

Abstract
Natriuretic peptides (NPs) are involved in many physiological processes, including regulation of vascular tone, sodium excretion, pressure-volume homeostasis, inflammatory responses and cellular growth. Two main receptors of NP, membrane-bound guanylyl cyclases A and B (GC-A and GC-B), mediate the effects of NP via generation of cGMP. NP-stimulated generation of cGMP can be modulated by intracellular processes, whose exact natures remain to be elucidated. Thus, serum and lysophosphatidic acid (LPA), by unknown pathways, have been shown to inhibit NP-induced generation of cGMP. Here we report that the nonreceptor tyrosine kinase Csk is an essential component of the intracellular modulation of atrial natriuretic peptide (ANP)-stimulated activation of GC-A. Genetic deletion of Csk (Csk−/−) in mouse embryonic fibroblasts blocked the inhibitory effect of both serum and LPA on ANP-stimulated generation of cGMP. Moreover, using a chemical rescue approach, we also demonstrate that the catalytic activity of Csk is required for its modulatory function. Our data demonstrate that Csk is involved in the control of cGMP levels, and that membrane-bound guanylyl cyclases can be critically modulated by other receptor-initiated intracellular signaling pathways.
Many cellular processes such as cell migration, smooth muscle contraction, cellular growth and proliferation are under the control of the second messenger cGMP (1). In eukaryotes, cGMP is synthesized by two distinctive classes of guanylyl cyclases: membrane-bound guanylyl cyclases and soluble guanylyl cyclases (2). Soluble guanylyl cyclases are regulated by intracellular nitric oxide. Among membrane-bound forms, the receptor guanylyl cyclases, GC-A and GC-B, represent the most widely expressed enzymes (3, 4). Their activity is primarily regulated through a set of natriuretic peptide hormones, namely atrial natriuretic peptide (ANP), brain type natriuretic peptide (BNP) and C-type natriuretic peptide (CNP) (5, 6). GC-A binds both ANP and BNP, while the specific ligand for GC-B is CNP. Binding of natriuretic peptides to the extracellular domains of GC-A and GC-B results in the activation of the receptors to produce cGMP (7). The most well studied physiological role of natriuretic peptides is the maintenance of cardiovascular pressure-volume homeostasis (8, 9). Natriuretic peptides lower the blood pressure, increase renal salt excretion, glomerular filtration rate and vascular smooth muscle relaxation, and antagonize all known actions of the renin-angiotensin-aldosterone system (8). In addition, natriuretic peptide receptor guanylyl cyclases have attracted a great deal of attention in recent years for their ability to modulate cell proliferation and cardiomyocyte hypertrophy (1, 10–13). Gene knockout experiments have shown that disruption of GC-A in mice results in a hypertensive and/or cardiac hypertrophic phenotype (12, 14, 15). Moreover, ANP has been shown to inhibit cardiomyocyte hypertrophy induced by growth factors and other stimuli through a cGMP dependent mechanism (16).
Both GC-A and GC-B consist of an extracellular ligand binding domain, a short membrane spanning domain, a kinase homology domain, a hinge region and a catalytic cyclase domain (2). Phosphorylation of the kinase homology domain is critical for the ligand-induced activation of GC-A and GC-B (17, 18). Extensive studies in the past have shown that desensitization of GC-A and GC-B involves dephosphorylation of the receptor without significant changes in the basal activity (17, 19). Protein kinase C (PKC) and PP2C family phosphatases have been implicated in the desensitization (20). In fibroblast cells, growth regulatory and mitogenic signals such as basic fibroblast growth factor (bFGF), platelet derived growth factor (PDGF) and serum have been shown to inhibit ANP-induced activation of GC-A through a mechanism that at least in part involves dephosphorylation (21). This study also suggests that tyrosine kinase receptor mediated pathways play a key role in the desensitization of GC-A. More recently it was reported that in NIH3T3 fibroblasts, serum, lysophosphatidic acid (LPA) and PDGF, all desensitized GC-B through an unknown mechanism (22).
Nonreceptor tyrosine kinase Csk (C-terminal Src kinase) was originally purified as a kinase capable of phosphorylating Src and other Src family kinases at their C-terminal tyrosine residues (23). Csk is ubiquitously expressed in mammalian cells, and is evolutionarily conserved from early-diverging metazoan Hydra to humans (24). Mice deficient in Csk exhibited developmental defects (25, 26). The Csk-deficient mouse embryos died around day 10 post gestation. Csk has Src-dependent and –independent physiological functions (27). Indeed, Csk is positively required for normal development of lymphoid cells. Csk deficiency blocks T- and B-cell differentiation as is the case with Src-family kinase deficiency (28).
We have previously demonstrated that activation of nonreceptor tyrosine kinases in response to G protein-coupled receptors such as receptors for LPA is a key step in the regulation of cellular growth, proliferation and cytoskeletal reorganization (29–34). Given that several studies have indicated that activation of nonreceptor tyrosine kinases is a critical event leading to the development of cardiomyocyte hypertrophy (35–37), we investigated the possibility of the existence of a nonreceptor tyrosine kinase mediated pathway in the regulation of GC-A activity. Herein we show that the nonreceptor tyrosine kinase Csk negatively modulates ANP-induced increases in GC-A activity and critically controls serum- and LPA-induced inhibition of ANP-stimulated GC-A activity in mouse embryonic fibroblasts. Furthermore, we demonstrate that the catalytic activity of Csk is required for this regulation.
