

Abstract
Patients with cancer can develop recurrent metastatic disease with latency periods that range from years even to decades. This pause can be explained by cancer dormancy, a stage in cancer progression in which residual disease is present but remains asymptomatic. Cancer dormancy is poorly understood, resulting in major shortcomings in our understanding of the full complexity of the disease. Here, I review experimental and clinical evidence that supports the existence of various mechanisms of cancer dormancy including angiogenic dormancy, cellular dormancy (G0–G1 arrest) and immunosurveillance. The advances in this field provide an emerging picture of how cancer dormancy can ensue and how it could be therapeutically targeted.
The vast majority of cancer-related deaths are due to metastatic tumour growth that impairs the function of vital organs1. Metastatic lesions invariably originate from disseminated tumour cells, which often undergo a period of dormancy2 (FIG. 1). Cancer recurrence after therapy and long periods of remission is frequent. For example, 20–45% of patients with breast or prostate cancer will relapse years or decades later3-5 (FIGS 1,2). In fact, most cancer types are associated with disseminated disease that after treatment might persist as minimal residual disease (FIG 2; TABLE 1). However, the lack of mechanistic insight into this stage has been a major shortcoming in our understanding of the full complexities of metastatic growth. Functional characterization of disseminated dormant tumour cells is important because these cells most probably contain the information about the future progression of the disease (that is, metastasis development). To fully understand dormancy, cells must be characterized during the dormant state. Therefore, given that these cells are present in a wide variety of cancers, information gathered from studies of cancer dormancy might be applicable to a large number of patients.
Figure 1. Tumour dormancy as a component of cancer progression.
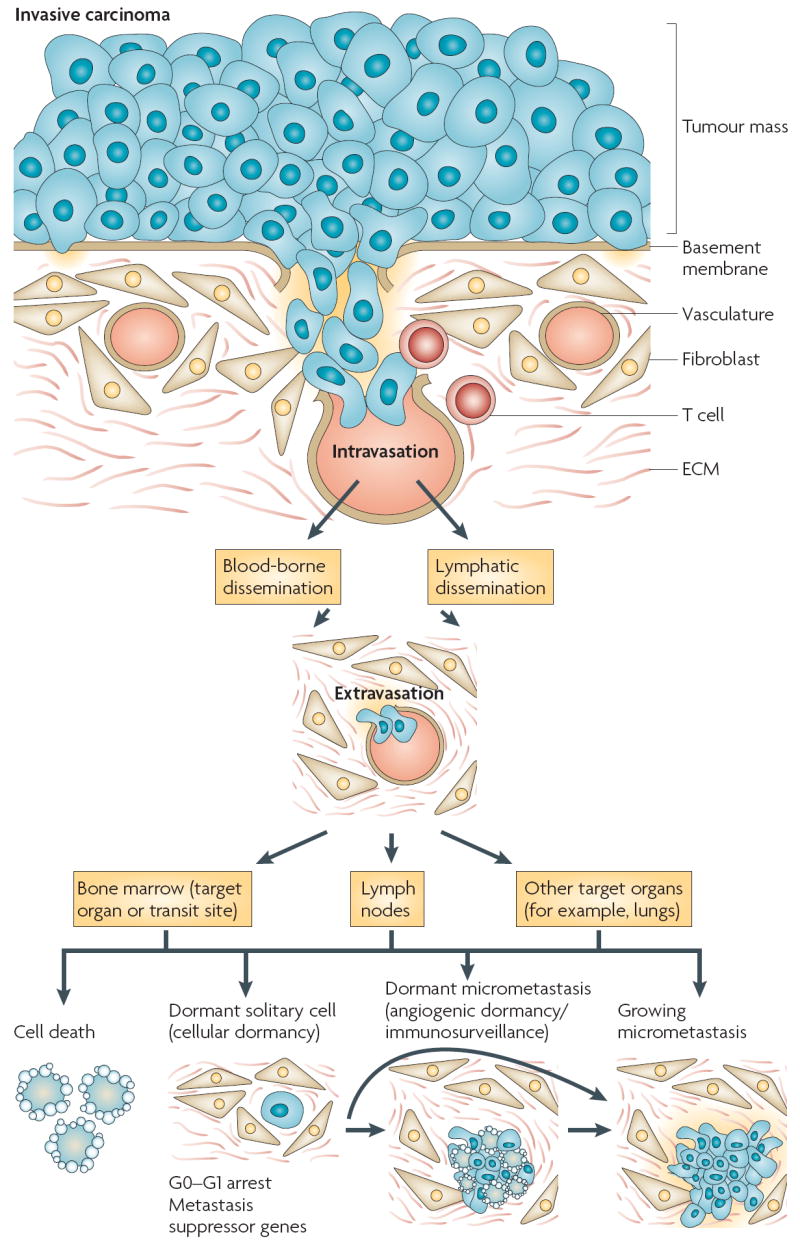
Tumour cells carrying genetic or epigenetic changes enabling motile and invasive properties can degrade the basement membrane and invade the underlying stroma. Invading tumour cells interact with fibroblasts or immune cells and the stromal matrix. Tumour cells (in cooperation with stromal cells) can degrade the extracellular matrix (ECM) and the vascular walls and intravasate (through either arterial or lymphatic routes). Tumour cells that arrest in the vasculature of the bone marrow can proliferate or remain dormant. Although the bone can be a target organ, it might also serve as a transit site from which cells can again disseminate, through as yet unknown mechanisms, to their final destination (that is, lungs, liver and so on, where they form metastases). Tumour cells in the bone marrow are not yet a metastasis but have the potential to become secondary lesions and they can also carry the information about the future progression of the disease2,8. Tumour cells can arrest in lymph nodes or in the target organ vasculature, where they can extravasate into the organ parenchyma. At this stage (this can also happen in the bone marrow or lymph nodes) intra- or extra-vascularly lodged tumour cells have four possible fates: they die (the vast majority of cells undergo apoptosis), they can enter a state of quiescence or dormancy, either as a single solitary cell or as a micrometastatic lesion that underwent a proliferative expansion and cannot recruit a vascular bed, or they can resume proliferation (growing micrometastases).
Figure 2. Manifestation of cancer dormancy.

a Tumour cells that have accumulated genetic and epigenetic changes that provide a growth advantage (solid blue line) form a primary tumour. After a treatment that results in tumour regression, residual disease can be detectable for long periods thereafter (dashed blue line). After this time, the tumour mass can increase again, but now in distant secondary organs (dashed red lines). b During the dormancy stage, sub-clinical disease might be due to dormant cells that have entered a G0–G1 arrest (cellular dormancy) and these cells might develop mechanisms to evade immune system recognition and eradication. c Angiogenic dormancy results from the balance between pro- and anti-angiogenic factors (such as vascular endothelial growth factor (VEGF) and thrombospondin (TSP), respectively). Genetic alterations in the pathways that maintain angiogenic dormancy or an exogenous angiogenic ‘spike’ might restore tumour growth. Oncogenic Ras can induce VEGF and repress TSP. By contrast, the stress-activated kinase p38 and the tumour suppressor p53 can induce TSP or repress VEGF. Loss of function of p53 and/or p38 might tip the balance towards enhanced angiogenesis. d Immunosurveillance. Proliferating tumour cells are kept at low numbers (sub-clinical) by an active immune system. This can be due to cytotoxic CD8+ T lymphocytes or anti-idiotypic antibodies against the B-cell receptor that arrest the tumour cells. An interruption of this state of dormancy might be due to tumour cell escape from immune system control by downregulation of specific tumour-associated antigens or by expression of co-stimulatory molecules that induce apoptosis of cytotoxic CD8+ T lymphocytes. It is unclear whether these forms of dormancy are mutually exclusive, although they are probably not, how long they last or whether they occur at different times. It is possible that cellular dormancy most frequently precedes the immunosurveillance or the angiogenic dormancy phase.
Table 1.
Examples of evidence for minimal residual disease in different types of cancer
| Cancer type* | Anatomical compartments where minimal residual disease has been detected‡ | References |
|---|---|---|
| Breast | Bone marrow , blood, lymph node | 3,127 |
| Gastric | Bone marrow | 80 |
| Colon | Bone marrow, liver | 128 |
| Head and neck | Bone marrow, lymph node | 129 |
| Neuroblastoma | Bone marrow | 130 |
| Leukaemia | Bone marrow, blood | 131,132 |
| Lymphoma | Bone marrow, blood | 132 |
| Prostate | Bone marrow, blood | 133-135 |
| Pancreatic | Bone marrow, blood, lymph node, peritoneal cavity | 136 |
| Melanoma | Bone marrow, blood, lymph node | 137,138 |
| Non-small-cell lung | Bone marrow, lymph node | 139 |
| Soft tissue sarcoma | Multiple sites | 140 |
This table is not intended to represent an exhaustive list of all cancers in which minimal residual disease has been detected but just provides some examples to illustrate the point that the phenomenon is widespread. Other cancers that are not listed here might also display minimal residual disease.
Detection in these anatomical compartments does not necessarily indicate prognostic value. Other anatomical sites might have minimal residual disease but they might not have been routinely inspected.
Cancer dormancy can be separated into mechanisms that antagonize the expansion of a dividing tumour cell population (tumour mass dormancy) and mechanisms that result in tumour cell growth arrest (tumour cell dormancy, or cellular dormancy) (FIG. 2). In the former, tumour cells usually divide but the lesion does not expand beyond a certain size because of either limitations in blood supply or an active immune system. Cellular dormancy can occur when tumour cells enter a state of quiescence (see BOX 1 for information on the relationship between quiescence, senescence and dormancy). These general mechanisms might explain the dormancy of residual cells that, following treatment, develop loco-regional or distant organ recurrences within different time frames.
Box 1 Cancer dormancy: senescence or quiescence programmes?
A common attempt to explain cellular dormancy is to catalogue it within known mechanisms of growth arrest such as senescence or quiescence. However, whether quiescence or senescence programmes drive tumour cell dormancy is still incompletely understood.
Senescence represents a tumour suppressive mechanism that becomes activated in normal cells in response to replicative or oncogenic stress; the latter activates replicative and oxidative stress104. Specific markers, such as senescence-associated β-galactosidase (SA-β-gal), upregulation of p16, activation of p53, development of senescence-associated heterochromatic foci and a persistent active metabolism despite a G1 arrest, are found in senescent cells105. Senescence might be an irreversible state depending on the machinery that is engaged104. Because dormancy occurs in tumour cells and is reversible, a permanent senescence does not seem to be a plausible mechanism. More importantly, successful tumour development must have escaped senescence106,107. However, perhaps some aspects of the senescence programme can still be invoked in malignant cells, indicated by the positive correlation with SA-β-gal and p16 staining in tumour samples from patients who have been treated with chemotherapy108. However, whether the tumour cells that undergo senescence are the source of the relapse is unknown. It will be important to determine if dormancy and senescence programmes intersect because the abundance of information on the latter might be applicable to understanding tumour cell dormancy.
Alternatively, cellular quiescence might be a better mechanistic fit. Quiescence is thought to be due to a G0–G1 arrest and most adult cells reside in this state89,109. A reversible quiescence might be observed in stem cells or in cells that withdraw into a G0 arrest because of lack of growth-promoting signals (such as serum withdrawal and contact inhibition, for example)89,110. During quiescence, cells pause several cellular activities and this can result in the activation of selective programmes that render cells refractory to differentiation111. Furthermore, quiescence that is induced by growth-factor withdrawal results in attenuation of translational and metabolic activity and the regulation of specific transcriptional programmes109,111,112.
