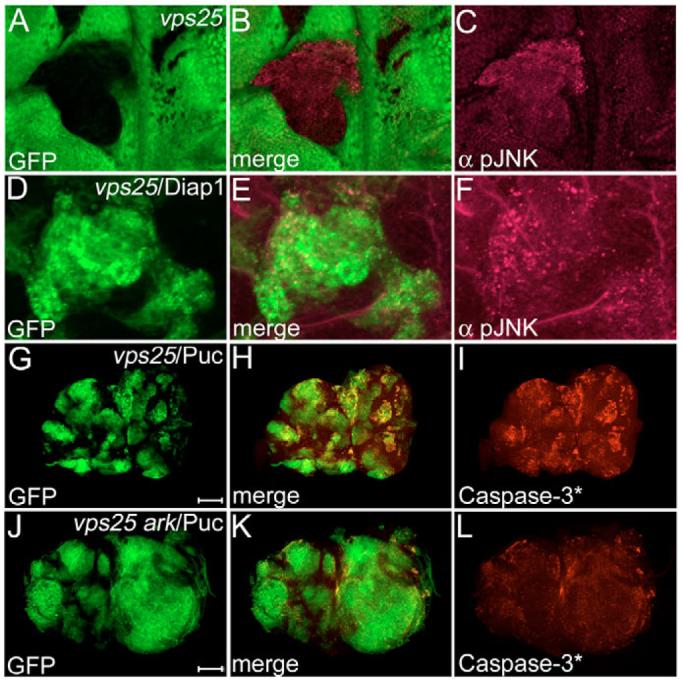
(A-C) Increased JNK activity in vps25 clones (genotype: ey-FLP; FRT42D vps25N55/FRT42D P[ubi-GFP]) shown by anti-pJNK labeling. Clones are marked by absence of GFP. (D-F) pJNK labeling of GFP-marked vps25/Diap1 mosaics (hs-FLP UAS-GFP/UAS-Diap1; FRT42D vps25N55/FRT42D tub-Gal80; tub-Gal4). (G-I) Caspase-3*-labeling of GFP-marked vps25/Puc mosaics (hs-FLP UAS-GFP; FRT42D vps25N55/FRT42D tub-Gal80; tub-Gal4/UAS-puc). Scale bar: 100 μm. (J-L) Caspase-3*-labeling of GFP-marked vps25 ark/Puc mosaics (hs-FLP UAS-GFP; FRT42D vps25N55 arkG8/FRT42D tub-Gal80; tub-Gal4/UAS- puc). Note, the clones marked with GFP are severely enlarged. Scale bar: 100 μm.
Hippo signaling, but not cell competition, controls apoptosis in vps25 clones
Which process controls the apoptotic phenotype of vps25 mutants? One possibility is cell competition. Cell competition was originally described in studies using Minute (M) mutations, in which faster growing cells (M+/M+) outcompete neighboring slow-growing cells (M–/M+) by inducing apoptosis (reviewed by Abrams, 2002). Thus, we analyzed vps25 clones in a M background. However, although vps25 clones are larger in a M background than in a wild-type background (Thompson et al., 2005), they are still Caspase-3* positive and undergo apoptosis (Fig. 7A-C). In addition, it was recently shown that Drosophila Myc (Dm – FlyBase) protein levels are crucial for cell competition (de la Cova et al., 2004; Moreno and Basler, 2004). An imbalance of Drosophila Myc protein levels between neighboring cells induces cell competition, outcompeting cells with lower Myc levels by apoptosis. However, expression of Drosophila Myc in vps25 clones (vps25/dMyc) does not significantly change Caspase-3* activity (Fig. 7D-F). These data illustrate that cell competition is not an important contributor for cell death in vps25 clones.
Fig. 7. Increased Hippo signaling, but not cell competition, controls apoptosis in vps25 clones.