MATERIALS AND METHODS
Materials
Imidazole, 3-isobutyl-methylxanthine (IBMX), Triton X-100, leupeptin, pepstatin, aprotinin, PMSF, EDTA, creatine phosphate, creatine phosphokinase, ATP, and GTP were purchased from Sigma Aldrich (St.Louis, MO). Calphostin C was from Calbiochem. Rat ANP1-28 and CNP were purchased from Peninsula Laboratories or Bachem Laboratories. Direct cGMP EIA system was purchased from Assay Designs Inc. All reagents used for cell culture were of tissue culture grade.
Cell Culture and Transient Transfection
All cell lines used for the experiments were maintained in DMEM supplemented with 10% fetal bovine serum (FBS), penicillin-streptomycin and L-glutamine at 37°C and 5% CO2. Cells, 80% confluent in 6 well dishes, were transfected with 2 μg plasmid DNA using Lipofectamine-2000 reagent following the manufacturers’ instructions (38). The transfection efficiency was 30~60% based on transfection with a control GFP plasmid. Briefly, one hour before transfection, cells were washed once with PBS and incubated in DMEM without serum and antibiotics. In two separate tubes, 2 μg of plasmid DNA and 6 μl of transfection reagent were dissolved in 100 μl each of DMEM. The solutions were allowed to stand at room temperature for 5 min. The two solutions were then gently mixed and the DNA-reagent mixture was allowed to stand at room temperature for 20 minutes. This mixture was added into the medium over the cells, and the plate was rocked five to six times to ensure uniform distribution of the reagent-DNA complex. Cells were incubated at 37°C and 5% CO2 for 5–6 hours. The medium was aspirated and replaced with fresh growth medium. Cells were allowed to grow at 37°C and 5% CO2 for 18–24 hours before switching into starvation medium (DMEM supplemented with penicillin-streptomycin and L-Glutamine).
Csk−/− cells and the stable Csk−/−/CskR318A cell line were described previously (30). The R318A mutant of human Csk cDNA in pcDNA3.1/hyg was transfected into Csk−/− cells. Stable cell lines (a pool of many clones) were selected with hygromycin.
Measurement of cGMP levels in whole cell extracts
Eighteen to twenty-four hours after transfection, the cells were washed once with PBS and switched into starvation medium. Sixteen to twenty-four hours after starvation, cells were treated for 30 min with DMEM medium containing 0.5 mM IBMX in the presence or absence of 10% serum or 10 μM LPA. This was followed by treatment with 200 nM ANP or vehicle for 3 min at 37°C (39, 40). The cells were then washed once with PBS at room temperature and lysed with 0.5% Triton X-100 containing 0.5 mM IBMX. The cGMP content present in the samples was determined by using the Direct cGMP EIA system following the acetylation method (Assay Designs). For comparison, cGMP levels obtained from separate experiments were normalized with respect to the amount of proteins present in the lysates. For assays in the presence of imidazole, 50 mM imidazole (pH 7.5) and 0.5 mM IBMX in growth medium was used (30). In some experiments, 200 nM PMA were used.
In vitro kinase assays
Purified Csk (final concentration 200 nM) or Src (500 nM) was incubated with 2.5 μg of purified GST-GC-A-intra (amino acid residues 495 to 1061) in 30 mM Hepes pH 7.5, 10 mM MgCl2, 5 mM MnCl2 and 100 nM sodium orthovanadate along with 5 μCi of [γ-32P] ATP (3000 Ci/mmol) (Perkin-Elmer) or 1 mM cold ATP for 15 minutes at 30°C (30, 41). The reaction was stopped by adding SDS-PAGE sample buffer and incubated at 90°C for 5 minutes. Samples were then separated on a 10% SDS-PAGE gel. The gels were dried and autoradiographed or transferred to a nitrocellulose membrane and western blotted with anti-phospho-tyrosine antibody (Cell Signaling). GST-CDB3 (2.5 μg) was used as a positive control (29, 34).
Western blots with anti-phospho-Ser/Thr antibody
Csk−/− cells grown in 10-cm plates were co-transfected with pAX-Neo-GC-A and pcDNA3.1/Hygromycin-Csk or with pAX-Neo-GC-A and pcDNA3.1/Hygromycin empty vector. After 24-hr starvation, cells were treated with 10 μM LPA or 1 μM PMA for 30 min. Cells from one 10-cm plate were lysed in 1 mL of RIPA buffer containing 50 mM Tris, pH 8, 150 mM NaCl, 1 mM EDTA, 5 mM n-dodecyl-D-maltoside, 50 mM NaF, 200 μM Na3VO4 and protease inhibitors (29, 42). The lysate was briefly sonicated and centrifuged at 13,000 rpm for 10 min. The supernatant was transferred to a fresh tube and pre-cleared by incubating with 30 μL of 50% protein A-agarose slurry. After centrifugation, the supernatant was collected and incubated with 1.5 μg of antiserum against GC-A (FabGennix Inc. International, Shreveport, LA) overnight at 4°C. Thirty μL of protein A-agarose were added and the mixture was incubated for an additional 3 hrs at 4°C. The agarose beads were collected by centrifugation and washed three times with RIPA buffer. Proteins were eluted with SDS sample buffer and subjected to western-blot analyses. Anti-phospho-serine/threonine antibodies were from BD Clontech.
