

Abstract
Inteins are naturally occurring protein elements that catalyze their own excision from within a larger protein together with the ligation of the flanking “extein” sequences. Previously we reported the directed evolution of an intein-based molecular switch in which intein splicing in yeast cells was made dependent on the cell-permeable small molecule 4-hydroxytamoxifen (4-HT). Here we show that these evolved inteins are effective means of rendering protein function and biological signaling pathway activation dependent on 4-HT in mammalian cells. We characterized the generality, speed, and dose-dependence of ligand-induced protein splicing in murine NIH3T3 cells and in human HEK293 cells. Evolved inteins were used to control in mammalian cells the function of Gli1 and a truncated form of Gli3, two transcriptional mediators of the Hedgehog signaling pathway. Finally, we show that a complex biological process such as osteoblast differentiation can be made dependent on 4-HT using the evolved intein system. Our findings suggest that evolved small-molecule-dependent inteins may serve as a general means of achieving gene-specific, dose-dependent, post-translational, and small-molecule-induced control over protein activity in mammalian systems.
Introduction
The study of mammalian signaling pathways and regulatory networks is a persistent challenge due to their complexity and to inherent limitations of methods used to study them. Although methods that enable inducible and highly specific control over a protein of interest without perturbing the structure of the protein or its regulatory genetic elements would be ideal, many approaches to modulating protein function in bacteria and yeast which meet some of these goals cannot readily be applied to mammalian systems.
Strategies for small-molecule-based regulation of protein activity in mammalian cells that can be applied to arbitrary proteins of interest include transcriptional control by regulatable promoters1,2 and genetic modification by small-molecule-responsive Cre recombinase fusion proteins.3,4,5,6 While both methods have been used successfully to probe biological systems, each has significant limitations. Promoter-based systems1,2 place expression of the protein of interest under the control of a non-native promoter. While the structure of the resulting protein is not affected by this change, the ability of endogenous factors to interact with the promoter can be compromised, potentially disrupting regulatory networks that normally control target protein expression. In addition, promoter-based systems typically require the expression of heterologous transcriptional activators or repressors, some of which can interact with the endogenous cellular machinery resulting in toxicity2 or immunogenicity.7
Recombinase-based systems require concurrent expression of a foreign small-molecule-dependent recombinase. In addition, because protein expression induced by genetic recombination requires that each individual cell undergo a relatively slow recombination event, cell lines expressing a variety of small-molecule dependent Cre recombinases typically only achieve target protein activation in ≤ 50% of cells after 48 hours following treatment with the small molecule3,4,6. More efficient transgenic cell lines have been developed that undergo higher levels of recombination, but their use requires longer periods of exposure or high doses of small molecule.5,8,9 An efficient small-molecule-dependent method of controlling the activity of any protein of interest in mammalian cells without requiring changes to the target protein's promoter or the concurrent expression of heterologous proteins may prove useful for probing protein function. In addition, control of proteins at the post-translational level, in contrast with the above approaches, may enhance the speed with which changes in activity can take place.
We recently described the directed evolution of an intein-based molecular switch that renders intein splicing and therefore target protein activity dependent on the presence of the cell-permeable small molecule 4-hydroxytamoxifen (4-HT) in yeast cells.10 Because the evolved intein-based switch is a 4-HT-dependent, self-excising protein element contained within a target protein, its use does not require promoter modification or any additional control elements.
Other groups have also reported the use of inteins to modulate protein activity in eukaryotic cells. Umezawa and coworkers have shown that splicing of the split Synechocystis sp. DnaE intein fused to the N- and C-terminal halves of luciferase generates active luciferase in mammalian cells, albeit in an unregulated manner.11 Perrimon and coworkers developed a temperature-sensitive Saccharomyces cerevisiae VMA intein and used it to control the activity of the yeast Gal80 transcription factor in both yeast and Drosophila melanogaster.12 Muir and coworkers engineered a split VMA intein in which each half of the intein is fused to FKBP or FRB domains.13,14,15 Rapamycin induces dimerization of the FKBP and FRB domains, which unites the intein halves and enables splicing. Rapamycin-dependent splicing in this systems has been shown to mediate the joining of two epitope tags in mammalian cells14 as well as the removal of an inhibitory peptide from a protein kinase A-split intein fusion using purified proteins in solution.15 Target proteins for which inhibitory peptides are not known or that are not stable when separated and expressed as two intein-fused halves, however, may prove difficult to manipulate using a split intein system. Regulated inteins have not yet been reported to control native protein function in mammalian cells.
Here we show that the evolved small-molecule-dependent intein10 confers 4-HT-dependence on the activities of proteins in murine and human cells in a dose-dependent and fairly rapid manner. We demonstrate that native mammalian proteins controlled by the evolved intein are able to perform their endogenous biological functions in mammalian cells. Further, we show that the evolved intein can render a complex process such as osteoblast differentiation dependent on 4-HT. Our results validate the use of evolved small-molecule-dependent inteins as tools to introduce post-translational control elements into signaling pathways in mammalian cells.
Experimental Section
General
4-hydroxytamoxifen, Leptomycin B, N-terminal Sonic Hedgehog peptide (nShh), anti-3X Flag antibody and anti-β-actin antibody were purchased from Sigma-Aldrich Anti-GFP antibody was purchased from Clontech Laboratories. Alexa Fluor 488 anti-mouse antibody was purchased from Invitrogen. Alexa Fluor 680 anti-rabbit and 800 anti-mouse antibodies were provided by Li-cor Biosciences. Restriction enzymes were purchased from New England Biolabs. Cell culture reagents were purchased from VWR and Invitrogen. Western blots were visualized and quantitated using an Odyssey imager (Li-cor Biosciences). FACS analysis was performed using a MoFlo cell sorter (DakoCytomation). Fluorescence microscopy was performed on a Zeiss Axioskop (Zeiss). SDS-PAGE and Western blotting were performed using standard protocols.
Plasmid construction
To create a stably transfected cell line expressing GFP-intein(3-2), the encoding gene was cloned into pcDNA3.1/Hygro (Invitrogen). The GFP-intein(3-2) gene was amplified by PCR with VENT polymerase (New England Biolabs) from p414Gal1-GFP-intein(3-2)10 using oligonucleotides 5′-GCCGCCCTTAAGATGGCAAGCAAAGGAGAAC-3′ and 5′-GCCGCCGCGGCCGCTTATTTGTAGAGCTCATCC-3′. The resulting fragment was treated with AflII and NotI (restriction sites underlined) and ligated into AflII- and NotI-digested pcDNA3.1/Hygro.