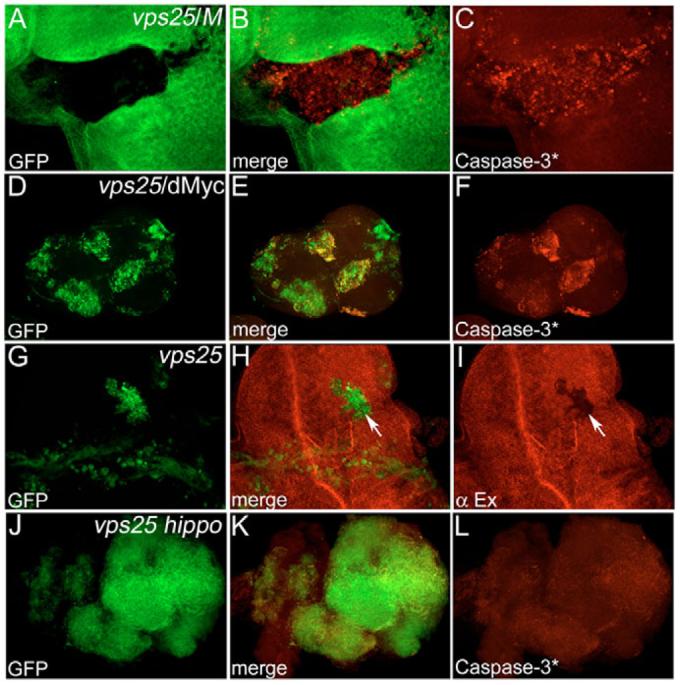
(A-C) Caspase-3*-labeling of vps25 mosaics (genotype: ey-FLP; FRT42D vps25N55/FRT42D arm-lacZ M(2)) in a Minute (M) background. Caspase-3* activity is unaffected. Clones are marked by the absence of β-Gal staining. (D-F) Caspase-3*-labeling of GFP-marked vps25/dMyc mosaics (hs-FLP UAS-GFP; FRT42 vps25N55/FRT42D tub-Gal80; tub-Gal4/UAS-dMyc). Caspase-3*-activity is unaffected. (G-I) Expanded (Ex) labeling of GFP-marked vps25 mosaics (hs-FLP UAS-GFP; FRT42D vps25N55/FRT42D tub-Gal80; tub- Gal4). Ex protein levels are reduced in vps25 clones (arrow), indicating increased Hippo activity. (J-L) Caspase-3*-labeling of GFP-marked vps25 hippo double mutant mosaics (hs-FLP UAS-GFP; FRT42D vps25N55 hippo3D/FRT42D tub-Gal80; tub-Gal4). Caspase-3* activity is blocked.
In recent years, the Hippo signaling pathway has emerged as an important regulator of tissue growth by controlling cell proliferation and apoptosis (Edgar, 2006; Harvey et al., 2003; Pantalacci et al., 2003; Udan et al., 2003; Vidal and Cagan, 2006; Wu et al., 2003). Thus, we tested whether Hippo activity is altered in vps25 clones. Expanded (Ex) is a useful marker for Hippo activity, and is inversely correlated with Hippo activity such that low Ex levels are indicative of high Hippo activity (Hamaratoglu et al., 2006). Ex protein levels are low in vps25 clones (Fig. 7G-I, arrow), indicating that they contain high Hippo activity. Interestingly, in vps25 hippo double mutants, Caspase-3* is almost completely blocked (Fig. 7J-L), suggesting that Hippo signaling either directly or indirectly controls apoptosis in vps25 mutant cells. The cause for increased Hippo signaling in vps25 clones is unknown. It is possible that a receptor that controls Hippo activity is deregulated at the vps25 endosome (see Discussion).
DISCUSSION
The inactivation of signaling pathways is as important for appropriate tissue homeostasis as its activation. Interference with the inactivation process often gives rise to malignant phenotypes, including cancer. Several strategies to restrict signaling exist, including receptor sequestration, receptor inactivation, production of inhibitory signaling proteins and inactivation of intracellular signaling proteins. The phenotypic analysis of vps25 mutants highlights the importance of receptor downregulation by endosomal protein sorting. Lack of vps25 function causes at least three phenotypes: non-autonomous proliferation, non-autonomous resistance to cell death and autonomous apoptosis. The cause of these phenotypes and the potential role of class E Vps proteins for tumorigenesis will be discussed.
Non-autonomous proliferation by Notch signaling in vps25 mosaics
Vps25 is a component of the ESCRT-II complex required for internalization of cell surface receptors into MVBs at the early endosome (Babst, 2005). The signal for protein sorting into MVBs is provided by mono-ubiquitylation (Haglund and Dikic, 2005; Hicke and Dunn, 2003). In yeast, vps25 mutants cause aberrant endosomal structures and the accumulation of ubiquitylated proteins (Katzmann et al., 2002). We, and others (Thompson et al., 2005; Vaccari and Bilder, 2005), have observed a similar phenotype in vps25 clones in Drosophila, suggesting the conserved function of vps25.