This Review discusses the relevance of cancer dormancy and how this state might manifest. Furthermore, the Review summarizes key discoveries from both experimental and clinical studies that shed light on how cancer dormancy is a part of disease progression and therapeutic response. It is important to note that the mechanisms that have been identified in experimental models have not fully been found to exist in patients, although all are highly plausible. For example, although there is clinical evidence for mechanisms of disseminated tumour cell growth arrest, there is limited documentation of angiogenic dormancy. By discussing the experimental and clinical evidence side by side this Review attempts, rather than to render these mechanisms as homologous, to provide potential mechanistic links between experimental and clinical research. Given the dearth of information on dormancy, it is crucial to take advantage of available mechanistic information, and novel discoveries and hypotheses from the experimental work reviewed here, as they might offer possibilities for translating our knowledge of cancer dormancy into the clinic.
Manifestations of cancer dormancy
It is thought that genetically fit tumour cells that emerge in the primary tumour (which is proposed to be a ‘late’ event in cancer progression) will be able to metastasize6,7. This was thought to be due to the time that must elapse for tumour cells to mutate and acquire traits that allow them to go through the different steps of the metastatic cascade (FIG. 1). However, recent studies suggest that tumour cell dissemination might occur early (that is, by less genetically progressed cells) and that disseminated tumour cells progress towards more aggressive phenotypes that will lead to metastatic growth in parallel with the primary tumour8. Were the disseminated tumour cells that remain after treatment to proliferate continuously, the relapse time would be expected much earlier than is actually observed9. Therefore, a pause in progression (that is, dormancy) has to be considered the most likely explanation for the discrepancy between the estimated and observed disease-free periods9. Tumour dormancy is observed in local recurrences or metastases. In the case of a primary tumour, the term commonly used is latency — the time that separates the carcinogenic insult from the clinical detection of the tumour. In all cases a portion of the latency period can be attributed to the slow accumulation of genetic alterations that lead to immortalization (that is, loss of TP53, RB1 (retinoblastoma 1), p16 (also known as cyclin-dependent kinase inhibitor 2A) and/or gain of telomerase, and so on) and transformation (that is, gain of Ras-activating mutations, ERBB2 (also known as HER2 or Neu) amplification, BRAF-activating mutations, and so on) after and/or during carcinogenesis6. However, after tumour cells are fully transformed it is not clear how much of their latency is attributable to tumour mass dormancy or cellular dormancy (FIG. 2).
At a glance.
In the clinic, tumour dormancy is observed in local recurrences or metastases. It usually refers to the time after treatment that a patient is asymptomatic but still carries local remnant or disseminated tumour cells that do not grow into overt lesions.
Tumour dormancy ensues when cancer cell proliferation is counteracted by other mechanisms such as apoptosis because of impaired vascularization or immunosurveillance, and cellular dormancy ensues when the cancer cells enter a growth arrest.
Cancer dormancy is a relevant problem because the majority of solid tumours and haematological malignancies undergo a period of dormancy that is characterized by years to decades of minimal residual disease. Because metastases always arise from disseminated tumour cells it is of importance to understand the biology of dormant tumour cells.
Several mechanisms can explain cancer dormancy. These include the disruption of crosstalk between growth factor and adhesion signalling, which prevents tumour cells from interpreting their microenvironment, leading to cellular tumour dormancy through a G0–G1 arrest or ‘differentiation’; the inability of a tumour cell population to recruit blood vessels despite active proliferation; and immunosurveillance, which can prevent residual tumour cell expansion.
The expression of genes that selectively suppress metastases might function by inducing dormancy. In addition, quiescent tumour ‘stem’ cells might be dormant tumour cells. Finally, dormant tumour cells seem to have active drug resistance mechanisms that might protect them from therapy.
The therapeutic opportunities that emerge from understanding dormancy include the possibility of inducing and/or maintaining the dormancy of tumour cells and inducing cell death in residual dormant cells by targeting their survival and drug-resistance mechanisms.
Studies of cancer dormancy might help determine whether a patient has dormant disease and what type of mechanism is active. These studies will be instrumental in identifying biomarkers of dormant cancer.
Dormancy of loco-regional lesions, disseminated tumour cells or micrometastases (FIGS 1,2), refers to the pause in cancer progression and the absence of clinical symptoms following treatment of the primary lesion10,11. In the case of disseminated tumour cells or micrometastases, it is in their final destinations (for example, bone marrow, lymph nodes, lungs or liver) that these cells are protractedly unable to resume growth. As mentioned above, a reason for this may be that dissemination might occur very early from ‘non-invasive’ lesions or in patients who are node negative2,8. Studies using comparative genomic hybridization (CGH) have shown that early dissemination of breast tumour cells with limited genetic abnormalities might be followed by progression and microevolution systemically8. These studies propose that, given that genetic abnormalities in diagnosed metastatic disease are quite homogeneous (that is, metastases share similar genetic abnormalities between patients), the stochastic and protracted nature of metastatic disease might depend on the rate at which genetic abnormalities progress in early disseminated tumour cells. Therefore, it is proposed that in the vast majority of patients a dormancy or ‘lead time’ (that is, the time it takes for genetic anomalies to accumulate) of ~5 years precedes relapse12. Alternatively, a true dormancy state, which most probably involves cellular tumour dormancy, might persist beyond 5 years in some patients despite the presence of additional genetic alterations in the disseminated tumour cells12. If early dissemination is applicable to other tumours it might explain a portion of the dormancy time of the disease. However, these cells must still survive during the ‘lead time’ and they seem to be growth arrested13. This suggests that although survival pathways are functional, the mechanisms that propel the growth of the primary lesion are insufficient for metastatic growth. Additionally, the microenvironment and host genetics (see below) might influence genetic progression. These data suggest that oncogenic signalling might not always be dominant and that other programmes (that is, ‘stem’ cell quiescence and stress signalling, or microenvironment restrictions (see below)) might overcome oncogenic signals, allowing tumour cell survival in a dormant state. The fact that disease-free periods in patients can last ~5 years and as much as 20–25 years (for example, in breast and oral cavity cancers)3,14, suggests that a pause in disease progression occurs often and might be explained by different forms of dormancy; this could be a feature of cancer progression (FIG. 2). When dormancy is induced and what mechanisms operate in different cancers is still unclear. However, answers to these questions are most likely to be found in the residual tumour cells during the remission periods after treatment of a diagnosed cancer. understanding the dormancy of remaining disseminated or local tumour cells is crucially important, as metastases or local recurrences, respectively, are invariably derived from these cells.
Tumour cell–microenvironment crosstalk
Disseminated tumour cells usually lodge in a non-orthotopic tissue microenvironment and their ability to interpret it can determine tumour cell fate15. Therefore, a failure to resume proliferation in these sites might result from stress signals that have been activated by a new non-permissive microenvironment or a niche that is conducive for quiescence (FIGS 2,3). By contrast, tumour cells that can remodel and/or reproduce a niche or orthotopic microenvironment that is conducive for expansion might be able to resume growth16-18. Data from different labs reviewed below reveal that deregulation of the reciprocal interaction between the microenvironment and tumour cells might decide between proliferation and growth arrest15,19,20. Some of these studies provide mechanistic evidence for cellular tumour dormancy (FIGS 2,3).
Figure 3. Signals that regulate cellular tumour dormancy.
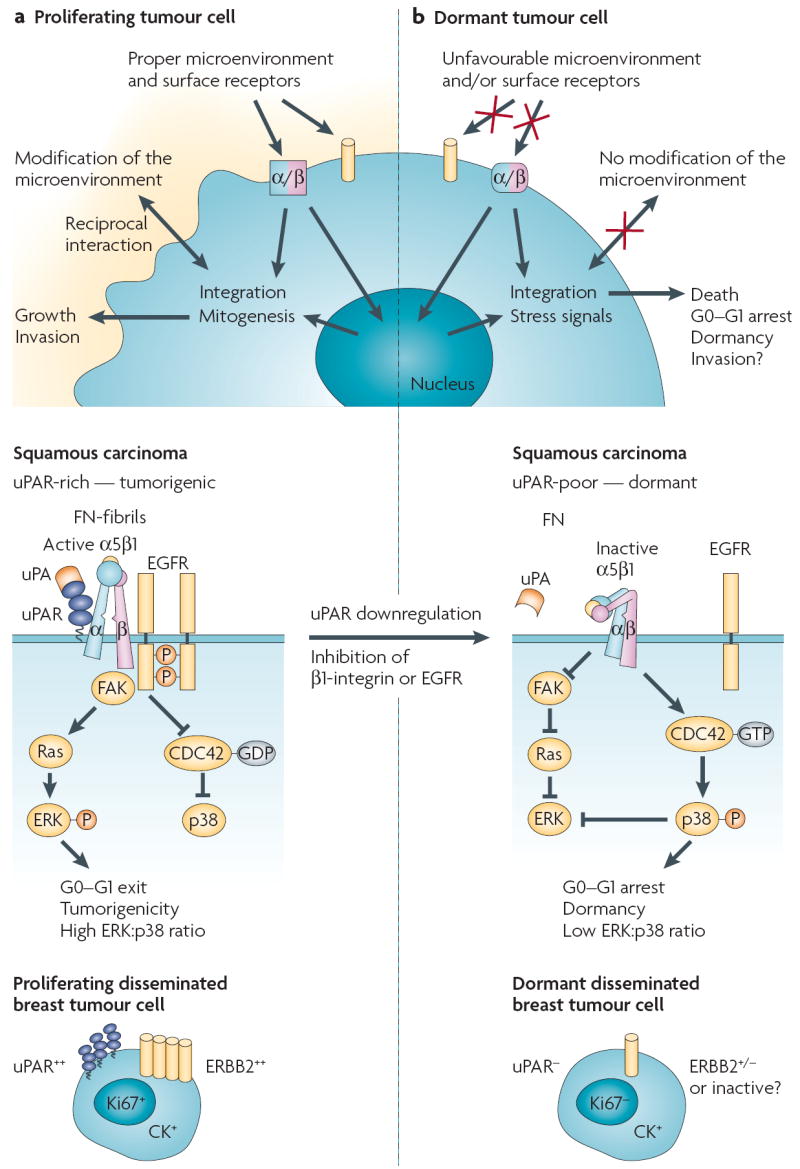
a A matching microenvironment and set of receptors allows metastatic cells to adapt and remodel their microenvironment to integrate growth-promoting signals. As an example (middle panel), signals derived from fibronectin (FN) and transduced by the uPAR (metastasis-associated urokinase receptor)–α5β1-integrin complex, focal adhesion kinase (FAK) and epidermal growth factor receptor (EGFR) can result in extracellular signal-regulated kinase (ERK) activation and p38 inactivation in an expanding tumour. In this scenario metastases might arise from disseminated tumour cells that acquire additional genetic abnormalities, re-activation of uPAR and mitogenic signalling (ERBB2 or EGFR) or from the expansion of tumour stem cells (not depicted). b Loss of a surface receptor (for example, uPAR, α5β1 integrin or EGFR) that transduces growth signals from the microenvironment (for example, fibronectin) results in stress signalling (low FAK–Ras–ERK, and high CDC42 (cell division cycle 42)–p38 activity; middle panel), which in turn might lead to dormancy. This is one example to illustrate the theme of crosstalk between the microenvironment and/or receptor signalling in cellular dormancy (for others see REFS 20,21,29,56,84,124,125). It is likely that other unidentified pathways are also involved. The bottom panels illustrate the possibility that some of the molecules that are found in experimental models (such as that shown in the middle panels) may also regulate the fate of disseminated tumour cells. Those disseminated tumour cells that are cytokeratin (CK) positive, have low uPAR expression and reduced ERBB2 signalling, might be dormant and negative for proliferation markers. Re-expression of uPAR and/or ERBB2 could allow these cells to escape dormancy.