C-terminally Flag-tagged Gli1-intein and Gli3T-intein expression vectors were constructed from p3XFlag-CMV-14-Gli1 and p3X-Flag-CMV-14-Gli3T. An XhoI restriction site was introduced into the Gli1 gene by silent mutation of the Ser 279 codon. An XhoI restriction site was removed from the Gli3T gene by silent mutation of the Leu 728 codon, while EcoRI and XhoI restriction sites were added by silent mutation of the Glu 512 and Ser 521 codons, respectively. All mutagenesis was performed using standard protocols for QuikChange site mutagenesis (Stratagene). Gene fragments encoding intein(3-2) and intein(2-4) were amplified by PCR from p414Gal1-GFP-intein(3-2) and p414Gal1-GFP-intein(2-4).10 Fragments were amplified using oligonucleotides 5′-GCCGCCGAATTCTGCCTTGCCGAGGGTACCCG-3′, and 5′-GCCGCCCTCGAGCAACCTCCCCAATGGCAGTTGTGCACGACAACC-3′ (for Gli1) or 5′-GCCGCCCTCGAGAACAATCAAGCCAGCGGCAGTTGTGCACGACAA-3′ (for Gli3T). Fragments were digested with EcoRI and XhoI and ligated into the appropriate EcoRI- and XhoI-treated vectors.
Genes encoding non-splicing Cys→Ala mutants of Gli1-intein(3-2) and Gli3T-intein(3-2) were constructed by mutating the Cys-encoding codon at the C-terminus of the intein (TGC) to one encoding Ala (GCC).
The genes encoding Flag-tagged Gli1, Gli1-intein(3-2) and Gli1-intein(2-4) were also cloned into pCAGGS16 for expression in 10T1/2 cells.
Cell culture
HEK293 cells were cultured in DMEM:F12 media with 10% fetal bovine serum while NIH3T3 and 10T1/2 cells were cultured in DMEM with 10% calf serum according to standard protocols. Transient transfections were performed using Lipofectamine2000 (Invitrogen) following the manufacturer's protocol. To generate a stably transfected cell line expressing GFP-intein, HEK293 cells were plated in a 10 cm dish coated with Matrigel (BD Biosciences). Cells were transfected with pcDNA3.1-GFP-intein(3-2) using the ProFection calcium phosphate transfection system (Promega) following the manufacturer's protocol. 72 hours after transfection, cells were replated onto three 10 cm dishes in media containing 150 μg/μL Hygromycin B (EMD Biosciences). Resistant clones were replated individually and analyzed by Western blot using an anti-GFP antibody.
FACS analysis
Stably transfected HEK293 cells expressing GFP-intein(3-2) were grown in 6-well plates with the specified concentrations of 4-HT for 24 hours. Cells were then trypsinized and resuspended in 500 μL PBS with 1% fetal bovine serum and 75 U/mL DNase (New England Biolabs). At least 104 cells were analyzed for each sample.
Luciferase assays
NIH3T3 cells were plated in a 24-well plate containing 5×104 cells per well. 18 hours after plating, cells were transiently transfected with a total of 800 ng DNA, including 300 ng ptcΔ136-GL3 luciferase reporter17 and 100 ng pSV-β-Galactosidase transfection control (Promega). The control transfection contained 100 ng p3XFlag vector with no encoding gene. Transfections to express Gli1, Gli1-intein(3-2), Gli1-intein(2-4) and non-splicing mutant Gli1-intein(3-2) contained 100 ng of the corresponding p3XFlag expression vector. Transfections to express Gli3T, Gli3T-intein(3-2), Gli3T-intein(2-4) and non-splicing mutant Gli3T-intein(3-2) contained 10 ng of the corresponding p3XFlag expression vector, as well as 100 ng p3XFlag-Gli1. pSK Bluescript (Stratagene) was added to each transfection mixture to bring the total DNA content to 800 ng. 48 hours after transfection, media was changed to DMEM with 0.5% calf serum, containing 4-HT where appropriate. After an additional 48 hours, cells were collected and assayed as previously described.17 Luciferase activity values were normalized to β-galactosidase activity values to correct for variations in numbers of transfected cells. Note that although NIH3T3 cells contain Gli1- and Gli3-encoding genes, the level of CMV promoter-driven overexpression of transfected Gli1 and Gli3T in this standard assay is much greater than endogenous Gli1 or Gli3 levels.
Immunohistochemistry
HEK293 cells were plated on Matrigel-coated glass coverslips in a 6-well plate, and transiently transfected with 4 μg of the specified p3XFlag expression vector. After growth in the absence or presence of 1 μM 4-HT for 48 hours, cells were fixed with methanol and treated with anti-3XFlag antibody, Alexa 488 anti-mouse antibody, and DAPI following standard protocols. For cells treated with Leptomycin B (LMB), 5 nM LMB was added 2 hours prior to fixation.
Alkaline phosphatase assays
10T1/2 cells were plated in 96-well plates containing 104 cells per well. 22 hours after plating, cells were transiently transfected with 200 ng of the appropriate pCAGGS expression vector or an empty pCAGGS vector as a control. 8 hours after transfection, fresh media was added that contained 1 μM 4-HT or 50 nM nShh where specified. Five days after transfection, cells were lysed by freeze-thaw and assayed using pNPP alkaline phosphate liquid substrate (Sigma). Reactions were incubated for 1 hour at 37° and measured by spectrophotometer.
Results
Evolved intein mediates small-molecule-dependent splicing of GFP in mammalian cells
Given the significant differences between yeast and mammalian cells, we first evaluated the ligand dependence and splicing ability of the previously yeast-evolved inteins10 in mammalian cells. We used the final and penultimate intein variants, intein(3-2) and intein(2-4), respectively, in the experiments described below. GFP-intein(3-2) consists of a FACS-optimized GFP mutant18 with Ala-intein(3-2)-Arg inserted in place of GFP residues 107-109 as previously described.10 We generated a stably transfected line of HEK293 cells that expresses GFP-intein(3-2) under the control of the CMV promoter and followed protein splicing in the presence and absence of 4-HT. In the absence of 4-HT, only unspliced GFP-intein(3-2) protein was detected by Western blot. In contrast, when these cells were grown in media containing 1 μM 4-HT, approximately 40% of GFP-containing protein was spliced GFP (Figure 1A).