RESULTS
LPA and serum do not exert an inhibitory effect on ANP-stimulated cGMP synthesis in Csk−/− cells
In order to investigate the possibility that GC-A may be regulated by tyrosine kinases, we used cultured mouse embryonic fibroblasts (MEF). MEF cells were grown to 80% confluence and serum starved overnight. Treatment of the cells with 200 nM ANP for 3 min at 37°C resulted in about a 30–60 fold increase in cGMP production, whereas similar concentrations of CNP resulted in only a modest increase in the intracellular cGMP content (Fig. 1A). The relatively lower elevation in intracellular cGMP levels when MEF cells were treated with CNP compared to that obtained when stimulated with similar concentrations of ANP shows that the majority of endogenously expressed NP receptors in MEF cells are GC-A. Previous reports showed that serum, LPA and PDGF were able to desensitize GC-B in fibroblast cells (4, 22). Therefore, we tested if endogenous GC-A in MEF cells is also inhibited by LPA and serum. Treatment of serum-starved MEF cells with 10% FBS or 10 μM LPA for 30 min resulted in about 60% inhibition of ANP-induced elevation in the intracellular cGMP levels (Fig. 1B). Serum and LPA also inhibited CNP-stimulated elevations in the intracellular cGMP levels in MEF cells to a similar extent (data not shown).
Figure 1.
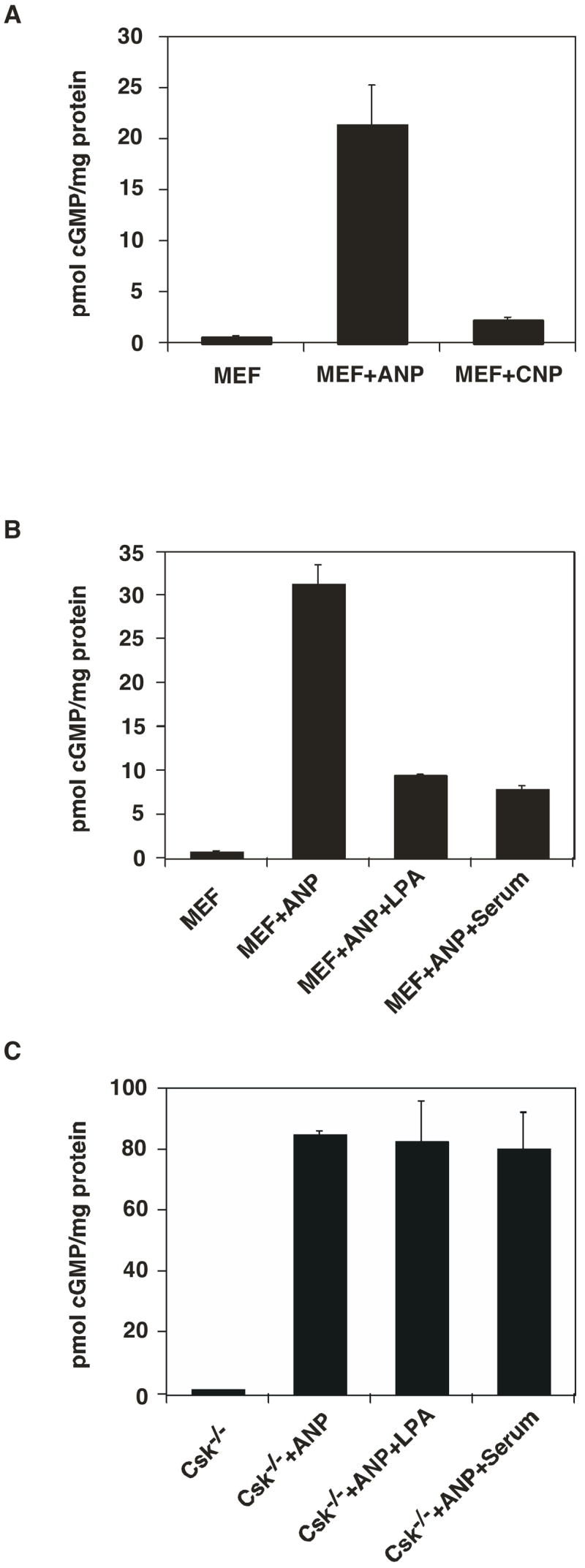
LPA and serum do not exert an inhibitory effect on ANP-stimulated cGMP synthesis in Csk−/− cells. A. Elevation in the intracellular cGMP levels in MEF cells in response to ANP and CNP treatment. MEF cells were cultured in 6 well dishes to 80% confluence. After serum starvation for 18–24 hours, cells were treated with starvation medium containing 0.5 mM IBMX for 30 minutes at 37°C, followed by treatment with 200 nM ANP or 200 nM CNP for 3 min at 37°C. Cells were washed once with PBS, lysed with 0.5% Triton X-100 containing 0.5 mM IBMX and cGMP content was determined. B. LPA (10 μM) and serum (10% FBS) attenuate ANP induced cGMP synthesis in MEF cells. C. ANP response is enhanced, and LPA and serum do not exert an inhibitory effect on ANP-stimulated cGMP synthesis in Csk−/− cells. Data represent the means ± S.D. of three experiments.