In some cases, cellular tumour dormancy might result from the ability of tumour cells to attain a ‘differentiated’ state. using reconstituted basement membrane assays it has been shown that breast cancer cells rapidly proliferate in a disorganized fashion with an absolute loss of tissue architecture21. Their growth and aberrant organization is dependent on β1-integrins and epidermal growth factor (eGF) signalling, as inhibition of these signals leads to the re-differentiation of these tumour cells into non-proliferating acinar structures21. Blocking of β1-integrin signalling in these tumour cells in vivo ablates tumour growth but it is not known whether this is due to induction of a protracted state of growth arrest. It is also unclear whether such mechanisms occur in dormant disseminated tumour cells in patients. nevertheless, these results indicate that, upon correcting their ability to interpret their microenvironment appropriately, tumour cells can undergo some degree of differentiation and enter a non-proliferative state21.
A similar switch between proliferation and growth arrest controlled by tumour cell–microenvironment crosstalk was observed in head and neck carcinoma19 (FIG. 3). In this model, the metastasis-associated urokinase receptor (uPAR) drives tumour growth by interacting and activating α5β1 integrins19. This complex in turn recruits focal adhesion kinase (FAK) and the eGF receptor (eGFR), which, in a fibronectin-dependent manner, propagates mitogenic signals through the Ras–extra-cellular signal-regulated kinase (eRK) pathway19,22,23. Blocking of uPAR, β1-integrins, FAK or eGFR, singly or in combination with each other, resulted in tumour suppression in vivo, which is due to induction of dormancy19. In these studies, although dormant tumour cells expressed the appropriate integrins and were surrounded by fibronectin, the downregulation of uPAR and loss of integrin function prevented these cells from transducing proliferative signals from the fibronectin-rich microenvironment24. Imaging and measurements of eRK signalling in vivo revealed that dormancy resulted from an almost complete inhibition of the Raf–MeK–eRK pathway and induction of a G0–G1 arrest as in quiescent cells18. Interestingly, activation of stress pathways is also required for dormancy induction. The disruption of the uPAR complex activates the p38 mitogen-activated protein kinase (MAPK) signalling pathway18,24,25. Furthermore, growing metastatic lesions show sustained eRK activity but greatly reduced p38 signalling, suggesting that proliferation in primary and secondary tumours requires a high eRK:p38 signalling ratio whereas the opposite favours cellular dormancy18.
Moreover, inhibition of p38α and/or β in dormant cells is sufficient to interrupt their growth arrest and resume proliferation in vivo18,24,25. Induction of dormancy by p38 activation required upregulation of p53 (but not p21, p15 or p16) (BOX 1), downregulation of Jun and attenuation of protein synthesis mediated by inhibition of eukaryotic initiation factor-2α (eIF2α) activity (REF. 26 and A. P. Adam, A. C. Ranganathan and J.A.A-G., unpublished observations). The possibility that p38 signalling might exert tumour suppressive functions on metastatic cells is in accordance with data showing that p38α/β activation is associated with tumour suppression by causing growth arrest, senescence or apoptosis27 (BOX 1). Whether these mechanisms are operational in patients is unclear. However, uPAR has been found to be a prognostic indicator and correlate with the recurrence of lesions after minimal residual disease in gastric cancer28 (see below). Therefore, it seems reasonable to suggest that strategies that are aimed at inducing and/or maintaining tumour cell dormancy should include concomitantly blocking receptor tyrosine kinases and uPAR, and/or integrin complexes to inhibit the tumour cell–microenvironment crosstalk (FIG. 3).
Tumour cell dormancy has also been described in a transgenic mouse model for breast cancer, in which polyoma middle-T (PyMT) antigen or eRBB2 signalling was studied in mammary gland tissue that was null for β1 integrin20. These studies show that whereas control littermates develop tumours at high frequency, the loss of β1 integrin results in tumour suppression. Additionally, the PyMT-positive tumour cells that lack β1 integrin expression were in a non-proliferating, dormant state20. Again, a loss of signals from the microenvironment favoured dormancy. loss of FAK-dependent signalling is attributed as a potential mechanism, consistent with previous results in squamous carcinoma cells22,23 (FIG. 3), in which FAK inhibition uncoupled α5β1 integrin from eGFR, leading to tumour cell dormancy.
Systemic signals that influence both stromal and tumour cells might also have a role in regulating dormancy. upon transplantation into virgin female mice, pregnancy-dependent tumours that have been induced by insertional mutations of mouse mammary tumour virus (MMTv) remain dormant for at least 300 days29. Stimulation by hormones that are produced during pregnancy or hormonal treatment caused these tumours to emerge from dormancy29. The ‘dormant’ state here is different from the main mechanisms described above as it is mostly due to a very slow rate of proliferation and it is not clear if cell death compensates for cell division. This slow-cycling but still dynamic tumour cell population seems to acquire new mutations that explain why subsequent transplants become hormone-independent. As in the above examples, growth-factor signalling was important for interruption of tumour dormancy as MMTv insertional mutations in fibroblast growth factor 3 (FGF3) and genes at the integration site 7 (Int7) locus correlated with the emergence of hormone-independent tumours29,30. Other pathways that depend on β1 integrin, including retinoic acid-induced protein 2, Rspo3 (a thrombospondin, type I, domain-2-containing protein) and endonexin, among others, might be required as the loci of these genes have been described as additional insertion sites in recurrent tumours29,30.
The above studies suggest that reduced tumour cell–microenvironment crosstalk might induce dormancy through quiescence or ‘differentiation’ of malignant cells. This might explain the behaviour of disseminated tumour cells in mouse models of experimental breast tumour metastasis10,11,31. In these studies, although most disseminated cancer cells die, there is a fraction that remain viable and do not proliferate in lungs and liver, but form tumours in the orthotopic mammary fat pad10,32. Similar results were obtained when detecting experimental brain metastases through the use of nano-ferromagnetic particles and magnetic resonance imaging (MRI)11. It is important to emphasize that in these early stages, solitary dormant tumour cells are surrounded by normal functioning tissue vasculature. Therefore, rather than invoking a mechanism that depends on neovascularization (see below), it is more likely that deficient crosstalk with the new microenvironment (such as those described above) and/or stress signalling (for example, JUN N-terminal kinase (JNK) and p38) explain the impaired proliferation of these cells. Interestingly, the identification of genes that selectively inhibit the growth of metastases, but not the primary tumour (that is, metastasis suppressor genes), revealed that they can inhibit mitogenic signals and activate stress signalling pathways33,34.
Metastasis suppressors and host genetics
Metastatic growth is crucially dependent on the ability of tumour cells to survive the stress of dissemination and the new microenvironment, as the majority (99.9% (REFS 10,35)) of disseminated tumour cells die (FIG. 1). The loss of metastasis suppressor gene expression or function36-38 is thought to explain the development of metastases39. Their function seems to be dominant in solitary cells that reach their target organs37. Genes of interest are those that curtail metastasis by inducing apoptosis or dormancy once the cells have lodged (intra- or extra-vascularly) at the secondary site39. KISS1 (kisspeptin) blocks metastases through the induction of prolonged dormancy of solitary cells40. This response is not mediated by its receptor, G-protein-coupled receptor 54, suggesting that either a paracrine signalling mechanism or an alternative receptor is important. The tetraspanin protein CD82 (also known as kangai-1), which has a role in adhesion signalling, inhibits mouse melanoma cell metastases when bound to the endothelial cell-expressed ligand DARC (Duffy antigen chemokine receptor, which is also known as CD234)36. In vitro analysis implicated tumour cell senescence that was induced by the CD82–DARC interaction as the inhibitory mechanism36. However, whether senescence is operational in vivo and whether it is reversible (dormancy implies reversibility) is unknown. This is important to determine, because it suggests that dormancy of metastases might be driven, at least in part, by mechanisms of reversible senescence (BOX 1).
Other metastasis suppressors such as nM23 (which is encoded by non-metastatic cells 1 (NME1) and NME2) seem to inhibit Ras signalling or, as in the case of MKK4 (MAPK kinase 4), can activate the JnK and p38 stress pathways34. In prostate cancer, MKK4 is lost and its suppressive function was found to operate through JnK activation41. In ovarian cancer, MKK4 is also lost42 and it exerts its metastasis suppressor function through the activation of p38 (REF. 38). The fate of these cells and the mechanism of dormancy are still unknown (that is, senescence, cellular dormancy, apoptosis, angiogenic switch or immunosurveillance). If JnK or p38 activation is exceedingly strong, cells might undergo apoptosis43. However, achieving p38 signalling levels that induce growth arrest without apoptosis25 might represent an adaptation or survival mechanism that favours subsequent re-growth. In many ways dormancy is a survival mechanism26,44 (see BOX 2 for information suggesting that cellular stress adaptation might recapitulate programmes of organismal stress adaptation). Therefore, mapping the pathways that metastasis suppressor genes regulate might offer targets for therapeutically preventing the development of metastases.
Box 2 Tumour dormancy as an evolutionary conserved process.
Dormancy is a phenomenon that is observed in cancer but also in whole organisms. For example, in Caenorhabditis elegans development can be paused at the dauer stage (that is, diapause) until the conditions are again propitious for successful development113. Similarly, a plant seed can be dormant until the appropriate signals (that is, light, humidity or salinity) favour germination114. The entry into a stage of diapause was shown to be a selective advantage that helps organisms adapt to stress with the ultimate goal of producing offspring. In C. elegans, the pathways that control the dauer stage (dauer formation (DAF) genes and the insulin-like growth factor-1 signalling pathway) are involved in lifespan extension115. Similarly, pathways that sense oxidative stress (such as the p38 mitogen-activated protein kinase pathway) were shown to induce a dauer stage and these larvae are more resistant to various types of stress such as nutritional deprivation116-119. Therefore, the theme of growth arrest or retardation coupled to stress resistance might be a basic programme of survival that is recapitulated by tumour cells and applicable to some of the models described here. If tumours progress following rules that apply to evolutionary and ecological processes120 then the acquisition of traits that allow for a pause in growth and an adaptation phase might fit this paradigm. It is possible that tumour cell dormancy recapitulates evolutionary conserved mechanisms of adaptation, robustness and resistance to stress that are imposed by dissemination and the ‘hostile’ microenvironment of the target or transit organs (the bone marrow stroma, for example)120,121. Testing whether pathways that control the dauer stage are activated in disseminated tumour cells might be informative about dormancy. For example, do disseminated tumour cells show nuclear localization of Forkhead transcription factors, which when inactivated induce growth arrest in mammalian cells and dauer in C. elegans, or enhanced expression or activation of pathways that are involved in lifespan extension, stress resistance and/or growth arrest pathways (such as the unfolded protein response and p38 pathway)116,119,122? This might be interesting to explore, as genes that are involved in lifespan extension seem to suppress tumour formation in C. elegans and humans115,123.
An exciting possibility is that metastatic efficiency might be modulated by polymorphisms that are present in the heterogeneous genome of the human population45. Crossing PyMT-expressing mice into different syngeneic strains and assessing both primary tumours and metastatic efficiency showed that, although primary tumour formation is not affected, metastatic efficiency is either down- or upregulated depending on the F1 genetic makeup. Therefore, the genetic background of the host might determine metastatic efficiency46. This led to the identification of a metastasis modifier locus (Mtes1), which encodes, among other genes, Sipa1 (signal-induced proliferation-associated gene 1), a RAP1-GTPase activating protein47. These studies reveal that Sipa1 is a metastasis-promoting gene and that polymorphisms in the PDZ protein–protein interaction domain might cause loss of function, reducing metastatic efficiency, which could be because of the induction of dormancy47. It will be exciting to determine whether polymorphisms in humans cause loss of function in metastasis modifier genes that, for example, suppress metastases through the induction of cellular dormancy (that is, growth arrest) or through the suppression of angiogenesis.