Figure 1.
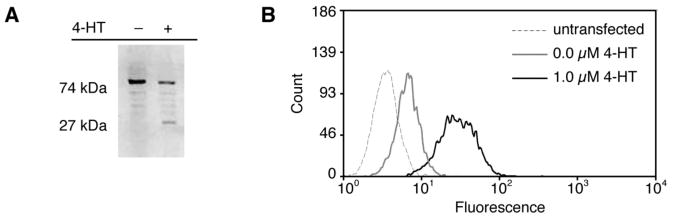
Evolved intein 3-2 mediates 4-HT-dependent splicing in mammalian cells. HEK293 cells stably expressing GFP-intein(3-2) were grown for 24 h in the absence or presence of 1 μM 4-HT. (A) Cells were lysed and analyzed by Western blot with an anti-GFP antibody. (B) Cells were analyzed by FACS and fluorescence histograms were compared to that of untransfected HEK293 cells.
To determine whether GFP generated by evolved intein-mediated splicing is active in mammalian cells, we used fluorescence activated cell sorting (FACS) to analyze cells stably expressing GFP-intein(3-2). When cells were grown in the absence of 4-HT, they exhibited less than a two-fold increase in fluorescence over the background fluorescence of untransfected cells. When grown in the presence of 1 μM 4-HT, however, these cells exhibited a ten-fold increase in fluorescence, indicating that the spliced GFP generated upon 4-HT treatment can adopt a conformation leading to fluorescence (Figure 1B). Together, these results demonstrate that evolved intein(3-2) can render target protein structure dependent on 4-HT in mammalian cells and that protein generated upon intein splicing can retain its activity.
4-HT-dependent splicing mediated by intein(3-2) is dose-dependent and occurs in hours
The dose-dependence of the evolved intein may be of particular value when probing proteins with concentration-dependent functions. In order to evaluate the dose-dependence of protein activation in mammalian cells, HEK293 cells stably expressing GFP-intein(3-2) were subjected to 0.25-2 μM 4-HT for 24-48 hours prior to analysis by fluorescence-activated cell sorting (FACS) (Figure 2A). A fairly linear dependence of median fluorescence on 4-HT concentration was observed over the range tested. Furthermore, for all samples, the level of fluorescence in each individual cell was relatively uniform across the cell population (Figure 2B). These results demonstrate that the evolved intein can mediate a dose-dependent response in which increasing amounts of 4-HT induce increasing concentrations of active protein within each individual cell. This graded response contrasts with the type of dose-dependence observed in Cre recombinase-based protein activation,9 in which altering the amount of small molecule changes the proportion of cells undergoing recombination, but does not result in intermediate levels of protein activity within individual cells.
Figure 2.
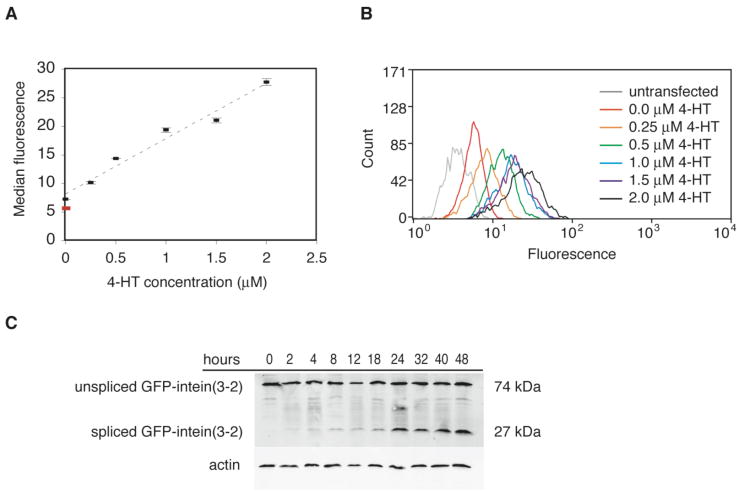
4-HT-dependent splicing of evolved intein is dose-dependent and takes place within hours. (A) HEK293 cells stably expressing GFP-intein(3-2) were grown for 24 hours in 0 to 2.0 μM 4-HT. Fluorescence was measured by FACS. The red spot represents the median fluorescence of untransformed cells. Data points and error bars reflect the mean and standard deviation, respectively, of three independent trials. (B) The fluorescence histograms of the cell populations described in (A) show that increasing concentrations of 4-HT induce increasing amounts of protein splicing throughout the cell populations. Individual histograms are normalized to a total cell count of 2 × 103. (C) HEK293 cells stably expressing GFP-intein(3-2) were treated with 1 μM 4-HT for the indicated time periods and analyzed by Western blot using an anti-GFP antibody.
In order to evaluate the speed of intein-mediated protein activation, we observed the time dependence of 4-HT responsiveness using GFP-intein(3-2). When cells stably expressing GFP-intein are treated with 4-HT, spliced protein can be detected by Western blot within 2 hours and reaches its maximum concentration within 24 hours (Figure 2C). In the absence of 4-HT, less than 0.5% of protein detected is spliced, while at 2 hours (the earliest time point measured), 5% of protein is spliced. Western blot quantitation of three separate trials (representative shown in Figure 2C) indicates that after 24 hours, the amount of spliced protein remains relatively constant at 30-40% of total protein detected. These findings indicate that intein(3-2) splicing in the presence of 4-HT takes place on a timescale of hours.
Evolved inteins mediate splicing of mammalian transcription factors
To determine whether evolved inteins could control the activity of native proteins in living mammalian cells, we inserted evolved inteins into the murine transcription factor Gli1 and into a C-terminally truncated form of the transcription factor Gli3 (Gli3T). Gli1 and Gli3 are key mammalian transcriptional mediators of Hedgehog signaling19 and play key roles in developmental processes including spinal cord patterning20 and limb development.21,22 Gli1 is a transcriptional activator,23 while Gli3T is an engineered version of the repressor form of the Gli3 transcription factor.24 Separate constructs were generated for both Gli1 and Gli3T containing either intein(3-2) or intein(2-4) preceding a zinc-coordinating cysteine in the highly conserved DNA binding domain present in both transcription factors (Cys 273 in Gli1, Cys 515 in Gli3T).25 In these constructs, protein splicing therefore generates Gli1 or Gli3T with primary structures that are identical to those of the native proteins.