The lack of appropriate protein sorting at early endosomes in vps25 clones causes the accumulation of cell surface receptors including N and Dl. Our genetic analysis using a dominant-negative N transgene (NDN; Fig. 4) suggests that the strong overgrowth phenotype of vps25 mosaics is largely due to inappropriate N signaling, which is known to induce proliferation non-autonomously through activation of the JAK/STAT pathway (Chao et al., 2004; Harrison et al., 1998; Reynolds-Kenneally and Mlodzik, 2005; Tsai and Sun, 2004).
It is unclear whether N exerts this function in a ligand-dependent manner. Dl protein also accumulates in vps25 clones, and endocytosis of Dl is required for N activation (Le Borgne et al., 2005). Thus, blocking MVB formation in vps25 clones may lead to the accumulation of active Dl, resulting in increased N activity. However, we also show that N is required for Dl accumulation in vps25 clones (Fig. 4I). Therefore, Dl accumulation is either directly or indirectly the result of increased N activity in vps25 clones. This conclusion infers that N activation occurs before Dl accumulation and would argue in favor of a ligand-independent mechanism for N activation in vps25 clones, although Dl may be required for maintaining N activity. N activity is also controlled by several proteolytic cleavages (Le Borgne et al., 2005), which lead to translocation of the intracellular domain of N to the nucleus where it regulates the expression of target genes. Thus, a potential ligand- independent mode of N activation may include inappropriate cleavage of N at the vps25 endosome. Further studies are needed to clarify this point.
Mutations in erupted, the vps23 homolog that encodes a component of ESCRT-I, give rise to similar phenotypes to those observed for vps25 (Moberg et al., 2005; Thompson et al., 2005; Vaccari and Bilder, 2005) (this study). However, in hrs mosaics in Drosophila, non-autonomous cell proliferation has not been observed, although signaling receptors including N and Dl accumulate in hrs clones (Jekely and Rorth, 2003; Lloyd et al., 2002). This is a puzzling observation as hrs encodes a class E Vps protein acting immediately upstream of the ESCRT complexes. It is possible that N and Dl are not in an environment in the hrs endosome that permits signaling. Alternatively, Jekely and Rorth (Jekely and Rorth, 2003) showed that hrs controls the steady-state levels of non-activated receptors at the plasma membrane. Although this function may apply to vps25, it may also indicate that there are inherent differences between the different class E proteins regarding protein sorting at the early endosome.
Suppression of GMR-hid by a non-autonomous increase of Diap1
Paradoxically, although vps25 clones die by apoptosis, we identified the vps25 alleles as being recessive suppressors of GMR-hid-induced cell death. Our analysis demonstrates that the wild-type tissue accounts for this suppression even though these cells are exposed to GMR-hid. Our initial explanation for this observation was that non-autonomous proliferation mediated by JAK/STAT signaling in vps25 mosaics overrides the apoptotic activity of GMR-hid. However, overexpression of Upd, the ligand of the JAK/STAT pathway, does not significantly suppress GMR-hid, although GMR-upd flies have a similar overgrowth phenotype to vps25 mosaics (Muller et al., 2005). This finding excludes non-autonomous proliferation for the suppression of GMR-hid by vps25. However, Diap1 protein levels are increased in tissue abutting vps25 clones (Fig. 5D-F). GMR-hid is sensitive to altered levels of Diap1 (Hay et al., 1995), suggesting that the increase of Diap1 outside of vps25 clones may account for the suppression of GMR-hid. Thus, in addition to non-autonomous proliferation, vps25 clones also increase the apoptotic resistance of adjacent wild-type tissue in a non-autonomous manner. The signaling pathway that can induce non-autonomous survival by increasing Diap1 protein levels is currently unknown.
Cell death in vps25 clones
Our data suggests that apoptosis in vps25 mutant tissue is not only executed via the Hid/Diap1/Dronc/Ark pathway. vps25 ark clones still died, suggesting that in addition to Ark at least one other cell death pathway is activated in vps25 clones. We have shown previously that a Dronc/Ark-independent cell death pathway exists in Drosophila, but we did not identify this pathway (Srivastava et al., 2006; Xu et al., 2005). Our data here implicate JNK as potential mediator of the alternative cell death pathway (Adachi-Yamada et al., 1999; Adachi-Yamada and O'Connor, 2002). vps25 ark/Puc mosaic eye discs are extremely overgrown and the clones occupy a large area of the disc. Caspase-3*-dependent apoptosis is blocked in these clones. Only at the clonal boundaries is Caspase-3* activity still detectable, suggesting that at the interface between vps25 clones and wild-type tissue a third potential apoptotic pathway is activated.