Angiogenic dormancy
once tumour cells emerge from quiescence and the tumour mass reaches a certain size that cannot be supported any longer by the normal tissue vasculature, it will become highly dependent on the availability of oxygen and nutrients. This leads to the development of a vascular bed through sensing low oxygen levels, and recruiting blood vessels (angiogenesis) — by inducing the expression of hypoxia-inducible factor 1 (HIF1), for example48 — and lymphatic vessels (lymphangiogenesis). Interestingly, tumour cells, although proliferation competent, might be unable to recruit new blood vessels or remodel the pre-existing vasculature as the tumour reaches a certain size49. This leads to a state of anoxia or hypoxia in the tumour mass that triggers cell death in the tumour cell population48. If cell death exceeds division, then these lesions would most probably regress. However, in tumours that are unable to recruit blood vessels the fraction of dying cells equals the dividing ones and there is no net increase in tumour mass over time50. This results in angiogenic dormancy (FIG. 2).
The ability of tumour cells to respond to hypoxia, promote neovascularization and interrupt angiogenic dormancy is known as the angiogenic switch (for excellent reviews see REFS 50,51). A balance between pro-angiogenic factors (such as vascular endothelial growth factor (veGF) and platelet-derived growth factor (PDGF)) and anti-angiogenic factors (thrombospondin, endostatin, vasculostatin and angiostatin) is thought to control this switch, which involves the regulation of transcriptional programmes48,52. Therefore, a tumour mass might enter this state if a tumour cell population with an active programme of proliferation lacks angiogenesis or hypoxia-response sub-programmes52. The balance between negative and positive angiogenesis factors can be regulated by the balance between genes and pathways that are involved in tumour promotion or suppression53-55 (FIG. 2). For example, active Ras signalling seems to oppose the induction of anti-angiogenic factors through a Rho and MYC-dependent pathway53. It is intuitively logical that pathways that regulate cell-cycle progression also regulate the expression of neovascularization factors. Why then are proliferating tumour cell populations unable to induce angiogenesis? It is possible that the distinct genetic alterations or dosage of a mutated gene (such as oncogenic Ras53) might be sufficient to favour cell-cycle progression, but might be below a threshold level for the angiogenic programme. A possible scenario can be envisioned in which growth arrest or slow cycling of early transformed cells might explain an initial pre-angiogenic phase of primary tumour dormancy18,56. once these cells progress and acquire new mutations that self-perpetuate cell-cycle progression, a second programme might be required to turn on the angiogenic switch51 (FIGS 2,4).
Figure 4. An integrated view of cancer metastasis dormancy.

Tumour cells that survive dissemination lodge in the target organ parenchyma. This new microenvironment most probably determines the fate of the disseminated tumour cells and could account for most of the dormancy time (Time #1). If the cells are not genetically progressed it is possible that they are unable to grow autonomously or transduce growth signals from the microenvironment, instead entering a quiescence-like phenotype. Stress from dissemination might contribute to activating growth arrest programmes. Even with genetic alterations, stress and/or microenvironment signals might impose a growth-suppressive programme. For tumour stem cells, a quiescent state might be a natural response to a microenvironment that lacks recruitment signals. Normal differentiated cells can remain growth arrested for years and solitary cells are found years after surgery, suggesting that a prolonged tumour cell arrest might be plausible. Upon exit from quiescence, tumour cells can fully progress into overt lesions. It is possible that before becoming overt lesions, dormancy might continue (Time #2) owing to the immune system preventing tumour expansion. The immune system can control pathogens during a lifetime. Therefore, it might prevent tumour mass expansion for long periods. After exit from quiescence or evasion of the immune system a tumour cell mass can enter angiogenic dormancy. Differentiated tissues such as the retinal pigment epithelium, which produces angiogenesis inhibitors (pigment epithelium-derived factor 126), can maintain the vasculature from expanding for long periods and therefore prevent diseases such as macular degeneration. However, it is still unclear how long (Time #3) this mechanism can be maintained in a genetically unstable proliferative tumour cell population, which probabilistically should be prone to accumulating new genetic alterations that activate the angiogenic switch.
Interruption of dormancy through the re-induction of angiogenesis is poorly understood (FIG. 2). For example, in a non-orthotopic mouse model of subcutaneous lymphoma growth caused by inducible MYC expression, sustained cancer regression can be observed upon oncogene de-induction57. However, in lymphomas that are p53 null, a disease-free period is invariably followed by relapse, which is inhibited by the expression of the p53 target gene thrombospondin 1, an anti-angiogenic factor57. Additional studies show that a brief and strong angiogenic stimulus might reverse angiogenic dormancy58. Human leukaemia cells (MOlT-3 cells) that are unable to form growing tumours in non-obese diabetic severe combined immunodeficient (NOD/SCID) mice but form dormant lesions can resume growth after a brief stimulation with VEGF or co-inoculation with angiogenic-competent Kaposi sarcoma cells. This stimulation was suppressed by exogenous angiogenesis inhibitors58. Growing and dormant tumour cells express similar levels of pro-angiogenic factors, suggesting that re-initiation of angiogenesis might need higher levels of angiogenic inducers than vascular maintenance. Alternatively, although not addressed by the authors58, downregulation of endogenous angiogenic inhibitors after the pro-angiogenic ‘spike’ might explain this result.
The above studies were carried out on primary tumour masses, but it is less clear whether angiogenic dormancy occurs in patient metastases or whether current anti-angiogenic therapies59 induce angiogenic dormancy. evidence from experimental models using lewis lung carcinoma and T241 fibrosarcoma cells in C57Bl6/J mice supports dormant metastases being in a balance between mitosis and apoptosis60. However, this must occur after a period of expansion because disseminated tumour cells can grow intravascularly, bypassing the need for new blood vessels61. In addition, clinical evidence for this mechanism is weak owing in part to the difficulty of detecting and molecularly characterizing micrometastatic lesions or accessing these samples after autopsy. Furthermore, there is evidence that solitary disseminated tumour cells in animal models and in patients remain growth arrested and do not express proliferation markers13,62 (see below). An unanswered question about angiogenic dormancy is how the angiogenic switch can remain inactive for years in a dynamically dividing tumour cell population that is prone to accumulating additional genetic changes that could activate this switch. Given that solitary tumour cells survive on the pre-existing vasculature, a cessation of proliferation triggered by signals that are determined by the new tissue microenvironment, as discussed previously, is the most probable explanation for their cellular dormancy32. Alternatively, a tumour mass, although angiogenesis competent, might be prevented from expanding and kept dormant by an active immune response63.
The immune system and tumour dormancy
The role of the immune system in controlling tumour growth has been recognized for decades63-65. Studies using syngeneic animals that are immunized with a subcutaneous implantation of tumour cells and then challenged by intraperitoneal injection of the same cells have shown that the immune system can target and kill most of the tumour cells in the challenge injection63,66. These responses are mostly mediated by cytotoxic CD8+ T lymphocytes, which induce cytolysis of the tumour cells63 (FIG. 2). However, it seems that some residual cells persist and this population might be kept clinically dormant by the immune system64,67. Therefore, the immune system prevents the expansion of proliferating tumour cells66. In other studies, interruption of tumour dormancy of mouse lymphoma and leukaemia was proposed to result from reduced expression of tumour-associated antigens and evasion of the immune system66,67. Studies of BCl1 mouse lymphoma that developed spontaneously in a BAlB/c mouse revealed that a humoral response that controls a regulatory network of anti-idiotypic antibodies activates B-cell receptor signalling, which inhibits proliferation and is followed by apoptosis or dormancy (for a comprehensive review see REF. 68). In this model dormancy was also shown to be mediated by CD8+ T cells69. By contrast, residual and dormant DA1-3b mouse acute myeloid leukaemia cells overexpressed B7H1 (also known as CD274 antigen and programmed cell death 1 ligand 1) or B7.1 (B-lymphocyte activation antigen B7) co-stimulatory molecules70. These were proposed to abrogate cytotoxic CD8+ T-cell-mediated responses allowing dormant leukaemia cells (that is, those in cellular dormancy) to evade recognition by the immune system70 (FIG. 2). However, it is not clear whether other mechanisms might be required because B7H1 does not mediate anti-tumour immunity in other models70.
Additional studies have shown that proliferating mouse lymphoma cells are kept at a low number in the bone marrow owing to persistent antigen and memory T cells that are able to coordinate a CD4+ and CD8+ T-cell-mediated response71-73. These results correlate with clinical studies showing that in the bone marrow of patients with breast cancer who carry cytokeratin-positive cells (breast cancer cells), a higher proportion of memory T cells among the CD4+ and CD8+ cells correlated with larger tumours72. These results suggest that in some situations the immune system might still be operating to suppress residual tumour cell expansion, whereas other mechanisms of dormancy favour immune-system evasion (FIG. 2). Additional studies are needed because it is still unclear whether immuno-surveillance is an operational mechanism controlling dormancy of non-virally induced tumours74. How the immunological response intersects with angiogenic dormancy or cellular dormancy is unknown. It is possible, although unknown, that the genes that control escape from dormancy (for example, quiescence or angiogenic-switch dependent) might also coordinate the ability of tumour cells to evade an immune response.
Clinical implications of cancer dormancy
Characterization of minimal residual disease revealed that these cells can remain dormant and perhaps some of the mechanisms described above (for example, enhanced uPAR and/or eRBB2 signalling) might explain the transition between dormancy and recurrence. Furthermore, the potential existence of tumour stem cells and drug resistance mechanisms, although less characterized in residual tumour cells, might help explain the biology behind dormancy of minimal residual disease (FIG. 3). of note is the fact that although the evidence from epithelial and haematopoietic cancer dormancy is discussed jointly, ultimately some of the mechanisms might diverge because of intrinsic differences between these tumour types. For example, mechanisms that depend on the regulation of uPAR and eRBB2 will most probably not be applicable to lymphoma dormancy. However, although the dormancy-initiating signals might be different in these cancers, it is possible that they might converge on similar general mechanisms (that is, transcriptional repression or methylation) to induce, for example, tumour cell quiescence.
Disseminated tumour cells, minimal residual disease and dormancy
Minimal residual disease is a significant problem as it is observed in many cancers and, in breast cancer for example, the relapse rate after >5 years is about 20% (REF. 75; TABLE 1). These residual tumour cells are usually found in circulation or in the bone marrow (FIGS 2,3), which are the most commonly inspected sites, but they most probably lodge in several organs that are not usually scrutinized. Minimal residual disease can also be detected in the lymph nodes or target organs for metastases, such as liver or lung1. The population of disseminated tumour cells in the bone marrow is of significance as increased counts of cells that express certain biomarkers, such as cytokeratin (see below), indicate poor prognosis even for tumours that do not usually metastasize to the bone (for example, colon cancer)1,76. Therefore, detecting these cells in bone marrow might be useful for predicting bone or other secondary organ metastases (for reviews see REFS 1,13).
Characterization of disseminated tumour cells has proven to be extremely difficult because of their low abundance (1 disseminated tumour cell per 106 bone marrow cells and 1–2 disseminated tumour cells per 20 ml of blood)13,77. Recent intriguing studies have shown that patients who are free of clinically detectable disease >20 years after treatment still have circulating disseminated tumour cells77. It was proposed that because tumour cells could not persist arrested in circulation for so long, a tumour stem cell population that sheds tumour cells into circulation might be responsible for this phenomenon77 (see below) (FIGS 2,3).