When HEK293 cells were transiently transfected with each of the Gli1-intein or Gli3T-intein fusions, 4-HT-dependent splicing was observed via Western blot (Figure 3A,B). The amount of spliced protein generated in the presence of 4-HT was approximately 50-60% of the total Gli1 or Gli3T protein detected, but only 0-10% in the absence of 4-HT. These results demonstrate that insertion of evolved intein(3-2) or intein(2-4) into Gli1 and Gli3T confers 4-HT-dependent control over each protein's primary structure.
Figure 3.
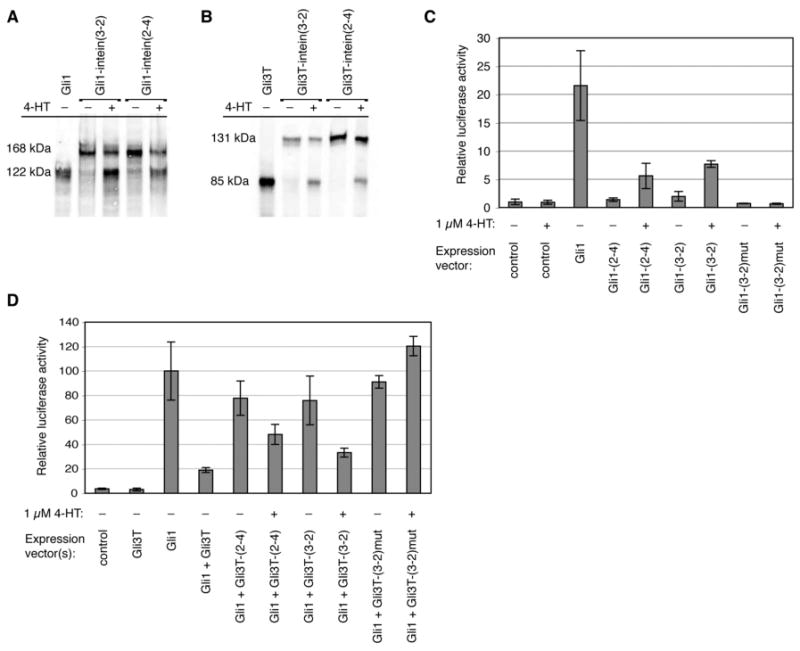
4-HT-dependent splicing of evolved inteins generates active Gli1 and Gli3T in mammalian cells. (A,B) HEK293 cells were transiently transfected with Flag-tagged Gli1- and Gli3T-intein expression vectors, grown for 24 h in the absence or presence of 1 μM 4-HT, and analyzed by Western blot using an anti-Flag antibody. (C) NIH3T3 cells were transiently transfected with the indicated expression vectors, as well as a luciferase reporter vector and a β-galactosidase transfection control vector. The control expression vector contains no gene, Gli1-(2-4) and Gli1-(3-2) refer to the Gli1-intein constructs, and Gli1-(3-2)mut refers to the Cys→Ala mutant of Gli1-intein(3-2). The luciferase activity resulting from the control vector in the absence of 4-HT is defined as 1. (D) NIH3T3 cells were transiently transfected with the indicated expression vectors, the luciferase reporter vector, and a β-galactosidase control vector. Gli3T-(2-4) and Gli3T-(3-2) refer to the Gli3T-intein constructs, and Gli3T-(3-2)mut refers to the Cys→Ala mutant of Gli3T-intein(3-2). Luciferase acitivty induced by Gli1 is defined as 100. For all luciferase assays, luciferase activity was normalized to β-galactosidase activity. Bar heights and error bars reflect the mean and standard deviation, respectively, of two or three independent trials.
4-HT-dependent splicing generates active Gli1 and Gli3T in mammalian cells
To analyze the activity of the Gli1- and Gli3T-intein fusions, we used an established luciferase-based reporter assay.17 Murine NIH3T3 cells were co-transfected with either Gli1-intein or Gli3T-intein expression vectors and a reporter construct containing the consensus Gli binding sequence from the promoter of patched, an endogenous target of Gli regulation,26 upstream of the firefly luciferase gene. Without 4-HT treatment, cells transfected with Gli1-intein(2-4) exhibited no significant increase in luciferase activity over cells transfected with a control expression vector that contains no gene (Figure 3C). Cells transfected with Gli1-intein(3-2) exhibited a two-fold increase in luciferase production over the control in the absence of 4-HT. In the presence of 1 μM 4-HT, Gli1-intein(2-4) induced approximately 25% as much luciferase production as wild-type Gli1, while Gli1-intein(3-2) induced approximately 35% as much. As controls, no luciferase production was observed in response to 4-HT in cells not expressing Gli1-intein protein (Figure 3C), and 4-HT treatment of cells transfected with wild-type Gli1 had no effect on luciferase production (data not shown). These results demonstrate that expression of Gli1-intein(2-4) or Gli1-intein(3-2) results in 4-HT-dependent transcriptional activation of the luciferase reporter.
To measure the activity of the intein-controlled Gli3T repressor, Gli3T-intein expression vectors were cotransfected with the reporter and a 10-fold excess of wild-type Gli1 expression vector (Figure 3D). Repression by native Gli3T decreases Gli1-induced luciferase production by 80% (down to 20% of its unrepressed level). In the presence of 4-HT, Gli3T-intein(2-4) and Gli3T-intein(3-2) decrease luciferase production by 50% and 70%, respectively, of its unrepressed level. Therefore, expression of Gli3T-intein(2-4) or Gli3T-intein(3-2) leads to 4-HT-dependent repression of luciferase production. Together, these results collectively show that the evolved inteins upon small molecule-triggered splicing generate Gli1 and Gli3T proteins that retain their native activities.
To determine whether protein splicing is necessary for the 4-HT-dependent activity of the Gli1- and Gli3T-intein fusions, we made mutants of Gli1- and Gli3T-intein(3-2) in which the nucleophilic cysteine at the C-terminus of the intein is mutated to alanine. An analogous mutation in the Pyrococcus GB-D Pol intein has been shown to completely eliminate splicing.27 The non-splicing mutant Gli1-intein(3-2)mut induces no luciferase production in response to 4-HT (Figure 3C), and the non-splicing Gli3T-intein(3-2)mut does not repress luciferase repression significantly either in the absence or in the presence of 4-HT (Figure 3D). These results indicate that the small molecule-dependent transcription factor activity of the Gli1- and Gli3T-inteins is contingent upon protein splicing. Furthermore, any background activity of the Gli1 and Gli3T-inteins in the absence of 4-HT is most likely due to low levels of protein splicing that occur in the absence of 4-HT, and not to any residual activity of unspliced protein.