Serum and LPA initiate downstream signaling including tyrosine kinase and G-protein mediated pathways (43–45). Previous studies from our laboratory have established that the nonreceptor tyrosine kinase Csk is essential for the cytoskeletal rearrangements induced by serum and LPA in MEF cells (30). Nonreceptor tyrosine kinases are also implicated in the regulation of sarcomeric organization as well as hypertrophic gene expression (37). Therefore we examined whether Csk plays a role in the regulation of ligand dependent activity of GC-A in MEF cells. For this purpose, we used embryonic fibroblasts derived from knockout mice that lack the nonreceptor tyrosine kinase Csk (Csk−/−) (30). Treatment of Csk−/− cells with 200 nM ANP resulted in about an 80–100 fold increase in cellular cGMP levels (Fig. 1C), whereas treatment with 200 nM CNP resulted in only a modest increase in the cGMP content (data not shown). The relatively lower elevation in the intracellular cGMP levels upon stimulation with CNP again shows that the majority of the receptors expressed in Csk−/− cells are GC-A. In contrast to the observation in MEF cells, treatment of Csk−/− cells with 10% FBS or 10 μM LPA for 30 min did not result in any statistically significant changes in ANP-stimulated cGMP production (Fig. 1C). These data strongly indicate that Csk is a negative modulator of the ANP-induced increase in cGMP, and is an essential mediator of serum- and LPA-induced attenuation of ANP-stimulated cGMP synthesis in MEF cells.
To further confirm this conclusion, we transiently transfected Csk−/− cells with a DNA plasmid encoding human Csk and measured the changes in ANP-induced cGMP production. Re-expression of Csk in Csk−/− cells resulted in about 3-fold lower ANP-stimulated cGMP levels compared to that observed in vector-transfected cells (Fig. 2). Moreover, re-expression of Csk rescued the inhibitory effect of LPA on ANP-induced cGMP production (Fig. 2).
Figure 2.
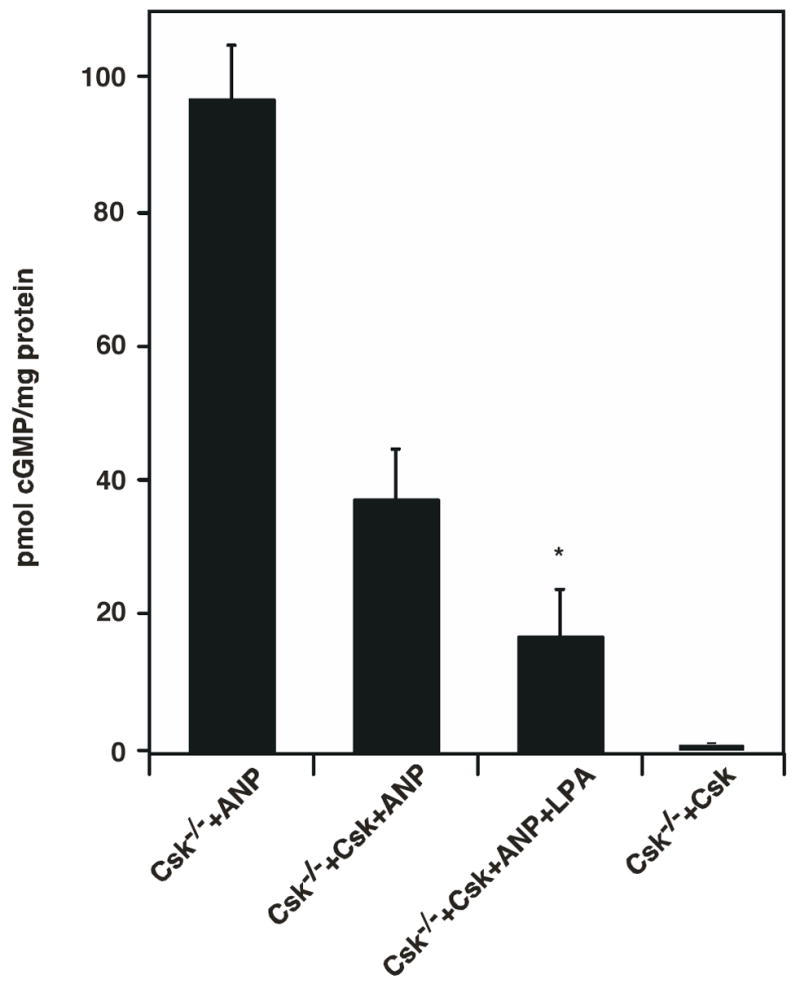
Transient re-expression of Csk in Csk−/− cells results in attenuation of ANP stimulated cGMP production and reconstitution of LPA inhibition. Csk−/− cells grown to 70%–80% confluence in 6 well dishes was transiently transfected with a vector plasmid (Column 1) or plasmids carrying human Csk (Columns 2 and 3) as described in experimental protocols. 16–18 hours after transfection, cells were serum starved overnight and treated for 30 min at 37°C and 5% CO2 with medium containing LPA and 0.5 mM IBMX. This was followed by treatment with 200 nM ANP at 37°C for 3 min. Medium was aspirated, cells were washed once with PBS, lysed with 0.5% Triton X-100 containing 0.5 mM IBMX and cGMP present in the samples was determined. * indicates that the cGMP level from Csk−/−+Csk+ANP is significant different from that from Csk−/−+Csk+ANP+LPA (t-test: p<0.05). The values shown represent the means ± S.D. of three experiments.