Disseminated tumour cells in the bone marrow can be examined for expression or overexpression of specific biomarkers, such as Ki67, p120 (also known as CTNND1), EGFR, ERBB2, extracellular matrix metallo-proteinase inducer (eMMPRIn) or uPAR; for genomic rearrangements (by CGH); or for mutations in specific genes (for example, KRAS and TP53)1,8. However, only a few studies have looked at stem cell markers and it is not clear if these serve as prognostic indicators78 (see below). In other studies, the absence or low proportion of disseminated tumour cells staining for markers such as Ki67 and proliferating cell nuclear antigen (PCnA)1,2,8,77,79 indicates that these cells are in G0–G1 arrest, which is indicative of quiescence as a form of cellular dormancy (FIG. 3). However, the mechanisms that are operational in these cells are poorly understood because simultaneous detection of multiple markers is usually difficult. In gastric cancer, the detection of uPAR in cytokeratin-positive disseminated tumour cells in the bone marrow predicted relapse28. By contrast, disseminated tumour cells that lacked uPAR staining predicted longer disease-free periods and better overall survival28. However, whether lack of uPAR staining correlated with the absence of proliferation markers is unknown.
On the basis of the data discussed above it is tempting to speculate that in disseminated tumour cells downregulation of uPAR might be responsible for their dormancy19,22,23,28,76,80,81 (FIG. 3). Similar findings were reported for prognosis and expression of eRBB2 in disseminated breast cancer cells13. It is possible that using the inducible systems for eRBB2 expression82, as well as other oncogenes (for example, MYC and Wnt57,83,84) some of these questions could be modelled in mice. However, it is unclear whether there is reduced eRBB2 expression, despite the presence of gene amplification in some cases, in disseminated tumour cells, so the relevance of the inducible systems is uncertain. Similarly, it is important to determine whether disseminated tumour cells that express eRBB2 also express proliferation markers. If not, then the mechanisms by which eRBB2-positive cells become growth arrested should be explored. If uPAR and eRBB2 upregulation serve as a switch to interrupt dormancy, these might be important targets for combination therapy of residual disease. Interestingly, the PLAUR (encoding uPAR) and ERBB2 genes were found to be co-amplified in disseminated breast cancer cells81. Recently, identification of the interaction site for α5β1 integrin with domain III of uPAR85 and the development of monoclonal antibodies that target this region19,86 has provided an excellent opportunity to target this mitogenic pathway85 in combination with anti-ERBB2 drugs (for example, trastuzumab, lapatinib and EKB-569). Together, the available data suggest that disseminated tumour cells can be found in a non-proliferating state and that some of the mechanisms that have been discovered in experimental models might aid in the understanding of disseminated tumour cell behaviour as well as providing new markers or targets for therapeutic applications.
Dormancy of normal and tumour stem cells
It is still unclear whether stem cells are indeed the cells that harbour the genetic alterations that cause cancer. However, properties of normal stem cells, such as their quiescent state within a niche that is both nurturing and protective87, might potentially explain tumour cell dormancy. Specific recruitment signals, such as tissue injury, activate stem cells to exit quiescence and proliferate88; tumour stem cells might also retain this property89,90. Therefore, current hypotheses suggest that metastases are the consequence of disseminated tumour stem cells that, after undergoing a state of quiescence, subsequently resume growth (FIG. 3). Recent studies report that disseminated breast cancer cells in the bone marrow are enriched for markers of breast tumour stem cells: ~65% of cells in the bone marrow were CD44+/CD24-/cytokeratin (CK)+ compared with <10% in the primary tumour78. However, the fate of these cells is unknown and whether only these cells are the source of relapse is also a mystery. It is also important to determine whether CD44+/CD24-/CK+ cells in the primary lesion, lymph nodes or bone marrow are in different proliferating states. The existence of tumour stem cell quiescence also implies that in those cells with defined genetic abnormalities that drive tumour progression (that is, oncogene addiction) the stem cell quiescence (cellular dormancy) programme might be dominant over oncogenic signalling. For example, interferon therapy can induce durable remission in a minority of patients who display residual Philadelphia chromosome (BCR–ABl oncogene)-positive chronic myeloid leukaemia (CMl) progenitors90. Furthermore, although genomic BCR–ABl-positive leukaemia cells can persist in CMl patients who exhibit complete cytogenetic remission, the BCR–ABl transcript levels are absent or extremely low90,91. These studies suggest that despite the presence of the oncogenic aberration(s), other signals can overcome oncogene expression in dormant cells. Although these are exciting hypotheses, much work needs to be done to understand whether tumour stem cells are indeed the origin of relapse.
Drug resistance of dormant cancer
It is assumed that dormant disease is chemotherapy resistant because dormant cells are not dividing1,9,92. However, it is still unclear whether this mechanism explains cancer cell drug resistance in patients or whether the active survival mechanisms that have been discovered in experimental models protect disseminated tumour cells. Induction of p21 or p27 in colon cancer cells causes G1 arrest and doxorubicin resistance in vitro 93. In vivo studies of green fluorescent protein (GFP)-tagged disseminated breast cancer cells showed that they are growth arrested and resistant to doxorubicin92. In addition, active mechanisms might protect dormant cells from chemotherapy. Breast cancer cells that re-differentiate after inhibition of β1 integrin activity are more resistant to chemotherapy94. Surprisingly, this is independent of proliferation but dependent on the level of polarity (appropriate polarity activates an anti-apoptotic β4-integrin–nuclear factor-κB (NFκB) pathway94) and differentiation (FIG. 3). In squamous carcinoma cells in which a low eRK:p38 activity ratio induces dormancy, activation and induction of the eIF2α kinase RnA-dependent protein kinase-like endoplasmic reticulum kinase (eIF2AK3, also known as PeRK) and the chaperone protein HSPA5, respectively, protected dormant cells from chemotherapy independently of proliferation. HSPA5 did so by inhibiting BAX (BCl2-associated protein X) activation26. Accordingly, HSPA5 has been shown to protect breast cancer cells from oestrogen starvation through inhibition of the pro- apoptotic proteins BIK (BCl2-interacting killer) and BAX95. Interestingly, in breast and prostate primary tumours, HSPA5 detection is prognostic for a shorter time to recurrence and a poor response to either adriamycin in patients with breast cancer or hormonal ablation in patients with prostate cancer96,97. Therefore, disseminated tumour cells expressing HSPA5 might be more refractory to treatment (FIG. 3). Finally, normal and tumour stem cells express ABC transporters such as ABCG2 (ATP-binding cassette G2)98,99. In neuroblastoma, cancer stem cells expressing ABCA3 are mitoxantrone resistant100. However, the link between ABC transporter expression in disseminated tumour cells, relapse and poor prognosis is still unknown. More detailed analysis of these mechanisms and detection of these markers in disseminated tumour cells will be important for the choice of therapy when patients are positively identified as bearing minimal residual disease.
Future directions
The future offers two exciting possibilities: first, to induce and/or maintain dormancy of tumour cells, and second, to induce cell death in residual dormant cells by targeting their survival and drug resistance mechanisms. It will also be important to determine whether a patient has dormant disease and what type of mechanism is active. The models described here will be instrumental in identifying the mechanisms and markers of dormant cancer. Cellular or serum biomarkers might help detect dormant disease, which will require detection of low abundance cells or circulating biomarkers. In addition, transcriptional profiles from dormant disseminated tumour cells or experimental models of dormancy might help determine whether primary tumours carry a cancer dormancy ‘signature’, which might have prognostic value101.
Many questions remain. For example, do the mechanisms of dormancy described in this article occur independently or are they all part of progression? (See FIG. 4 for an integration hypothesis.) Is dormancy a phenomenon of less progressed cancers? For example, are patients who relapse within months devoid of any dormant disease because of highly progressed tumour cells that can adapt and grow in the new microenvironment, while less progressed disseminated tumour cells can only adapt and survive in a dormant state? Do different types of tumour undergo dormancy through particular mechanisms? Can tumour-initiating (stem) cells undergo angiogenic dormancy and/or can they evade immuno-surveillance? What happens to survival of tumour cells that are highly dependent on an oncogene102 when, despite carrying oncogenic abnormalities, they become quiescent? Finally, it will be important to use the available mouse models of cancer to model more accurately aspects of human cancer progression including, but not limited to, early dissemination, surgery of a tumour mass, chemotherapy, residual disease and spontaneous, rather than experimental (that is, intravenously injected cells), metastases. The importance and urgency in understanding cancer dormancy is highlighted by a recent report on a u.S. national Cancer Institute-organized workshop on tumour cell dormancy103. The future understanding of cancer dormancy will most likely lead to a shift in how we think about cancer progression and in consequence how we diagnose and treat the disease.
Acknowledgments
I apologize to those authors whose work I could not cite directly because of space constraints. I would like to thank the members of my laboratory and L. Ossowski (Mount Sinai School of Medicine) for stimulating discussions and critical reading of the manuscript. I also thank D. Schewe for discussions and help with the figures. This work is supported by a grant from the Samuel Waxman Cancer Research Foundation Tumour Dormancy Program and the NIH/National Cancer Institute grant CA109182 to J.A.A.G.
- Disseminated tumour cells
Tumour cells that have physically separated from the primary tumour and spread to other anatomical locations through circulation. In this Review these tumour cells are not yet considered micrometastases as they have not yet expanded to form a small population of cells.
- Minimal residual disease
Remnant tumour cells that are left after treatment and that cannot be detected by conventional clinical testing. These cells can persist in the primary site or as disseminated tumour cells in proliferative and/or dormant phases.
- Dormant
Applies to cells in a non-dividing state whose physiological functions become paused or quiescent. In several organisms, such as Caenorhabditis elegans, dormant is a synonym for diapause (pause in development). Plant seeds are also dormant during the latency phase before germination.
- Tumour mass dormancy
Cancer cell proliferation that is counterbalanced by apoptosis owing to poor vascularization (see angiogenic dormancy) or by an immune response. In this case the cancer cells are never truly inactive, but rather are incapable of expanding beyond a certain number.
- Cellular dormancy
When normal cells enter the G0 phase of the cell cycle and have low metabolism. This might apply to cancer cells that enter a G0–G1 arrest. This form of dormancy is clinically asymptomatic and the tumour cells are truly inactive.
- Angiogenic dormancy
Occurs when cancer cell proliferation is counterbalanced by apoptosis owing to poor vascularization. In this case, the cancer cells are incapable of expanding beyond a certain number and although clinically asymptomatic the tumour cells are not truly inactive.
- Genetically fit
The concept of evolutionary genetic fitness. Having the appropriate traits and properties to survive and grow in a particular environment.
- Immortalization
Cells with genetic alterations that become insensitive to the Hayflick limit (the number of times a normal cell can divide in vitro before dying or entering senescence). Immortalized cells evade senescence and proliferate limitlessly in culture but are unable to form tumours.
- Transformation
Cells that have been immortalized but now acquire additional genetic and epigenetic alterations that allow them to form primary tumours.
- Micrometastasis
A small group of tumour cells, derived from a disseminated tumour cell, that has grown in secondary organs but is too small to be seen or detected by available methods. These lesions might also be clinically asymptomatic.
- Orthotopic
The growth of transplanted cells or tissues in the normal anatomical position and tissue of origin.
- Niche
A term borrowed from ecology, it refers to a unique and optimal tissue microenvironment with defined nurturing and positional cues in a given anatomical location that allows a cell or group of cells to survive and function.
- Reconstituted basement membrane assays
Assays that take advantage of extracellular matrix gels that are derived from a cancer cell line or collagen-I gels. Cells embedded in these matrices can recapitulate the tissue organization that is observed in the tissue of origin.
- Acinar structures
Sac-like structures that are formed by secretory epithelial cells that have defined apico-basal polarity.