Our data show that cell competition is not sufficient to induce cell death in vps25 clones. By contrast, given the extremely large size of cell death-inhibited vps25 clones (Fig. 6J), it appears that vps25 clones have no intrinsic growth disadvantage, and have the capability to overgrow and outcompete the surrounding wild-type tissue if cell death is blocked. Thus, cell competition does not contribute significantly to the apoptotic phenotype of vps25 clones.
We show that Hippo signaling is increased in vps25 clones (reviewed by Edgar, 2006; Vidal and Cagan, 2006). Hippo signaling can induce cell death, and, consistently, hippo mutants block cell death in vps25 clones. It is unknown how Hippo signaling is activated in vps25 clones. However, in analogy to N, a putative receptor that controls Hippo signaling may be deregulated in vps25 clones and triggers Hippo signaling. This receptor is currently unknown, but has been postulated previously (Hamaratoglu et al., 2006). However, it should be pointed out that ESCRT components have additional functions outside of MVB protein sorting. Certain ESCRT-II members have been shown to bind to the transcriptional elongation factor ELL in order to derepress transcription by RNA polymerase II (Kamura et al., 2001; Schmidt et al., 1999). Thus, in the absence of Vps25, transcriptional control of components of the Hippo pathway may be deregulated and contribute to cell death.
In summary, our data suggest that impaired ESCRT function leads to the accumulation of N and Dl, and possibly of a receptor controlling the Hippo pathway. These receptors control non-autonomous proliferation and autonomous apoptosis, respectively. In addition, we postulate a signaling pathway that induces non-autonomous cell survival by controlling Diap1 protein levels. Further characterization of the vps25 mutant phenotype may help to identify the postulated receptor of the Hippo pathway and the cell survival signaling pathway.
vps25: a model for human cancer?
Human ESCRT components, most notably TSG101 (Vps23p), have been implicated in tumor suppression. NIH3T3 cells, depleted of Tsg101 by an antisense approach, formed colonies on soft agar and produced metastatic tumors in nude mice (Li and Cohen, 1996). However, the conditional Tsg101 knockout in mouse mammary glands did not cause the formation of tumors over a period of two years, making a role of TSG101 as tumor suppressor controversial (Wagner et al., 2003). However, Tsg101 mutant cells are very sensitive to apoptotic death (Wagner et al., 2003), implying that they die before they become harmful to the organism.
The phenotypical characterization of vps25 mutants in Drosophila provides an explanation for the failure to confirm TSG101 as tumor suppressor. vps25 clones need to survive over extended periods of time in order to sustain growth. Even though they induce non-autonomous proliferation, after they have died, N signaling is turned off and proliferation stops. Furthermore, the size of the adult eye of vps25 mosaics is only slightly increased when compared with wild type, and does not match the strong overgrowth phenotype of larval imaginal discs, which can be twice as large as wild-type discs (Fig. 1G-I). Thus, as long as vps25 clones are not resistant to their own apoptotic death, tissue repair during pupal stages may partially regress the size of the imaginal disc back to almost normal. Instead, it appears that inhibition of cell death is the triggering event for a tumorous phenotype of vps25 clones. vps25/Diap1 and vps25 ark/Puc clones can make up a large fraction of the tissue of imaginal discs, and the entire discs can be five times as large as wild-type discs.
Tumorigenesis requires multiple genetic alterations that transform normal cells progressively into malignant cancer cells (Hanahan and Weinberg, 2000). Thus, additional genetic ‘hits’ may be necessary to inhibit apoptosis of Tsg101 mutant cells, which may then be able to induce a similar growth phenotype to that observed for vps25. Thus, although a tumor suppressor function for Tsg101 was not confirmed in a mouse model, it still is possible that Tsg101 and other mammalian ESCRT members have tumor suppressor properties.
Acknowledgments
We thank Georg Halder and Jennifer Childress for advice and reagents; David Stein, Hermann Steller, Hyung-Don Ryoo, Hugo Bellen, Graeme Mardon, David Bilder, Richard Sorrentino and the Bloomington stock center for fly stocks and antibodies; and the MD Anderson DNA Analysis Core Facility for sequencing (supported by Core Grant #CA16672 from the NCI). A.B. is a fellow of the MD Anderson Research Trust. This work was supported by grants from the NIH (GM068016) and the Robert A. Welch Foundation (G1496) to A.B.