The catalytic activity of Csk is essential for its regulation of receptor guanylyl cyclase activity
In order to determine if the catalytic activity of Csk is essential for its inhibitory effect on GC-A, we used Csk−/− cells stably expressing the Csk R318A mutant whose catalytic activity is impaired but can be chemically rescued and activated by small organic compounds such as imidazole (30, 46). Using this mutant cell line, we previously demonstrated that the catalytic activity of Csk is required for LPA- and serum-induced cytoskeletal reorganization in MEF cells (30). As shown in Fig. 3A, ANP-stimulated cGMP production in CskR318A cells was up to ten-fold lower in the presence than in the absence of imidazole. Imidazole had no effect on the EC50 of ANP for GC-A (EC50 ~28 nM). As control, treatment of Csk−/− cells (without the CskR318A mutant) with imidazole did not result in a statistically significant change in ANP-stimulated cGMP production (Fig. 3B). Transient expression of CskR318A in Csk−/− cells (without imidazole addition) did not lead to any significant changes in ANP-stimulated cGMP synthesis (Fig. 3B). These results demonstrate that the catalytic activity of Csk is essential for its observed inhibitory effect on ANP-stimulated cGMP synthesis in MEF cells.
Figure 3.
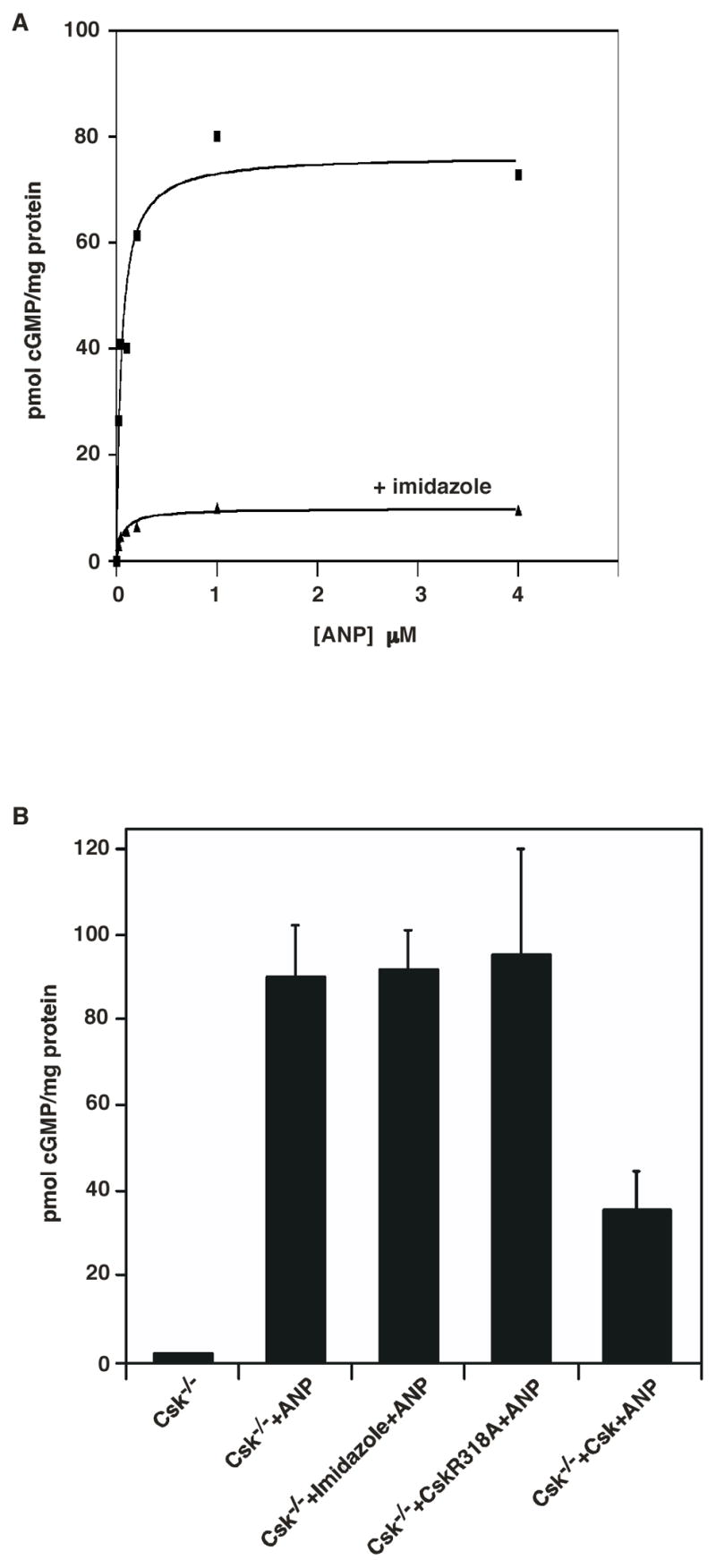
The catalytic activity of Csk is essential for its regulation of receptor guanylyl cyclase activity. A. Csk−/− cells stably expressing the CskR318A mutant were grown to 80% confluence in 6 well dishes. Following serum starvation overnight, cells were treated with medium containing LPA and 0.5 mM IBMX for 30 min at 37°C in the presence (▼) or absence (■) of 50 mM imidazole. This was followed by treatment with indicated concentrations of ANP for 3 min at 37°C. Cells were then washed once with PBS, lysed with 0.5% Triton X-100 containing 0.5 mM IBMX and cGMP content present in the samples was determined. B. Treatment of Csk−/− cells with imidazole (Column 3) or transient expression of CskR318A in Csk−/− cells (without imidazole addition) (Column 4) did not lead to significant changes in ANP-stimulated cGMP production. Data are representative of three similar experiments.