- Fibronectin
An extracellular matrix molecule that is a main constituent of normal tissues but is also present in serum. The α5β1 integrin is its most specific receptor (other integrins can also bind it) and it can serve as a survival or mitogenic factor.
- CD8+ T lymphocytes
Immune cells from the T-cell lineage specialized in killing target cells (that is, virus-infected cells). These responses are usually mediated by class-I major histocompatibility complexes and cytotoxicity is delivered by a repertoire of molecules that are contained in secretory vesicles.
- Neovascularization
The formation of new blood vessels that create new pathways for blood flow during normal tissue development or remodelling, or during pathological conditions such as tumour growth or retinopathy.
- Angiogenic switch
When a small tumour mass senses the lack of appropriate blood supply and activates a series of transcriptional programmes that allows them to produce signals that will recruit new blood vessels.
- Adaptation
Mechanisms that allow normal or tumour cells to respond to a determined stress with a programme that allows them to correct and survive the imposed stress signals.
- Dauer stage
German for ‘enduring’. It is an alternative larval stage in which development is arrested in response to environmental or hormonal cues in nematodes such as Caenorhabditis elegans. It allows larvae to survive harsh conditions for long periods until the environment is propitious to resume development.
- Unfolded protein response
A cellular response to stress that is unique to the endoplasmic reticulum (ER), and which senses the misfolding of proteins in the ER. It activates a series of pathways that help the cells survive proteotoxicity that is caused by unfolded proteins or activate mechanisms of cell death.
- Humoral response
The component of the immure response that is mediated by secreted antibodies.
- Anti-idiotypic antibodies
Antibodies that are directed towards the hypervariable regions of an antibody.
- Oestrogen starvation
Occurs when cells that are usually dependent on oestrogen for normal cellular functions are deprived of this hormone. This is usually done in cell culture by leaving oestrogen out of the tissue culture medium.
- ABC transporters
ATP-dependent protein pumps that extrude numerous compounds from the cell cytoplasm. These can be exogenous molecules such as drugs, or physiological membrane constituents such as cholesterol.
References
- 1.Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nature Rev Cancer. 2004;4:448–456. doi: 10.1038/nrc1370. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt-Kittler O, et al. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci U S A. 2003;100:7737–7742. doi: 10.1073/pnas.1331931100.First evidence that breast cancer cells can disseminate in a far less progressed genomic state than previously thought. Genomic aberrations that make them metastatic are acquired after this step.
- 3.Karrison TG, Ferguson DJ, Meier P. Dormancy of mammary carcinoma after mastectomy. J Natl Cancer Inst. 1999;91:80–85. doi: 10.1093/jnci/91.1.80. [DOI] [PubMed] [Google Scholar]
- 4.Pfitzenmaier J, et al. Telomerase activity in disseminated prostate cancer cells. BJU Int. 2006;97:1309–1313. doi: 10.1111/j.1464-410X.2006.06194.x. [DOI] [PubMed] [Google Scholar]
- 5.Weckermann D, et al. Disseminated cytokeratin positive tumor cells in the bone marrow of patients with prostate cancer: detection and prognostic value. J Urol. 2001;166:699–703. [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 7.Talmadge JE, Wolman SR, Fidler IJ. Evidence for the clonal origin of spontaneous metastases. Science. 1982;217:361–363. doi: 10.1126/science.6953592.First evidence that spontaneous metastases are clonal in origin and that they probably are derived from different progenitor cells.
- 8.Schardt JA, et al. Genomic analysis of single cytokeratin-positive cells from bone marrow reveals early mutational events in breast cancer. Cancer Cell. 2005;8:227–239. doi: 10.1016/j.ccr.2005.08.003.This paper genetically characterized disseminated single breast tumour cells and determined that cells with normal karyotypes can disseminate, suggesting that dissemination might occur very early.
- 9.Demicheli R. Tumour dormancy: findings and hypotheses from clinical research on breast cancer. Semin Cancer Biol. 2001;11:297–306. doi: 10.1006/scbi.2001.0385. [DOI] [PubMed] [Google Scholar]
- 10.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nature Rev Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 11.Heyn C, et al. In vivo MRI of cancer cell fate at the single-cell level in a mouse model of breast cancer metastasis to the brain. Magn Reson Med. 2006;56:1001–1010. doi: 10.1002/mrm.21029. [DOI] [PubMed] [Google Scholar]
- 12.Klein CA, Hölzel D. Systemic cancer progression and tumor dormancy: mathematical models meet single cell genomics. Cell Cycle. 2006;5:1788–1798. doi: 10.4161/cc.5.16.3097. [DOI] [PubMed] [Google Scholar]
- 13.Lacroix M. Significance, detection and markers of disseminated breast cancer cells. Endocr Relat Cancer. 2006;13:1033–1067. doi: 10.1677/ERC-06-0001. [DOI] [PubMed] [Google Scholar]
- 14.Kovacs AF, Ghahremani MT, Stefenelli U, Bitter K. Postoperative chemotherapy with cisplatin and 5-fluorouracil in cancer of the oral cavity and the oropharynx--long-term results. J Chemother. 2003;15:495–502. doi: 10.1179/joc.2003.15.5.495. [DOI] [PubMed] [Google Scholar]
- 15.Boudreau N, Bissell MJ. Extracellular matrix signaling: integration of form and function in normal and malignant cells. Curr Opin Cell Biol. 1998;10:640–646. doi: 10.1016/s0955-0674(98)80040-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wicha MS. Cancer stem cells and metastasis: lethal seeds. Clin Cancer Res. 2006;12:5606–5607. doi: 10.1158/1078-0432.CCR-06-1537. [DOI] [PubMed] [Google Scholar]
- 17.Kaplan RN, Rafii S, Lyden D. Preparing the “soil”: the premetastatic niche. Cancer Res. 2006;66:11089–11093. doi: 10.1158/0008-5472.CAN-06-2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aguirre-Ghiso JA, Ossowski L, Rosenbaum SK. Green fluorescent protein tagging of extracellular signal-regulated kinase and p38 pathways reveals novel dynamics of pathway activation during primary and metastatic growth. Cancer Res. 2004;64:7336–7345. doi: 10.1158/0008-5472.CAN-04-0113.Provides evidence that spontaneous or forced downregulation of uPAR induces a state of tumor cell dormancy through decreased ERK signalling and G0–G1 arrest.
- 19.Aguirre Ghiso JA, Kovalski K, Ossowski L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J Cell Biol. 1999;147:89–104. doi: 10.1083/jcb.147.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.White DE, et al. Targeted disruption of β1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer Cell. 2004;6:159–170. doi: 10.1016/j.ccr.2004.06.025.Provides evidence that mouse mammary epithelial cells transformed with an oncogene require β1 integrin for tumour progression. Absence of this integrin results in a state of tumour cell dormancy.
- 21.Weaver VM, et al. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137:231–245. doi: 10.1083/jcb.137.1.231.Provides evidence that correcting the crosstalk between the tumour cell and the microenvironment can result in loss of malignancy associated with a more differentiated phenotype.
- 22.Liu D, Aguirre Ghiso J, Estrada Y, Ossowski L. EGFR is a transducer of the urokinase receptor initiated signal that is required for in vivo growth of a human carcinoma. Cancer Cell. 2002;1:445–457. doi: 10.1016/s1535-6108(02)00072-7.Shows that loss of uPAR expression causes tumour cell dormancy by impairing EGFR activation by integrins that transduce mitogenic signals from the extracellular matrix molecule fibronectin.
- 23.Aguirre Ghiso JA. Inhibition of FAK signaling activated by urokinase receptor induces dormancy in human carcinoma cells in vivo. Oncogene. 2002;21:2513–2524. doi: 10.1038/sj.onc.1205342. [DOI] [PubMed] [Google Scholar]
- 24.Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol Biol Cell. 2001;12:863–879. doi: 10.1091/mbc.12.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK) Cancer Res. 2003;63:1684–1695. [PubMed] [Google Scholar]
- 26.Ranganathan AC, Zhang L, Adam AP, Aguirre-Ghiso JA. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006;66:1702–1711. doi: 10.1158/0008-5472.CAN-05-3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bulavin DV, Fornace AJ., Jr p38 MAP kinase’s emerging role as a tumor suppressor. Adv Cancer Res. 2004;92:95–118. doi: 10.1016/S0065-230X(04)92005-2. [DOI] [PubMed] [Google Scholar]
- 28.Heiss MM, et al. Individual development and uPA-receptor expression of disseminated tumour cells in bone marrow: a reference to early systemic disease in solid cancer. Nature Med. 1995;1:1035–1039. doi: 10.1038/nm1095-1035.Shows that patients with tumour relapse had an increase or higher number of disseminated tumour cells in bone marrow aspirates, whereas patients without recurrence had negative or low tumour cell counts. uPAR expression on disseminated tumour cells was correlated with increasing tumour cell counts and poor prognosis.
- 29.Gattelli A, et al. Progression of pregnancy-dependent mouse mammary tumors after long dormancy periods. Involvement of Wnt pathway activation. Cancer Res. 2004;64:5193–5199. doi: 10.1158/0008-5472.CAN-03-3992. [DOI] [PubMed] [Google Scholar]
- 30.Gattelli A, Zimberlin MN, Meiss RP, Castilla LH, Kordon EC. Selection of early-occurring mutations dictates hormone-independent progression in mouse mammary tumor lines. J Virol. 2006;80:11409–11415. doi: 10.1128/JVI.00234-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Naumov GN, et al. Cellular expression of green fluorescent protein, coupled with high-resolution in vivo videomicroscopy, to monitor steps in tumor metastasis. J Cell Sci. 1999;112(Pt 12):1835–1842. doi: 10.1242/jcs.112.12.1835. [DOI] [PubMed] [Google Scholar]
- 32.Naumov GN, et al. Persistence of solitary mammary carcinoma cells in a secondary site: a possible contributor to dormancy. Cancer Res. 2002;62:2162–2168.This paper used an experimental model to show that solitary dormant cells that persist in target organs might be the source of dormancy and recurrence.
- 33.Steeg PS. Metastasis suppressors alter the signal transduction of cancer cells. Nature Rev Cancer. 2003;3:55–63. doi: 10.1038/nrc967. [DOI] [PubMed] [Google Scholar]
- 34.Steeg PS, Ouatas T, Halverson D, Palmieri D, Salerno M. Metastasis suppressor genes: basic biology and potential clinical use. Clin Breast Cancer. 2003;4:51–62. doi: 10.3816/cbc.2003.n.012. [DOI] [PubMed] [Google Scholar]
- 35.Mehlen P, Puisieux A. Metastasis: a question of life or death. Nature Rev Cancer. 2006;6:449–458. doi: 10.1038/nrc1886. [DOI] [PubMed] [Google Scholar]
- 36.Bandyopadhyay S, et al. Interaction of KAI1 on tumor cells with DARC on vascular endothelium leads to metastasis suppression. Nature Med. 2006;12:933–938. doi: 10.1038/nm1444. [DOI] [PubMed] [Google Scholar]
- 37.Rinker-Schaeffer CW, O’Keefe JP, Welch DR, Theodorescu D. Metastasis suppressor proteins: discovery, molecular mechanisms, and clinical application. Clin Cancer Res. 2006;12:3882–3889. doi: 10.1158/1078-0432.CCR-06-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hickson JA, et al. The p38 kinases MKK4 and MKK6 suppress metastatic colonization in human ovarian carcinoma. Cancer Res. 2006;66:2264–2270. doi: 10.1158/0008-5472.CAN-05-3676. [DOI] [PubMed] [Google Scholar]
- 39.Kauffman EC, Robinson VL, Stadler WM, Sokoloff MH, Rinker-Schaeffer CW. Metastasis suppression: the evolving role of metastasis suppressor genes for regulating cancer cell growth at the secondary site. J Urol. 2003;169:1122–1133. doi: 10.1097/01.ju.0000051580.89109.4b. [DOI] [PubMed] [Google Scholar]
- 40.Nash KT, et al. Requirement of KISS1 secretion for multiple organ metastasis suppression and maintenance of tumor dormancy. J Natl Cancer Inst. 2007;99:309–321. doi: 10.1093/jnci/djk053.First evidence that a metastasis suppressor gene functions though the induction of dormancy.