The high sensitivity and quantitative nature of the luciferase assay enables a more detailed comparison of the activities of intein(3-2) and intein(2-4). Consistent with their properties in yeast,10 intein(3-2) generates greater quantities of spliced protein in the presence of 4-HT than intein(2-4), although the activity of either intein-containing transcription factor in the presence of 4-HT is lower than the activity of the wild-type transcription factor (Figure 3C,D). Intein(2-4) leads to a smaller amount of background splicing in the absence of 4-HT than intein(3-2) in mammalian cells. This background splicing causes a two-fold increase in luciferase production over the control in cells expressing Gli1-intein(3-2), but no observable increase in cells expressing Gli1-intein(2-4). The background splicing of Gli3T-intein(2-4) and Gli3T-intein(3-2) results in repression of Gli1-induced luciferase production by 20% and 25%, respectively. This background repression is surprising given that no spliced protein is observed in the absence of 4-HT by Western blot. Gli3T may be a sufficiently potent transcriptional repressor that very low concentrations of active Gli3T may produce significant measurable effects in the luciferase reporter assay.
Spliced Gli1-intein and Gli3T-intein localize identically to their native counterparts
Next we examined the subcellular localization of Gli1 and Gli3T containing the evolved intein. HEK293 cells were transiently transfected with Flag-tagged Gli1, Gli3T, Gli1-intein(3-2) or Gli3T-intein(3-2), then fixed and stained with anti-Flag antibody (Figure 4). Normally the repressor form of Gli3 is localized to the nucleus,28 while Gli1 undergoes nuclear export as part of its regulatory mechanism and can therefore appear in both the nucleus and cytoplasm.29 Consistent with these observations, Gli1 expressed in transfected cells appears broadly distributed throughout the nucleus and cytoplasm (Figure 4A), but addition of the small molecule Leptomycin B (LMB), a potent inhibitor of nuclear export, leads to accumulation of Gli1 in the nucleus (Figure 4B). Gli3T expressed in transfected cells is nuclearly localized (Figure 4C).
Figure 4.
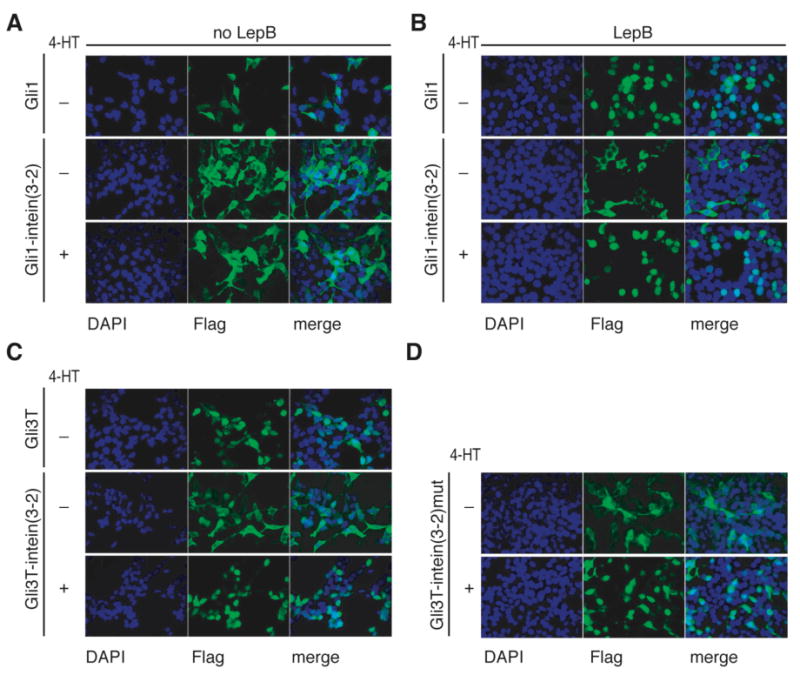
Gli1-intein(3-2) and Gli3T-intein(3-2) localize identically to native protein in the presence of 4-HT. (A,B) HEK293 cells were transiently transfected with Flag-tagged Gli1 or Gli1-intein(3-2) and grown in the absence or presence of 1 μM 4-HT for 48 hours. (B) Two hours prior to fixation, 5 nM LMB was added. (C,D) HEK293 cells were transiently transfected with Flag-tagged Gli3T, Gli3T-intein(3-2), or non-splicing Gli3T-intein(3-2)mut, and grown in the absence or presence of 1 μM 4-HT for 48 hours. All samples were fixed and stained with an anti-Flag antibody (false colored green) and DAPI, which stains nuclei (false colored blue).
In cells transfected with Gli1-intein(3-2) or Gli3T-intein(3-2), protein produced in the absence of 4-HT is cytoplasmically sequestered, in contrast with the localization of native Gli1 and Gli3T (Figures 4A,C). We attribute cytoplasmic sequestration in the absence of 4-HT to the interaction of Hsp90 with the mutated estrogen receptor ligand-binding domain that exists within the evolved intein,30 even though in yeast cells we demonstrated that this interaction is not solely responsible for the 4-HT-dependence of protein splicing.10 When cells transfected with Gli1-intein(3-2) were grown in the presence of 4-HT, protein appeared broadly distributed (Figure 4A). However, when Gli1-intein(3-2) transfected cells were grown in the presence of 4-HT and treated with LMB, nuclear localization was observed (Figure 4B). When cells transfected with Gli3T-intein(3-2) were grown in the presence of 4-HT, the resulting protein localized to the nucleus (Figure 4C). The ability of Gli1- and Gli3T-intein(3-2) to localize correctly within the cell in the presence of 4-HT further suggests that the Gli1 and Gli3T generated upon 4-HT-dependent splicing retain their native properties.
To determine whether protein splicing is necessary for the correct localization of proteins containing the evolved intein, the localization of the non-splicing Cys→Ala Gli3T-intein(3-2)mut was examined (Figure 4D). As with Gli3T-intein(3-2), the non-splicing mutant protein is cytoplasmically localized in the absence of 4-HT and nuclearly localized in the presence of 4-HT. Therefore, nuclear localization of Gli3T-intein(3-2) is not contingent upon protein splicing but is instead likely dependent on the abrogation of interactions with Hsp90 upon treatment with 4-HT.