References
- Abrams JM. Competition and compensation: coupled to death in development and cancer. Cell. 2002;110:403–406. doi: 10.1016/s0092-8674(02)00904-2. [DOI] [PubMed] [Google Scholar]
- Adachi-Yamada T, O'Connor MB. Morphogenetic apoptosis: a mechanism for correcting discontinuities in morphogen gradients. Dev. Biol. 2002;251:74–90. doi: 10.1006/dbio.2002.0821. [DOI] [PubMed] [Google Scholar]
- Adachi-Yamada T, Fujimura-Kamada K, Nishida Y, Matsumoto K. Distortion of proximodistal information causes JNK-dependent apoptosis in Drosophila wing. Nature. 1999;400:166–169. doi: 10.1038/22112. [DOI] [PubMed] [Google Scholar]
- Babst M. A protein's final ESCRT. Traffic. 2005;6:2–9. doi: 10.1111/j.1600-0854.2004.00246.x. [DOI] [PubMed] [Google Scholar]
- Bergmann A, Yang AY, Srivastava M. Regulators of IAP function: coming to grips with the grim reaper. Curr. Opin. Cell Biol. 2003;15:717–724. doi: 10.1016/j.ceb.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Brown S, Hu N, Hombria JC. Identification of the first invertebrate interleukin JAK/STAT receptor, the Drosophila gene domeless. Curr. Biol. 2001;11:1700–1705. doi: 10.1016/s0960-9822(01)00524-3. [DOI] [PubMed] [Google Scholar]
- Cashio P, Lee TV, Bergmann A. Genetic control of programmed cell death in Drosophila melanogaster. Semin. Cell Dev. Biol. 2005;16:225–235. doi: 10.1016/j.semcdb.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Chao JL, Tsai YC, Chiu SJ, Sun YH. Localized Notch signal acts through eyg and upd to promote global growth in Drosophila eye. Development. 2004;131:3839–3847. doi: 10.1242/dev.01258. [DOI] [PubMed] [Google Scholar]
- de Gassart A, Geminard C, Hoekstra D, Vidal M. Exosome secretion: the art of reutilizing nonrecycled proteins? Traffic. 2004;5:896–903. doi: 10.1111/j.1600-0854.2004.00223.x. [DOI] [PubMed] [Google Scholar]
- de la Cova C, Abril M, Bellosta P, Gallant P, Johnston LA. Drosophila myc regulates organ size by inducing cell competition. Cell. 2004;117:107–116. doi: 10.1016/s0092-8674(04)00214-4. [DOI] [PubMed] [Google Scholar]
- Demirov DG, Freed EO. Retrovirus budding. Virus Res. 2004;106:87–102. doi: 10.1016/j.virusres.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Edgar BA. From cell structure to transcription: hippo forges a new path. Cell. 2006;124:267–273. doi: 10.1016/j.cell.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Felder S, Miller K, Moehren G, Ullrich A, Schlessinger J, Hopkins CR. Kinase activity controls the sorting of the epidermal growth factor receptor within the multivesicular body. Cell. 1990;61:623–634. doi: 10.1016/0092-8674(90)90474-s. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Gaitan M. Signal dispersal and transduction through the endocytic pathway. Nat. Rev. Mol. Cell Biol. 2003;4:213–224. doi: 10.1038/nrm1053. [DOI] [PubMed] [Google Scholar]
- Grether ME, Abrams JM, Agapite J, White K, Steller H. The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev. 1995;9:1694–1708. doi: 10.1101/gad.9.14.1694. [DOI] [PubMed] [Google Scholar]
- Gruenberg J, Stenmark H. The biogenesis of multivesicular endosomes. Nat. Rev. Mol. Cell Biol. 2004;5:317–323. doi: 10.1038/nrm1360. [DOI] [PubMed] [Google Scholar]
- Haglund K, Dikic I. Ubiquitylation and cell signaling. EMBO J. 2005;24:3353–3359. doi: 10.1038/sj.emboj.7600808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaratoglu F, Willecke M, Kango-Singh M, Nolo R, Hyun E, Tao C, Jafar-Nejad H, Halder G. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat. Cell Biol. 2006;8:27–36. doi: 10.1038/ncb1339. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Harrison DA, McCoon PE, Binari R, Gilman M, Perrimon N. Drosophila unpaired encodes a secreted protein that activates the JAK signaling pathway. Genes Dev. 1998;12:3252–3263. doi: 10.1101/gad.12.20.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey KF, Pfleger CM, Hariharan IK. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell. 2003;114:457–467. doi: 10.1016/s0092-8674(03)00557-9. [DOI] [PubMed] [Google Scholar]