- 41.Vander Griend DJ, et al. Suppression of metastatic colonization by the context-dependent activation of the c-Jun NH2-terminal kinase kinases JNKK1/MKK4 and MKK7. Cancer Res. 2005;65:10984–10991. doi: 10.1158/0008-5472.CAN-05-2382. [DOI] [PubMed] [Google Scholar]
- 42.Nakayama K, et al. Homozygous deletion of MKK4 in ovarian serous carcinoma. Cancer Biol Ther. 2006;5:630–634. doi: 10.4161/cbt.5.6.2675. [DOI] [PubMed] [Google Scholar]
- 43.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 44.Ranganathan AC, Adam AP, Zhang L, Aguirre-Ghiso JA. Tumor cell dormancy induced by p38(SAPK) and ER-stress signaling: an adaptive advantage for metastatic cells? Cancer Biol Ther. 2006;5:729–735. doi: 10.4161/cbt.5.7.2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hunter K. Host genetics influence tumour metastasis. Nature Rev Cancer. 2006;6:141–146. doi: 10.1038/nrc1803. [DOI] [PubMed] [Google Scholar]
- 46.Crawford NP, et al. Germline polymorphisms in SIPA1 are associated with metastasis and other indicators of poor prognosis in breast cancer. Breast Cancer Res. 2006;8:R16. doi: 10.1186/bcr1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park YG, et al. Sipa1 is a candidate for underlying the metastasis efficiency modifier locus Mtes1. Nature Genet. 2005;37:1055–1062. doi: 10.1038/ng1635.Provides evidence that constitutional genetic polymorphisms in a gene can affect tumour metastasis.
- 48.Semenza GL. Targeting HIF-1 for cancer therapy. Nature Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 49.Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15–18. doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- 50.Naumov GN, Akslen LA, Folkman J. Role of angiogenesis in human tumor dormancy: animal models of the angiogenic switch. Cell Cycle. 2006;5:1779–1787. doi: 10.4161/cc.5.16.3018. [DOI] [PubMed] [Google Scholar]
- 51.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nature Rev Cancer. 2003;3:401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 52.Naumov GN, et al. A model of human tumor dormancy: an angiogenic switch from the nonangiogenic phenotype. J Natl Cancer Inst. 2006;98:316–325. doi: 10.1093/jnci/djj068. [DOI] [PubMed] [Google Scholar]
- 53.Watnick RS, Cheng YN, Rangarajan A, Ince TA, Weinberg RA. Ras modulates Myc activity to repress thrombospondin-1 expression and increase tumor angiogenesis. Cancer Cell. 2003;3:219–231. doi: 10.1016/s1535-6108(03)00030-8. [DOI] [PubMed] [Google Scholar]
- 54.Volpert OV, Alani RM. Wiring the angiogenic switch: Ras, Myc, and Thrombospondin-1. Cancer Cell. 2003;3:199–200. doi: 10.1016/s1535-6108(03)00056-4. [DOI] [PubMed] [Google Scholar]
- 55.Okajima E, Thorgeirsson UP. Different regulation of vascular endothelial growth factor expression by the ERK and p38 kinase pathways in v-ras, v-raf, and v-myc transformed cells. Biochem Biophys Res Commun. 2000;270:108–111. doi: 10.1006/bbrc.2000.2386. [DOI] [PubMed] [Google Scholar]
- 56.Guba M, et al. A primary tumor promotes dormancy of solitary tumor cells before inhibiting angiogenesis. Cancer Res. 2001;61:5575–5579. [PubMed] [Google Scholar]
- 57.Giuriato S, et al. Sustained regression of tumors upon MYC inactivation requires p53 or thrombospondin-1 to reverse the angiogenic switch. Proc Natl Acad Sci USA. 2006;103:16266–16271. doi: 10.1073/pnas.0608017103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Indraccolo S, et al. Interruption of tumor dormancy by a transient angiogenic burst within the tumor microenvironment. Proc Natl Acad Sci USA. 2006;103:4216–4221. doi: 10.1073/pnas.0506200103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ruegg C, Mutter N. Anti-angiogenic therapies in cancer: achievements and open questions. Bull Cancer. 2007;94:753–762. [PubMed] [Google Scholar]
- 60.Holmgren L, O’Reilly MS, Folkman J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nature Med. 1995;1:149–153. doi: 10.1038/nm0295-149.Shows in a mouse model that the balance between apoptosis and mitosis caused by lack of vascularization can induce angiogenic dormancy.
- 61.Al-Mehdi AB, et al. Intravascular origin of metastasis from the proliferation of endothelium-attached tumor cells: a new model for metastasis. Nature Med. 2000;6:100–102. doi: 10.1038/71429.Shows in a mouse model that disseminated tumour cells can start growing intravascularly into metastases, bypassing any initial need for blood vessels.
- 62.Pantel K, Otte M. Disseminated tumor cells: diagnosis, prognostic relevance, and phenotyping. Recent Results Cancer Res. 2001;158:14–24. doi: 10.1007/978-3-642-59537-0_2. [DOI] [PubMed] [Google Scholar]
- 63.Finn OJ. Human tumor antigens, immunosurveillance, and cancer vaccines. Immunol Res. 2006;36:73–82. doi: 10.1385/IR:36:1:73. [DOI] [PubMed] [Google Scholar]
- 64.Weinhold KJ, Miller DA, Wheelock EF. The tumor dormant state. Comparison of L5178Y cells used to establish dormancy with those that emerge after its termination. J Exp Med. 1979;149:745–757. doi: 10.1084/jem.149.3.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nature Rev Cancer. 2005;5:263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 66.Weinhold KJ, Goldstein LT, Wheelock EF. The tumor dormant state. Quantitation of L5178Y cells and host immune responses during the establishment and course of dormancy in syngeneic DBA/2 mice. J Exp Med. 1979;149:732–744. doi: 10.1084/jem.149.3.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matsuzawa A, Takeda Y, Narita M, Ozawa H. Survival of leukemic cells in a dormant state following cyclophosphamide-induced cure of strongly immunogenic mouse leukemia (DL811) Int J Cancer. 1991;49:303–309. doi: 10.1002/ijc.2910490227. [DOI] [PubMed] [Google Scholar]
- 68.Rabinovsky R, Uhr JW, Vitetta ES, Yefenof E. Cancer dormancy: lessons from a B cell lymphoma and adenocarcinoma of the prostate. Adv Cancer Res. 2007;97:189–202. doi: 10.1016/S0065-230X(06)97008-0. [DOI] [PubMed] [Google Scholar]
- 69.Farrar JD, et al. Cancer dormancy. VII. A regulatory role for CD8+ T cells and IFN-γ in establishing and maintaining the tumor-dormant state. J Immunol. 1999;162:2842–2849. [PubMed] [Google Scholar]
- 70.Saudemont A, Quesnel B. In a model of tumor dormancy, long-term persistent leukemic cells have increased B7-H1 and B7.1 expression and resist CTL-mediated lysis. Blood. 2004;104:2124–2133. doi: 10.1182/blood-2004-01-0064. [DOI] [PubMed] [Google Scholar]
- 71.Mahnke YD, Schwendemann J, Beckhove P, Schirrmacher V. Maintenance of long-term tumour-specific T-cell memory by residual dormant tumour cells. Immunology. 2005;115:325–336. doi: 10.1111/j.1365-2567.2005.02163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Feuerer M, et al. Enrichment of memory T cells and other profound immunological changes in the bone marrow from untreated breast cancer patients. Int J Cancer. 2001;92:96–105. [PubMed] [Google Scholar]
- 73.Muller M, et al. EblacZ tumor dormancy in bone marrow and lymph nodes: active control of proliferating tumor cells by CD8+ immune T cells. Cancer Res. 1998;58:5439–5446. [PubMed] [Google Scholar]
- 74.Willimsky G, Blankenstein T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature. 2005;437:141–146. doi: 10.1038/nature03954. [DOI] [PubMed] [Google Scholar]
- 75.Marches R, Scheuermann RH, Uhr J. Cancer dormancy from mice to man: a review. Cell Cycle. 2006;5:1772–1778. doi: 10.4161/cc.5.16.2995. [DOI] [PubMed] [Google Scholar]
- 76.Allgayer H, et al. Urokinase plasminogen activator receptor (uPA-R.): one potential characteristic of metastatic phenotypes in minimal residual tumor disease. Cancer Res. 1997;57:1394–1399. [PubMed] [Google Scholar]
- 77.Meng S, et al. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res. 2004;10:8152–8162. doi: 10.1158/1078-0432.CCR-04-1110.Provides evidence that patients with breast cancer who have been free of disease for 22 years still carry circulating tumour cells in their blood. It is proposed that an occult replicating population is present and sheds cells into the circulation, which eventually die.
- 78.Balic M, et al. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin Cancer Res. 2006;12:5615–5621. doi: 10.1158/1078-0432.CCR-06-0169. [DOI] [PubMed] [Google Scholar]
- 79.Riethmuller G, Klein CA. Early cancer cell dissemination and late metastatic relapse: clinical reflections and biological approaches to the dormancy problem in patients. Semin Cancer Biol. 2001;11:307–311. doi: 10.1006/scbi.2001.0386. [DOI] [PubMed] [Google Scholar]
- 80.Heiss MM, et al. Minimal residual disease in gastric cancer: evidence of an independent prognostic relevance of urokinase receptor expression by disseminated tumor cells in the bone marrow. J Clin Oncol. 2002;20:2005–2016. doi: 10.1200/jco.2002.08.003. [DOI] [PubMed] [Google Scholar]
- 81.Meng S, et al. uPAR and HER-2 gene status in individual breast cancer cells from blood and tissues. Proc Natl Acad Sci USA. 2006;103:17361–17365. doi: 10.1073/pnas.0608113103.Provides evidence that circulating tumour cells and tumour tissues from patients with cancer display co-amplification of uPAR and ERBB2.
- 82.Moody SE, et al. Conditional activation of Neu in the mammary epithelium of transgenic mice results in reversible pulmonary metastasis. Cancer Cell. 2002;2:451–461. doi: 10.1016/s1535-6108(02)00212-x. [DOI] [PubMed] [Google Scholar]
- 83.Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4:199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- 84.Gestl SA, Leonard TL, Biddle JL, Debies MT, Gunther EJ. Dormant wnt-initiated mammary cancer can participate in reconstituting functional mammary glands. Mol Cell Biol. 2007;27:195–207. doi: 10.1128/MCB.01525-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chaurasia P, et al. A region in urokinase plasminogen receptor domain III controlling a functional association with alpha5beta1 integrin and tumor growth. J Biol Chem. 2006;281:14852–14863. doi: 10.1074/jbc.M512311200. [DOI] [PubMed] [Google Scholar]
- 86.Hoyer-Hansen G, et al. Urokinase-catalysed cleavage of the urokinase receptor requires an intact glycolipid anchor. Biochem J. 2001;358:673–679. doi: 10.1042/0264-6021:3580673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shen Q, et al. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science. 2004;304:1338–1340. doi: 10.1126/science.1095505. [DOI] [PubMed] [Google Scholar]
- 88.Tumbar T, et al. Defining the epithelial stem cell niche in skin. Science. 2004;303:359–363. doi: 10.1126/science.1092436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pelayo R, et al. Cell cycle quiescence of early lymphoid progenitors in adult bone marrow. Stem Cells. 2006;24:2703–2713. doi: 10.1634/stemcells.2006-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Talpaz M, et al. Persistence of dormant leukemic progenitors during interferon-induced remission in chronic myelogenous leukemia. Analysis by polymerase chain reaction of individual colonies. J Clin Invest. 1994;94:1383–1389. doi: 10.1172/JCI117473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kitzis A, et al. Persistence of transcriptionally silent BCR-ABL rearrangements in chronic myeloid leukemia patients in sustained complete cytogenetic remission. Leuk Lymphoma. 2001;42:933–944. doi: 10.3109/10428190109097712. [DOI] [PubMed] [Google Scholar]
- 92.Naumov GN, et al. Ineffectiveness of doxorubicin treatment on solitary dormant mammary carcinoma cells or late-developing metastases. Breast Cancer Res Treat. 2003;82:199–206. doi: 10.1023/B:BREA.0000004377.12288.3c.Provides evidence that solitary non-dividing dormant tumour cells are resistant to chemotherapy.