Evolved inteins render Gli-mediated osteoblast differentiation dependent on 4-HT
The above results demonstrate that 4-HT-dependent splicing can be used to generate protein that is both active and correctly localized, but do not reveal if the amounts of active protein generated are sufficient to fulfill the proteins' endogenous roles in their native biological contexts. We therefore next evaluated the function of an intein-controlled mammalian protein in its endogenous context. To achieve this goal, we used the well-characterized mouse embryonic 10T1/2 cell line as a test system for the biological function of Gli1-intein(3-2) and Gli1-intein(2-4).
Activation of the Hedgehog signaling pathway causes 10T1/2 cells to differentiate into osteoblasts, which can be quantitatively measured by the production of alkaline phosphatase.31 In nature, the Hedgehog pathway is activated by the presence of Sonic Hedgehog (Shh) protein, which binds to an extracellular receptor to initiate a signaling cascade that results in the modulation of Gli transcription factor activity. As Gli1 is a downstream component of the Hedgehog pathway, transfection of 10T1/2 cells with a construct expressing Gli1 induces osteoblast differentiation in the absence of Shh. When cells are transfected with Gli1-intein(3-2) or Gli1-intein(2-4), however, osteoblast differentiation becomes dependent on 4-HT in the absence of Shh (Figure 5). Importantly, in the presence of 4-HT, both Gli1-intein(2-4) and Gli1-intein(3-2) promote differentiation to the same extent as wild-type Gli1, even though the intein fusions generate only 25% and 35% as much activity, respectively, as measured by the luciferase reporter assay. These results demonstrate that the quantities and activity of Gli1 generated by 4-HT-mediated splicing of Gli1-intein are sufficient to fulfill its endogenous role in osteoblast differentiation to an extent comparable to that of wild-type Gli1, and in a manner that is no longer dependent on Shh.
Figure 5.
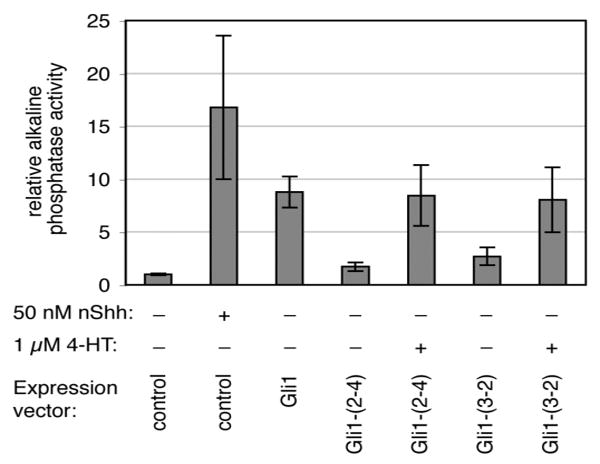
Gli1-intein renders osteoblast differentiation dependent on 4-HT. 10T1/2 cells were transiently transfected and grown for 4 days in the absence or presence of 1 μM 4-HT. The control vector contains no gene, and Gli1-(2-4) and Gli1-(3-2) refer to the Gli1-intein constructs. Differentiation was evaluated by measurement of alkaline phosphatase activity. Differentiation induced by exogenous Shh peptide is shown for comparison. Bar heights and error bars reflect the mean and standard deviation, respectively, of three independent trials.
Discussion
Comparison of evolved intein properties in yeast and mammalian cells
In this work we have shown that two evolved intein variants are capable of rendering mammalian protein activity dependent on a cell-permeable small molecule. In general, the properties of the inteins in mammalian cells parallel their properties in yeast. Protein splicing is dependent on 4-HT in both systems, and intein(3-2) generates more spliced product in the presence of 4-HT than intein(2-4). A graded dose-dependent response to 4-HT is also observed in both yeast and mammalian cells. These similarities of behavior further underscore the generality of the evolved inteins even though the environment of the mammalian cell is significantly different than the yeast cell environment used to evolve the inteins.
One difference observed between the evolved intein's behavior in yeast and its behavior in mammalian cells is that spliced GFP appears to accumulate slower in mammalian cells expressing GFP-intein(3-2) than in yeast expressing the same protein. While the onset of splicing is rapid in mammalian cells, only 5% of detected protein is spliced after two hours. In yeast, this percentage is significantly higher.10 This discrepancy may be due to differences in the stability of the unspliced protein, its folding ability, or its rate of degradation in the two cellular contexts. Although the accumulation of spliced protein over a 24-hour period is consistent with the product accumulation observed using an engineered rapamycin-dependent trans intein system in mammalian cells,14 further optimization of the evolved intein's activity in mammalian cells may be possible through additional rounds of directed evolution.
Basis of ligand-dependent activation of Gli1-intein and Gli3T-intein
The cytoplasmic sequestration of Gli1-intein(3-2) and Gli3T-intein(3-2) in the absence of 4-HT complicates the analysis of the mechanism of their evolved ligand-dependent activities. In contrast to GFP, Gli1 and Gli3T are only active in the nucleus. Therefore, it could be argued that cytoplasmic sequestration rather than the presence of the intein is responsible for the lack of Gli1 and Gli3T activity in the absence of 4-HT. Indeed, the Hsp90-mediated cytoplasmic sequestration of protein fusions containing hormone receptor ligand-binding domains has served as a basis for previously engineered small-molecule dependent proteins,32 including the Cre recombinase variants described above.3,4,5,6 However, the above results indicate that a non-splicing evolved intein mutant leads to cytoplasmically sequesteration in the absence of 4-HT, and localization identical to that of native protein in the presence of 4-HT (Figure 4D). Importantly, non-splicing Gli1-intein or Gli3T-intein mutants demonstrate no activity by luciferase assay either in the presence or absence of 4-HT (Figures 3C and 3D). These results indicate that while 4-HT releases proteins containing the evolved intein from cytoplasmic sequestration, splicing is required for protein activity. Therefore, 4-HT-dependent splicing of the evolved intein rather than a 4-HT-mediated change in localization is responsible for 4-HT-dependent activity of Gli1- and Gli3T-inteins in mammalian cells. This conclusion is consistent with our earlier studies in yeast cells in which we proposed that Hsp90 complexation may stabilize the unspliced protein but is not responsible for the ligand-dependence of splicing.