Attenuation of ANP-stimulated cGMP synthesis by Csk does not require PKC activity
Since GC-A is known to be heterologously desensitized by PKC possibly working through a phosphatase to dephosphorylate GC-A, we used a general PKC inhibitor, Calphostin C, to determine if inhibition of GC-A by Csk is occurring through a PKC dependent pathway. As shown in Fig. 4A, Calphostin C blocked phorbol myristate acetate (PMA, a pharmacological activator of PKC) induced inhibition of GC-A, but not LPA-initiated inhibition (Fig. 4A). Treatment of Csk−/− cells with Calphostin C has no effect on ANP-induced cGMP production (Fig. 4B). Expression of CskR318A mutant in Csk−/− cells (without addition of imidazole) did not suppress ANP stimulation of GC-A, confirming the impaired catalytic activity of CskR318A mutant. Addition of imidazole rescued the catalytic activity of CskR318A, leading to the inhibition of ANP-induced cGMP production. Moreover, treatment of CskR318A cells with imidazole in the presence Calphostin C resulted in no significant change in the magnitude of Csk inhibition on ANP-induced cGMP production (Fig. 4B). This implies that the observed inhibitory effect of Csk on ANP-induced cGMP synthesis in MEF cells is not mediated by PKC.
Figure 4.
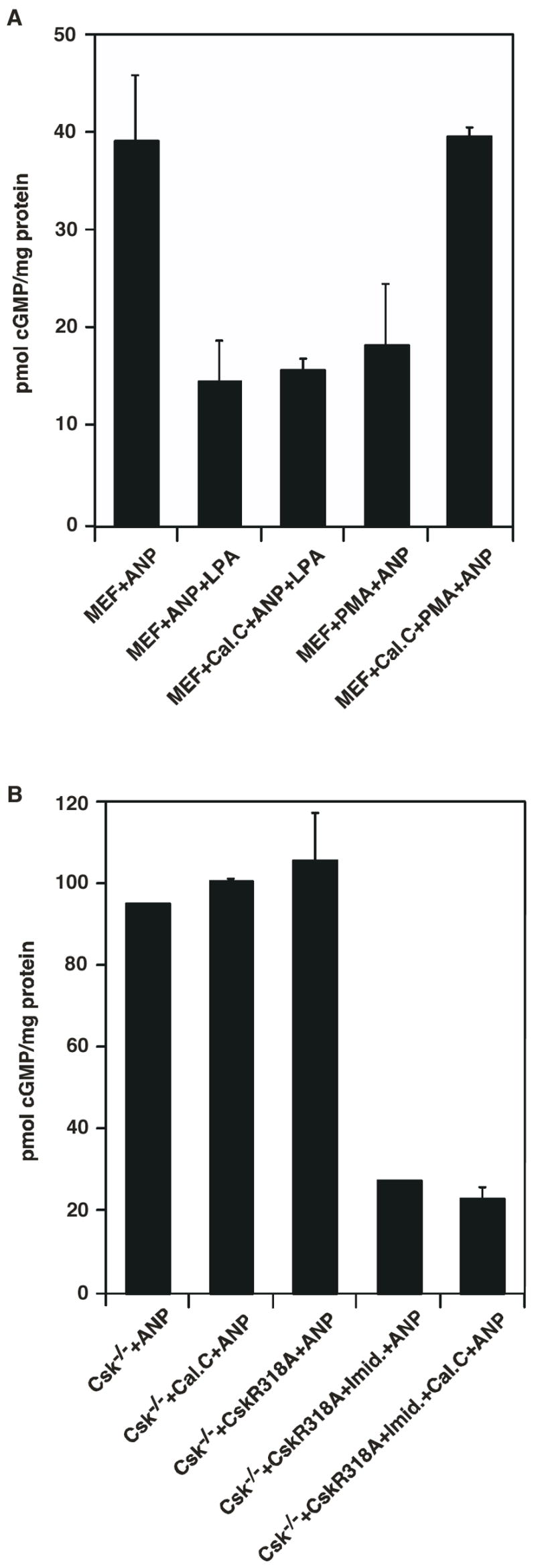
Attenuation of ANP-stimulated cGMP synthesis by Csk does not require PKC activity. A. MEF cells were serum-starved overnight. After pretreatment with Calphostin C or vehicle, cells were treated with ANP, LPA then ANP, or PMA then ANP for 3 min. Cells were then washed once with PBS, lysed with 0.5% Triton X-100 containing 0.5 mM IBMX and cGMP content present in the samples was determined. B. Csk−/− cells grown to 70–80% confluence in 6 well dishes were transiently transfected with CskR318A or empty vector (pcDNA3.1). 16–18 hours after transfection, cells were serum-starved overnight. After pretreatment with Calphostin C or vehicle, cells were treated with growth medium containing 50 mM imidazole and 0.5 mM IBMX for 30 min at 37°C. This was followed by treatment with 200 nM ANP for 3 min at 37°C and 5% CO2. Cells were then washed once with PBS, lysed with 0.5% Triton X-100 containing 0.5 mM IBMX and cGMP content present in the samples was determined. Data represent the means ± S.D. of three experiments.