- Hay BA, Wassarman DA, Rubin GM. Drosophila homologs of baculovirus inhibitor of apoptosis proteins function to block cell death. Cell. 1995;83:1253–1262. doi: 10.1016/0092-8674(95)90150-7. [DOI] [PubMed] [Google Scholar]
- Hicke L, Dunn R. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu. Rev. Cell Dev. Biol. 2003;19:141–172. doi: 10.1146/annurev.cellbio.19.110701.154617. [DOI] [PubMed] [Google Scholar]
- Hierro A, Sun J, Rusnak AS, Kim J, Prag G, Emr SD, Hurley JH. Structure of the ESCRT-II endosomal trafficking complex. Nature. 2004;431:221–225. doi: 10.1038/nature02914. [DOI] [PubMed] [Google Scholar]
- Holley CL, Olson MR, Colon-Ramos DA, Kornbluth S. Reaper eliminates IAP proteins through stimulated IAP degradation and generalized translational inhibition. Nat. Cell Biol. 2002;4:439–444. doi: 10.1038/ncb798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh JR, Guo M, Hay BA. Compensatory proliferation induced by cell death in the Drosophila wing disc requires activity of the apical cell death caspase Dronc in a nonapoptotic role. Curr. Biol. 2004;14:1262–1266. doi: 10.1016/j.cub.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Jekely G, Rorth P. Hrs mediates downregulation of multiple signalling receptors in Drosophila. EMBO Rep. 2003;4:1163–1168. doi: 10.1038/sj.embor.7400019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamura T, Burian D, Khalili H, Schmidt SL, Sato S, Liu WJ, Conrad MN, Conaway RC, Conaway JW, Shilatifard A. Cloning and characterization of ELL-associated proteins EAP45 and EAP20. a role for yeast EAP-like proteins in regulation of gene expression by glucose. J. Biol. Chem. 2001;276:16528–16533. doi: 10.1074/jbc.M010142200. [DOI] [PubMed] [Google Scholar]
- Katzmann DJ, Odorizzi G, Emr SD. Receptor downregulation and multivesicular-body sorting. Nat. Rev. Mol. Cell Biol. 2002;3:893–905. doi: 10.1038/nrm973. [DOI] [PubMed] [Google Scholar]
- Krempler A, Henry MD, Triplett AA, Wagner KU. Targeted deletion of the Tsg101 gene results in cell cycle arrest at G1/S and p53-independent cell death. J. Biol. Chem. 2002;277:43216–43223. doi: 10.1074/jbc.M207662200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuranaga E, Kanuka H, Igaki T, Sawamoto K, Ichijo H, Okano H, Miura M. Reaper-mediated inhibition of DIAP1-induced DTRAF1 degradation results in activation of JNK in Drosophila. Nat. Cell Biol. 2002;4:705–710. doi: 10.1038/ncb842. [DOI] [PubMed] [Google Scholar]
- Le Borgne R, Bardin A, Schweisguth F. The roles of receptor and ligand endocytosis in regulating Notch signaling. Development. 2005;132:1751–1762. doi: 10.1242/dev.01789. [DOI] [PubMed] [Google Scholar]
- Le Roy C, Wrana JL. Signaling and endocytosis: a team effort for cell migration. Dev. Cell. 2005;9:167–168. doi: 10.1016/j.devcel.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci. 2001;24:251–254. doi: 10.1016/s0166-2236(00)01791-4. [DOI] [PubMed] [Google Scholar]
- Lewis EB, Bacher F. Method of feeding ethyl methanesulfonate (EMS) to Drosophila males. Drosoph. Inf. Serv. 1968;43:193. [Google Scholar]
- Li L, Cohen SN. Tsg101: a novel tumor susceptibility gene isolated by controlled homozygous functional knockout of allelic loci in mammalian cells. Cell. 1996;85:319–329. doi: 10.1016/s0092-8674(00)81111-3. [DOI] [PubMed] [Google Scholar]
- Li L, Li X, Francke U, Cohen SN. The TSG101 tumor susceptibility gene is located in chromosome 11 band p15 and is mutated in human breast cancer. Cell. 1997;88:143–154. doi: 10.1016/s0092-8674(00)81866-8. [DOI] [PubMed] [Google Scholar]