- 93.Schmidt M, et al. Differential roles of p21(Waf1) and p27(Kip1) in modulating chemosensitivity and their possible application in drug discovery studies. Mol Pharmacol. 2001;60:900–906. doi: 10.1124/mol.60.5.900. [DOI] [PubMed] [Google Scholar]
- 94.Weaver VM, et al. beta4 integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell. 2002;2:205–216. doi: 10.1016/s1535-6108(02)00125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fu Y, Li J, Lee AS. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 2007;67:3734–3740. doi: 10.1158/0008-5472.CAN-06-4594. [DOI] [PubMed] [Google Scholar]
- 96.Lee E, et al. GRP78 as a novel predictor of responsiveness to chemotherapy in breast cancer. Cancer Res. 2006;66:7849–7853. doi: 10.1158/0008-5472.CAN-06-1660. [DOI] [PubMed] [Google Scholar]
- 97.Pootrakul L, et al. Expression of stress response protein Grp78 is associated with the development of castration-resistant prostate cancer. Clin Cancer Res. 2006;12:5987–5993. doi: 10.1158/1078-0432.CCR-06-0133. [DOI] [PubMed] [Google Scholar]
- 98.Scharenberg CW, Harkey MA, Torok-Storb B. The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood. 2002;99:507–512. doi: 10.1182/blood.v99.2.507. [DOI] [PubMed] [Google Scholar]
- 99.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nature Rev Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 100.Hirschmann-Jax C, et al. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci USA. 2004;101:14228–14233. doi: 10.1073/pnas.0400067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Roepman P, et al. An expression profile for diagnosis of lymph node metastases from primary head and neck squamous cell carcinomas. Nature Genet. 2005;37:182–186. doi: 10.1038/ng1502. [DOI] [PubMed] [Google Scholar]
- 102.Weinstein IB. Cancer. Addiction to oncogenes — the Achilles heal of cancer. Science. 2002;297:63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 103.Vessella RL, Pantel K, Mohla S. Tumor cell dormancy: an NCI workshop report. Cancer Biol Ther. 2007;6 doi: 10.4161/cbt.6.9.4828. [DOI] [PubMed] [Google Scholar]
- 104.Beausejour CM, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003;22:4212–4222. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Collado M, Serrano M. The power and the promise of oncogene-induced senescence markers. Nature Rev Cancer. 2006;6:472–476. doi: 10.1038/nrc1884. [DOI] [PubMed] [Google Scholar]
- 106.Xue W, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sarkisian CJ, et al. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nature Cell Biol. 2007;9:493–505. doi: 10.1038/ncb1567. [DOI] [PubMed] [Google Scholar]
- 108.Roninson IB. Tumor cell senescence in cancer treatment. Cancer Res. 2003;63:2705–2715. [PubMed] [Google Scholar]
- 109.Zetterberg A, Larsson O. Kinetic analysis of regulatory events in G1 leading to proliferation or quiescence of Swiss 3T3 cells. Proc Natl Acad Sci USA. 1985;82:5365–5369. doi: 10.1073/pnas.82.16.5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang J, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- 111.Coller HA, Sang L, Roberts JM. A new description of cellular quiescence. PLoS Biol. 2006;4:e83. doi: 10.1371/journal.pbio.0040083.A comprehensive gene-profiling analysis of quiescence programmes induced by three different stimuli.
- 112.Larsson O, Zetterberg A, Engstrom W. Cell-cycle-specific induction of quiescence achieved by limited inhibition of protein synthesis: counteractive effect of addition of purified growth factors. J Cell Sci. 1985;73:375–387. doi: 10.1242/jcs.73.1.375. [DOI] [PubMed] [Google Scholar]
- 113.Wang J, Kim SK. Global analysis of dauer gene expression in Caenorhabditis elegans. Development. 2003;130:1621–1634. doi: 10.1242/dev.00363. [DOI] [PubMed] [Google Scholar]
- 114.Koornneef M, Bentsink L, Hilhorst H. Seed dormancy and germination. Curr Opin Plant Biol. 2002;5:33–36. doi: 10.1016/s1369-5266(01)00219-9. [DOI] [PubMed] [Google Scholar]
- 115.Pinkston JM, Garigan D, Hansen M, Kenyon C. Mutations that increase the life span of C. elegans inhibit tumor growth. Science. 2006;313:971–975. doi: 10.1126/science.1121908. [DOI] [PubMed] [Google Scholar]
- 116.Kondo M, et al. The p38 signal transduction pathway participates in the oxidative stress-mediated translocation of DAF-16 to Caenorhabditis elegans nuclei. Mech Ageing Dev. 2005;126:642–647. doi: 10.1016/j.mad.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 117.Fukuyama M, Rougvie AE, Rothman JHC. elegans DAF-18/PTEN mediates nutrient-dependent arrest of cell cycle and growth in the germline. Curr Biol. 2006;16:773–779. doi: 10.1016/j.cub.2006.02.073. [DOI] [PubMed] [Google Scholar]
- 118.Long X, et al. TOR deficiency in C. elegans causes developmental arrest and intestinal atrophy by inhibition of mRNA translation. Curr Biol. 2002;12:1448–1461. doi: 10.1016/s0960-9822(02)01091-6. [DOI] [PubMed] [Google Scholar]
- 119.Solomon A, et al. Caenorhabditis elegans OSR-1 regulates behavioral and physiological responses to hyperosmotic environments. Genetics. 2004;167:161–170. doi: 10.1534/genetics.167.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Merlo LM, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nature Rev Cancer. 2006;6:924–935. doi: 10.1038/nrc2013. [DOI] [PubMed] [Google Scholar]
- 121.Kitano H. Cancer as a robust system: implications for anticancer therapy. Nature Rev Cancer. 2004;4:227–235. doi: 10.1038/nrc1300.Suggests that inducing genuine dormancy (that is, cellular dormancy) is important to prevent the tumour heterogeneity from increasing and limiting robustness of cancer as a system (that is, the ability to maintain stable functioning despite perturbations, such as therapy).
- 122.Viswanathan M, Kim SK, Berdichevsky A, Guarente L. A role for SIR-2.1 regulation of ER stress response genes in determining C. elegans life span. Dev Cell. 2005;9:605–615. doi: 10.1016/j.devcel.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 123.Matheu A, et al. Delayed ageing through damage protection by the Arf/p53 pathway. Nature. 2007;448:375–379. doi: 10.1038/nature05949. [DOI] [PubMed] [Google Scholar]
- 124.Naumov GN, MacDonald IC, Chambers AF, Groom AC. Solitary cancer cells as a possible source of tumour dormancy? Semin Cancer Biol. 2001;11:271–276. doi: 10.1006/scbi.2001.0382. [DOI] [PubMed] [Google Scholar]
- 125.Shachaf CM, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431:1112–1117. doi: 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- 126.Dawson DW, et al. Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science. 1999;285:245–248. doi: 10.1126/science.285.5425.245. [DOI] [PubMed] [Google Scholar]
- 127.Kvalheim G, Naume B, Nesland JM. Minimal residual disease in breast cancer. Cancer Metastasis Rev. 1999;18:101–108. doi: 10.1023/a:1006216504892. [DOI] [PubMed] [Google Scholar]
- 128.Merrie AE, Yun K, van Rij AM, McCall JL. Detection and significance of minimal residual disease in colorectal cancer. Histol Histopathol. 1999;14:561–569. doi: 10.14670/HH-14.561. [DOI] [PubMed] [Google Scholar]
- 129.Gath HJ, Brakenhoff RH. Minimal residual disease in head and neck cancer. Cancer Metastasis Rev. 1999;18:109–126. doi: 10.1023/a:1006268621730. [DOI] [PubMed] [Google Scholar]
- 130.Cheung IY, Feng Y, Vickers A, Gerald W, Cheung NK. Cyclin D1, a novel molecular marker of minimal residual disease, in metastatic neuroblastoma. J Mol Diagn. 2007;9:237–241. doi: 10.2353/jmoldx.2007.060130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Roberts WM, et al. Measurement of residual leukemia during remission in childhood acute lymphoblastic leukemia. N Engl J Med. 1997;336:317–323. doi: 10.1056/NEJM199701303360501. [DOI] [PubMed] [Google Scholar]
- 132.Bruggemann M, Pott C, Ritgen M, Kneba M. Significance of minimal residual disease in lymphoid malignancies. Acta Haematol. 2004;112:111–119. doi: 10.1159/000077566. [DOI] [PubMed] [Google Scholar]
- 133.Schwarzenbach H, et al. Detection of tumor-specific DNA in blood and bone marrow plasma from patients with prostate cancer. Int J Cancer. 2007;120:1465–1471. doi: 10.1002/ijc.22470. [DOI] [PubMed] [Google Scholar]
- 134.Morgan TM, Lange PH, Vessella RL. Detection and characterization of circulating and disseminated prostate cancer cells. Front Biosci. 2007;12:3000–3009. doi: 10.2741/2290. [DOI] [PubMed] [Google Scholar]
- 135.Pfitzenmaier J, et al. The detection and isolation of viable prostate-specific antigen positive epithelial cells by enrichment: a comparison to standard prostate-specific antigen reverse transcriptase polymerase chain reaction and its clinical relevance in prostate cancer. Urol Oncol. 2007;25:214–220. doi: 10.1016/j.urolonc.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 136.Kienle P, Koch M. Minimal residual disease in gastrointestinal cancer. Semin Surg Oncol. 2001;20:282–293. doi: 10.1002/ssu.1046. [DOI] [PubMed] [Google Scholar]
- 137.Max N, Keilholz U. Minimal residual disease in melanoma. Semin Surg Oncol. 2001;20:319–328. doi: 10.1002/ssu.1050. [DOI] [PubMed] [Google Scholar]
- 138.Blaheta HJ, et al. Detection of melanoma cells in sentinel lymph nodes, bone marrow and peripheral blood by a reverse transcription-polymerase chain reaction assay in patients with primary cutaneous melanoma: association with Breslow’s tumour thickness. Br J Dermatol. 2001;145:195–202. doi: 10.1046/j.1365-2133.2001.04334.x. [DOI] [PubMed] [Google Scholar]
- 139.Hosch SB, Scheunemann P, Izbicki JR. Minimal residual disease in non-small-cell lung cancer. Semin Surg Oncol. 2001;20:278–281. doi: 10.1002/ssu.1045. [DOI] [PubMed] [Google Scholar]
- 140.Willeke F, Sturm JW. Minimal residual disease in soft-tissue sarcomas. Semin Surg Oncol. 2001;20:294–303. doi: 10.1002/ssu.1047. [DOI] [PubMed] [Google Scholar]