Csk does not phosphorylate GC-A
To learn the biochemical mechanism by which Csk mediates the LPA inhibitory effect on GC-A, we first tested the possibility that Csk might directly phosphorylate GC-A leading to the inhibition of GC-A activity. We purified recombinant Csk and GST-fusion of the intracellular domain of GC-A (residue 495 to the C-terminal end) from E. coli (30) (Fig. 5A). In the presence of ATP, Csk phosphorylated an exogenous purified substrate GST-CDB3 or auto-phosphorylated itself (29) (Fig. 5B). We noticed that, in the presence of a phosphorylatable exogenous substrate, Csk would phosphorylate the exogenous substrate with little auto-phosphorylation. In the absence of a phosphorylatable exogenous substrate, Csk autophosphorylated. These protein tyrosine phosphorylation events were monitored with an anti-phospho-tyrosine antibody. However, Csk did not phosphorylate GST-GC-A-intra (Fig. 5B). Therefore, Csk could not directly phosphorylate GC-A.
Figure 5.
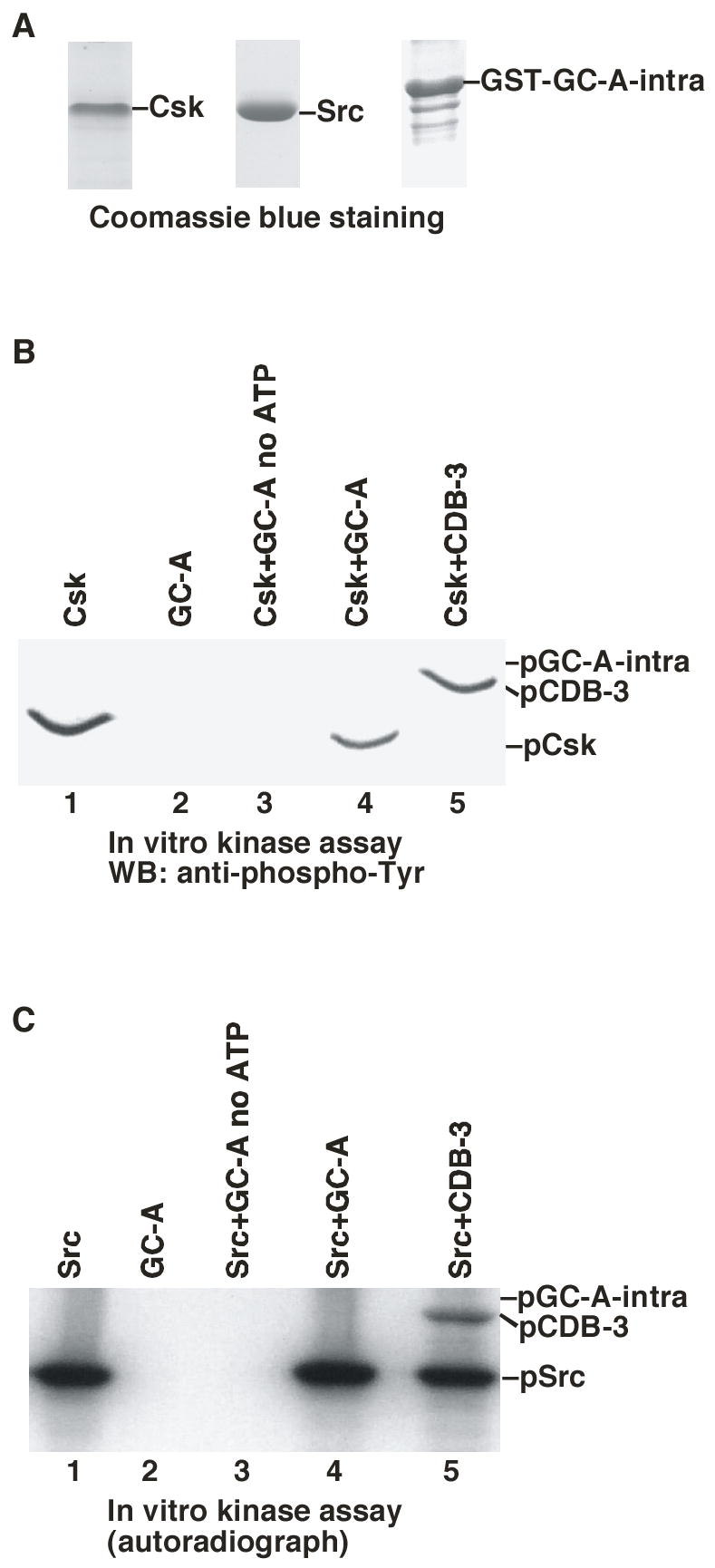
Csk does not directly phosphorylate GC-A. A. Coomassie blue staining of purified recombinant Csk, Src and GST-GC-A intra (the intracellular domain of GC-A). B. In vitro kinase assay of Csk using GST-CDB3 and GST-GC-A as exogenous substrates. The in vitro kinase assay was performed in the presence of ATP and the results were analyzed with SDS-PAGE and western blotted with anti-phospho-Tyr antibody. C. In vitro kinase assay of Src using GST-CDB3 and GST-GC-A-intra as substrates. The kinase assay was performed in the presence of γ-32P-ATP and the results were analyzed by SDS-PAGE and autoradiography.
Csk was originally purified as a tyrosine kinase capable of phosphorylating the C-terminal tyrosine residue of Src-family tyrosine kinases (23). We next sought to examine whether Src could directly phosphorylate GC-A. We purified recombinant Src from Sf9 insect cells (34) (Fig. 5A). In the presence of [γ-32P]ATP, Src autophosphorylated itself and the exogenous substrate GST-CDB3, but not GST-GC-A-intra (Fig. 5C). Furthermore, immunoprecipitated GC-A from HEK-293 cells and MEF cells was not tyrosine phosphorylated when the immunoprecipitates were probed with anti-phospho-tyrosine antibodies (data not shown). Hence, these data demonstrate that direct tyrosine phosphorylation of GC-A is not the mechanism by which Csk mediates LPA’s inhibitory effect.