- Lin SY, Chen YJ, Chang JG. Multiple truncated transcripts of TSG101 in gastrointestinal cancers. J. Gastroenterol. Hepatol. 1998;13:1111–1114. doi: 10.1111/j.1440-1746.1998.tb00585.x. [DOI] [PubMed] [Google Scholar]
- Lloyd TE, Atkinson R, Wu MN, Zhou Y, Pennetta G, Bellen HJ. Hrs regulates endosome membrane invagination and tyrosine kinase receptor signaling in Drosophila. Cell. 2002;108:261–269. doi: 10.1016/s0092-8674(02)00611-6. [DOI] [PubMed] [Google Scholar]
- Meier P, Silke J, Leevers SJ, Evan GI. The Drosophila caspase DRONC is regulated by DIAP1. EMBO J. 2000;19:598–611. doi: 10.1093/emboj/19.4.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DT, Cagan RL. Local induction of patterning and programmed cell death in the developing Drosophila retina. Development. 1998;125:2327–2335. doi: 10.1242/dev.125.12.2327. [DOI] [PubMed] [Google Scholar]
- Moberg KH, Schelble S, Burdick SK, Hariharan IK. Mutations in erupted, the Drosophila ortholog of mammalian tumor susceptibility gene 101, elicit non-cell-autonomous overgrowth. Dev. Cell. 2005;9:699–710. doi: 10.1016/j.devcel.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Moreno E, Basler K. dMyc transforms cells into super-competitors. Cell. 2004;117:117–129. doi: 10.1016/s0092-8674(04)00262-4. [DOI] [PubMed] [Google Scholar]
- Muller P, Kuttenkeuler D, Gesellchen V, Zeidler MP, Boutros M. Identification of JAK/STAT signalling components by genome-wide RNA interference. Nature. 2005;436:871–875. doi: 10.1038/nature03869. [DOI] [PubMed] [Google Scholar]
- Newsome TP, Asling B, Dickson BJ. Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development. 2000;127:851–860. doi: 10.1242/dev.127.4.851. [DOI] [PubMed] [Google Scholar]
- Odorizzi G, Babst M, Emr SD. Fab1p PtdIns(3)P 5-kinase function essential for protein sorting in the multivesicular body. Cell. 1998;95:847–858. doi: 10.1016/s0092-8674(00)81707-9. [DOI] [PubMed] [Google Scholar]
- Pantalacci S, Tapon N, Leopold P. The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat. Cell Biol. 2003;5:921–927. doi: 10.1038/ncb1051. [DOI] [PubMed] [Google Scholar]
- Perez-Garijo A, Martin FA, Morata G. Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development. 2004;131:5591–5598. doi: 10.1242/dev.01432. [DOI] [PubMed] [Google Scholar]
- Pornillos O, Garrus JE, Sundquist WI. Mechanisms of enveloped RNA virus budding. Trends Cell Biol. 2002;12:569–579. doi: 10.1016/s0962-8924(02)02402-9. [DOI] [PubMed] [Google Scholar]
- Raiborg C, Rusten TE, Stenmark H. Protein sorting into multivesicular endosomes. Curr. Opin. Cell Biol. 2003;15:446–455. doi: 10.1016/s0955-0674(03)00080-2. [DOI] [PubMed] [Google Scholar]
- Raymond CK, Howald-Stevenson I, Vater CA, Stevens TH. Morphological classification of the yeast vacuolar protein sorting mutants: evidence for a prevacuolar compartment in class E vps mutants. Mol. Biol. Cell. 1992;3:1389–1402. doi: 10.1091/mbc.3.12.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds-Kenneally J, Mlodzik M. Notch signaling controls proliferation through cell-autonomous and non-autonomous mechanisms in the Drosophila eye. Dev. Biol. 2005;285:38–48. doi: 10.1016/j.ydbio.2005.05.038. [DOI] [PubMed] [Google Scholar]
- Ryoo HD, Bergmann A, Gonen H, Ciechanover A, Steller H. Regulation of Drosophila IAP1 degradation and apoptosis by reaper and ubcD1. Nat. Cell Biol. 2002;4:432–438. doi: 10.1038/ncb795. [DOI] [PubMed] [Google Scholar]
- Ryoo HD, Gorenc T, Steller H. Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev. Cell. 2004;7:491–501. doi: 10.1016/j.devcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Schmidt AE, Miller T, Schmidt SL, Shiekhattar R, Shilatifard A. Cloning and characterization of the EAP30 subunit of the ELL complex that confers derepression of transcription by RNA polymerase II. J. Biol. Chem. 1999;274:21981–21985. doi: 10.1074/jbc.274.31.21981. [DOI] [PubMed] [Google Scholar]