The possibility exists that intein splicing observed in the absence of 4-HT may be triggered by endogenous estrogen. However, growth in estrogen-free media did not affect the levels of background splicing of Gli1- and Gli3-intein(3-2) observed by Western blot (data not shown). The responsiveness of the evolved inteins to estrogen and estrogen analogs other than 4-HT will be the subject of future experiments, and selectivity for 4-HT over estrogen could be optimized through further directed evolution.
Application of evolved ligand-dependent inteins to the study of mammalian signaling pathways
The demonstration that an evolved intein inserted into Gli1 renders osteoblast differentiation dependent on 4-HT suggests the possibility of target protein-specific, post-translational, small-molecule-dependent, dose-dependent, and temporally controlled protein activation in a transgenic mammalian system. Such a system would enable control of target protein function to be decoupled from natural upstream signaling partners. For example, a transgenic cell expressing Gli1-intein(3-2) in place of Gli1 will not activate Gli1 in response to Shh, but will do so in response to 4-HT. Achieving small-molecule-dependent control over the function of a specific protein in order to study cellular processes is a central goal of chemical genetics.33 Traditional chemical genetic approaches require the discovery of a small molecule activator or repressor of each protein of interest. The use of the evolved intein confers 4-HT-dependence on proteins of interest in mammalian cells without requiring small-molecule synthesis and discovery, albeit with the additional significant requirement of genetic intervention.
The evolved intein is a particularly attractive tool for the study of signaling pathways because it requires minimal disruption of regulatory networks. No changes to the promoter or other regulatory regions of the gene of interest are required. Many signaling pathways involve feedback inhibition loops in which a downstream product of the pathway represses transcription of an upstream protein to attenuate the signal. An inducible promoter can replace the endogenous promoter of an upstream signaling protein in order to activate the signaling pathway on demand, but natural feedback transcriptional repression in this case cannot occur, precluding deactivation of the pathway by its normal regulatory mechanism. In contrast, the evolved intein exerts control over protein activity at the post-translational level. As a result, feedback regulation of protein-intein expression can proceed normally because no changes to regulatory promoter elements are required.
Because our results indicate that the evolved intein can exhibit a linear dose-dependent response to varying concentrations of 4-HT in mammalian cells, it is well suited for studying the response of a pathway to different levels of target protein activity. For example, transcription factors involved in stem cell differentiation have been shown to induce different cell fates based on modest changes to their cellular concentrations. Smith and coworkers showed that a 50% increase in the expression of Oct-4, a transcription factor involved in regulating pluripotency in embryonic stem cells, induces differentiation to endoderm or mesoderm, while a 50% decrease in expression causes de-differentiation to trophectoderm.34 Likewise, Yamamoto and coworkers reported that complete inactivation of GATA-1, a transcription factor required for erythroid differentiation, both inhibits differentiation and induces apoptosis in erythroid progenitor cells, while expression of GATA-1 at 5% of wild-type levels inhibits differentiation without inducing apoptosis.35 While these studies required the generation of multiple cell lines to achieve different levels of protein activity, the use of the evolved intein may enable regulation of the level of target protein activity in a single cell line simply by altering the concentration of 4-HT in the media.
Conclusion
In conclusion, we have shown that the evolved 4-HT-dependent intein can function as a tool for the relatively rapid, post-translational manipulation of protein function in mammalian cells. We inserted evolved inteins into three target proteins to create 4-HT-dependent GFP, Gli1, and Gli3T, all of which exhibited native-like function in the presence of 4-HT. Expression of Gli1-intein fusions in embryonic mouse cells enables 4-HT to induce a cellular response that mimics the effect of Hedgehog pathway activation. These results collectively suggest that evolved small-molecule-dependent inteins may provide a general method for achieving small-molecule-dependent control over protein activity in mammalian systems. The ability to control protein activity in a post-translational, rapid, dose-dependent, and gene-specific manner without requiring non-native promoters or heterologous control elements could prove valuable in the study of mammalian proteins and signaling pathways.
Acknowledgments
Reporter plasmid ptcΔ136-GL3 was provided by Professor Norbert Perrimon. We thank Professors Gavin Macbeath and Stuart Schreiber, and the Bauer Center for Genomic Research for use of instrumentation. George Kenty and Professor Doug Melton generously provided FACS equipment and assistance. We thank Mariana Mihalusova for molecular biology assistance. Work in DRL's laboratory was supported by National Institutes of Health/National Institute of General Medical Sciences R01GM065400 and the Howard Hughes Medical Institute. Work in APM's laboratory was supported by a grant from the National Institutes of Health (NS033642). CMY was supported by a National Defense Science and Engineering Graduate Fellowship; SJR was supported by fellowships from the National Health and Medical Research Council of Australia (#301299) and the Arthritis Foundation (#401683); SAV was supported by the Helen Hay Whitney Foundation.