- Sen A, Reddy GV, Rodrigues V. Combinatorial expression of Prospero, Seven-up, and Elav identifies progenitor cell types during sense-organ differentiation in the Drosophila antenna. Dev. Biol. 2003;254:79–92. doi: 10.1016/s0012-1606(02)00021-0. [DOI] [PubMed] [Google Scholar]
- Seto ES, Bellen HJ. The ins and outs of Wingless signaling. Trends Cell Biol. 2004;14:45–53. doi: 10.1016/j.tcb.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Srivastava M, Scherr M, Lackey M, Xu D, Chen Z, Lu J, Bergmann A. ARK, the Apaf-1 related killer in Drosophila, requires diverse domains for its apoptotic activity. Cell Death Diff. 2006 doi: 10.1038/sj.cdd.4401931. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Pan J, Bubley G, Balk SP. Frequent abnormalities of TSG101 transcripts in human prostate cancer. Oncogene. 1997;15:3121–3125. doi: 10.1038/sj.onc.1201521. [DOI] [PubMed] [Google Scholar]
- Teo H, Perisic O, Gonzalez B, Williams RL. ESCRT-II, an endosome-associated complex required for protein sorting: crystal structure and interactions with ESCRT-III and membranes. Dev. Cell. 2004;7:559–569. doi: 10.1016/j.devcel.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Thompson BJ, Mathieu J, Sung HH, Loeser E, Rorth P, Cohen SM. Tumor suppressor properties of the ESCRT-II complex component Vps25 in Drosophila. Dev. Cell. 2005;9:711–720. doi: 10.1016/j.devcel.2005.09.020. [DOI] [PubMed] [Google Scholar]
- Tsai YC, Sun YH. Long-range effect of upd, a ligand for Jak/STAT pathway, on cell cycle in Drosophila eye development. Genesis. 2004;39:141–153. doi: 10.1002/gene.20035. [DOI] [PubMed] [Google Scholar]
- Udan RS, Kango-Singh M, Nolo R, Tao C, Halder G. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat. Cell Biol. 2003;5:914–920. doi: 10.1038/ncb1050. [DOI] [PubMed] [Google Scholar]
- Vaccari T, Bilder D. The Drosophila tumor suppressor vps25 prevents nonautonomous overproliferation by regulating notch trafficking. Dev. Cell. 2005;9:687–698. doi: 10.1016/j.devcel.2005.09.019. [DOI] [PubMed] [Google Scholar]
- Vidal M, Cagan RL. Drosophila models for cancer research. Curr. Opin. Genet. Dev. 2006;16:10–16. doi: 10.1016/j.gde.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Wagner KU, Krempler A, Qi Y, Park K, Henry MD, Triplett AA, Riedlinger G, Rucker IE, Hennighausen L. Tsg101 is essential for cell growth, proliferation, and cell survival of embryonic and adult tissues. Mol. Cell. Biol. 2003;23:150–162. doi: 10.1128/MCB.23.1.150-162.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Huang J, Dong J, Pan D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell. 2003;114:445–456. doi: 10.1016/s0092-8674(03)00549-x. [DOI] [PubMed] [Google Scholar]
- Xu D, Li Y, Arcaro M, Lackey M, Bergmann A. The CARD-carrying caspase Dronc is essential for most, but not all, developmental cell death in Drosophila. Development. 2005;132:2125–2134. doi: 10.1242/dev.01790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Rubin GM. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development. 1993;117:1223–1237. doi: 10.1242/dev.117.4.1223. [DOI] [PubMed] [Google Scholar]
- Yoo SJ, Huh JR, Muro I, Yu H, Wang L, Wang SL, Feldman RM, Clem RJ, Muller HA, Hay BA. Hid, Rpr and Grim negatively regulate DIAP1 levels through distinct mechanisms. Nat. Cell Biol. 2002;4:416–424. doi: 10.1038/ncb793. [DOI] [PubMed] [Google Scholar]
- Zhai RG, Hiesinger PR, Koh TW, Verstreken P, Schulze KL, Cao Y, Jafar-Nejad H, Norga KK, Pan H, Bayat V, et al. Mapping Drosophila mutations with molecularly defined P element insertions. Proc. Natl. Acad. Sci. USA. 2003;100:10860–10865. doi: 10.1073/pnas.1832753100. [DOI] [PMC free article] [PubMed] [Google Scholar]