References
- 1.Rossi FM, Blau HM. Recent advances in inducible gene expression systems. Curr Op Biotech. 1998;9:451–456. doi: 10.1016/s0958-1669(98)80028-1. [DOI] [PubMed] [Google Scholar]
- 2.Saez E, No D, West A, Evans RM. Inducible gene expression in mammalian cells and transgenic mice. Curr Op Biotech. 1997;8:608–616. doi: 10.1016/s0958-1669(97)80037-7. [DOI] [PubMed] [Google Scholar]
- 3.Feil R, Brocard J, Mascrez B, LeMeur M, Metzger D, Chambon P. Ligand-activated site-specific recombination in mice. Proc Nat Acad Sci. 1996;93:10887–10890. doi: 10.1073/pnas.93.20.10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kellendonk C, Tronche F, Monaghan AP, Angrand PO, Stewart F, Schultz G. Regulation of Cre recombinase activity by the synthetic steroid RU 486. Nucleic Acids Res. 1996;24:1404–1411. doi: 10.1093/nar/24.8.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, Riesterer C, Ayral AM, Sablitzky F, Littlewood TD, Reth M. Inducible site-directed recombination in mouse embryonic stem cells. Nucleic Acids Res. 1996;24:543–548. doi: 10.1093/nar/24.4.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 1998;8:1323–1326. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- 7.Lena AM, Giannetti P, Sporeno E, Ciliberto G, Savino R. Immune responses against tetracycline-dependent transactivators affect long-term expression of mouse erythropoietin delivered by a helper-dependent adenoviral vector. J Gene Med. 2005;7:1086–1096. doi: 10.1002/jgm.758. [DOI] [PubMed] [Google Scholar]
- 8.Indra AK, Warot X, Brocard J, Bornet JM, Xiao JH, Chambon P, Metzger D. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acids Res. 1999;27:4324–4327. doi: 10.1093/nar/27.22.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;344:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- 10.Buskirk AR, Ong YC, Gartner ZJ, Liu DR. Directed evolution of ligand dependence: small-molecule-activated protein splicing. Proc Nat Acad Sci. 2004;101:10505–10510. doi: 10.1073/pnas.0402762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ozawa T, Kaihara A, Sato M, Tachihara K, Umezawa Y. Split luciferase as an optical probe for detecting protein-protein interactions in mammalian cells based on protein splicing. Anal Chem. 2001;73:2516–2521. doi: 10.1021/ac0013296. [DOI] [PubMed] [Google Scholar]
- 12.Zeidler MP, Tan C, Bellaiche Y, Cherry S, Häder S, Gayko U, Perrimon N. Temperature-sensitive control of protein activity by conditionally splicing inteins. Nat Biotech. 2004;22:871–876. doi: 10.1038/nbt979. [DOI] [PubMed] [Google Scholar]
- 13.Mootz HD, Muir TW. Protein splicing triggered by a small molecule. J Am Chem Soc. 2002;124:9044–9045. doi: 10.1021/ja026769o. [DOI] [PubMed] [Google Scholar]
- 14.Mootz HD, Blum ES, Tyszkiewicz AB, Muir TW. Conditional protein splicing: a new tool to control protein structure and function in vitro and in vivo. J Am Chem Soc. 2003;125:10561–10569. doi: 10.1021/ja0362813. [DOI] [PubMed] [Google Scholar]
- 15.Mootz HD, Blum ES, Muir TW. Activation of an autoregulated protein kinase by conditional protein splicing. Angew Chem Int Ed. 2004;43:5189–5192. doi: 10.1002/anie.200460941. [DOI] [PubMed] [Google Scholar]
- 16.Niwa H, Yamamura KI, Miyazaki JI. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 17.Nybakken K, Vokes SA, Lin TY, McMahon AP, Perrimon N. A genome-wide RNA interference screen in Drosophila melanogaster cells for new components of the Hh signaling pathway. Nat Genet. 2005;37:1323–1332. doi: 10.1038/ng1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescenct protein (GFP) Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- 19.Koebernick K, Pieler T. Gli-type zinc finger proteins as bipotential transducers of Hedgehog signaling. Differentiation. 2002;70:68–76. doi: 10.1046/j.1432-0436.2002.700201.x. [DOI] [PubMed] [Google Scholar]
- 20.Bai CB, Stephen D, Joyner AJ. All mouse ventral spinal cord patterning by Hedghog is Gli dependent and involves an activator function of Gli3. Dev Cell. 2004;6:103–115. doi: 10.1016/s1534-5807(03)00394-0. [DOI] [PubMed] [Google Scholar]
- 21.te Welscher P, Zuniga A, Kuijper S, Drenth T, Goedemans HJ, Meijlink F, Zeller R. Progression of vertebrate limb development through SHH-mediated counteraction of GLI3. Science. 2002;298:827–830. doi: 10.1126/science.1075620. [DOI] [PubMed] [Google Scholar]
- 22.Barna M, Pandolfi PP, Niswander L. Gli3 and Plzf cooperate in proximal limb patterning at early stages of limb development. Nature. 2005;436:277–281. doi: 10.1038/nature03801. [DOI] [PubMed] [Google Scholar]
- 23.Yoon JW, Liu CZ, Yang JT, Swart R, Iannaccone P, Walterhouse D. GLI activates transcription through a herpes simplex viral protein 16-like activation domain. J Biol Chem. 1998;273:3496–3501. doi: 10.1074/jbc.273.6.3496. [DOI] [PubMed] [Google Scholar]
- 24.Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100:423–434. doi: 10.1016/s0092-8674(00)80678-9. [DOI] [PubMed] [Google Scholar]
- 25.Pavletich NP, Pabo CO. Crystal structure of a five-finger GLI-DNA complex: new perspectives on zinc fingers. Science. 1993;261:1701–1707. doi: 10.1126/science.8378770. [DOI] [PubMed] [Google Scholar]
- 26.Ingham PW. Transducing Hedgehog: the story so far. EMBO. 1998;17:3505–3511. doi: 10.1093/emboj/17.13.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu MQ, Perler FB. The mechanism of protein splicing and its modulation by mutation. EMBO. 1996;15:5146–5153. [PMC free article] [PubMed] [Google Scholar]
- 28.Shin SH, Kogerman P, Lindström E, Toftgárd R, Biesecker LG. Gli3 mutations in human disorders mimic Drosophila Cubitus interruptus protein functions and localization. Proc Natl Acad Sci. 1999;96:2880–2884. doi: 10.1073/pnas.96.6.2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kogerman P, Grimm T, Kogerman L, Krause D, Undén AB, Sandstedt B, Toftgård R, Zaphiropoulous PG. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat Cell Biol. 1999;1:312–319. doi: 10.1038/13031. [DOI] [PubMed] [Google Scholar]
- 30.Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18:306–360. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- 31.Ruiz i Altaba A. Gli proteins encode context-dependent positive and negative functions: implications for development and disease. Development. 1998;126:3205–3216. doi: 10.1242/dev.126.14.3205. [DOI] [PubMed] [Google Scholar]
- 32.Picard D. Posttranslational regulation of proteins by fusions to steroid-binding domains. Methods Enzymol. 2000;327:385–401. doi: 10.1016/s0076-6879(00)27291-1. [DOI] [PubMed] [Google Scholar]
- 33.Mayer TU. Chemical genetics: tailoring tools for cell biology. Trends Cell Biol. 2003;13:270–277. doi: 10.1016/s0962-8924(03)00077-1. [DOI] [PubMed] [Google Scholar]
- 34.Niwa H, Miyazaki JI, Smith AG. Quantitative expression of Oct3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat Genet. 2000;24:372–376. doi: 10.1038/74199. [DOI] [PubMed] [Google Scholar]
- 35.Pan X, Ohneda O, Ohneda K, Lindeboom F, Iwata F, Shimizu R, Nagano M, Suwabe N, Philipsen S, Lim KC, Engel JD, Yamamoto M. Graded levels of GATA-1 expression modulate survival, proliferation, and differentiation of erythroid progenitors. J Biol Chem. 2005;280:22385–22394. doi: 10.1074/jbc.M500081200. [DOI] [PubMed] [Google Scholar]