LPA decreased the Ser/Thr phosphorylation of GC-A in the presence of Csk
Previously it has been shown that activation of protein kinase C (by PMA) led to the activation of protein Ser/Thr phosphatase to decrease the Ser/Thr phosphorylation of GC-A and inhibition of GC-A activity (17). Furthermore, it is known that Ser/Thr phosphorylation of GC-A is required for ANP activation of GC-A (17). Therefore, there is a correlation of a decrease in Ser/Thr phosphorylation of GC-A and a decrease of GC-A activity. Although we had shown that Csk-mediated inhibition does not involve PKC (Fig. 4), we investigated whether Csk-mediated inhibition involves a decrease in Ser/Thr phosphorylation of GC-A (Fig. 6). We treated starved Csk−/−/Csk cells with LPA or PMA. GC-A protein was immunoprecipitated with anti-GC-A antibody and western blotted with anti-phospho-Ser/Thr antibody (Fig. 6 A and B). Both LPA and PMA induced a decrease of Ser/Thr phosphorylation of GC-A, indicating that LPA-induced inhibition of GC-A involves a decrease in Ser/Thr phosphorylation of GC-A, similar to protein kinase C activation. Similar results were obtained with MEF cells (data not shown). More importantly, this LPA-induced reduction of Ser/Thr phosphorylation of GC-A was dependent on Csk. In Csk−/− cells, while PMA induced a decrease in GC-A Ser/Thr phosphorylation, LPA treatment did not (Fig. 6 C and D). These results demonstrate that Csk is required for LPA-induced reduction of Ser/Thr phosphorylation of GC-A. Furthermore, comparing the percentage of Ser/Thr phosphorylated GC-A in total GC-A, there was a 50% increase in GC-A Ser/Thr phosphorylation in Csk−/− cells (Fig. 6 B and D). This result is consistent with the higher ANP-induced activity of GC-A in Csk−/− cells than MEF cells (Fig. 1). Therefore, the molecular mechanism by which Csk mediates LPA-induced GC-A inhibition likely involves a reduction of Ser/Thr phosphorylation of GC-A.
Figure 6.
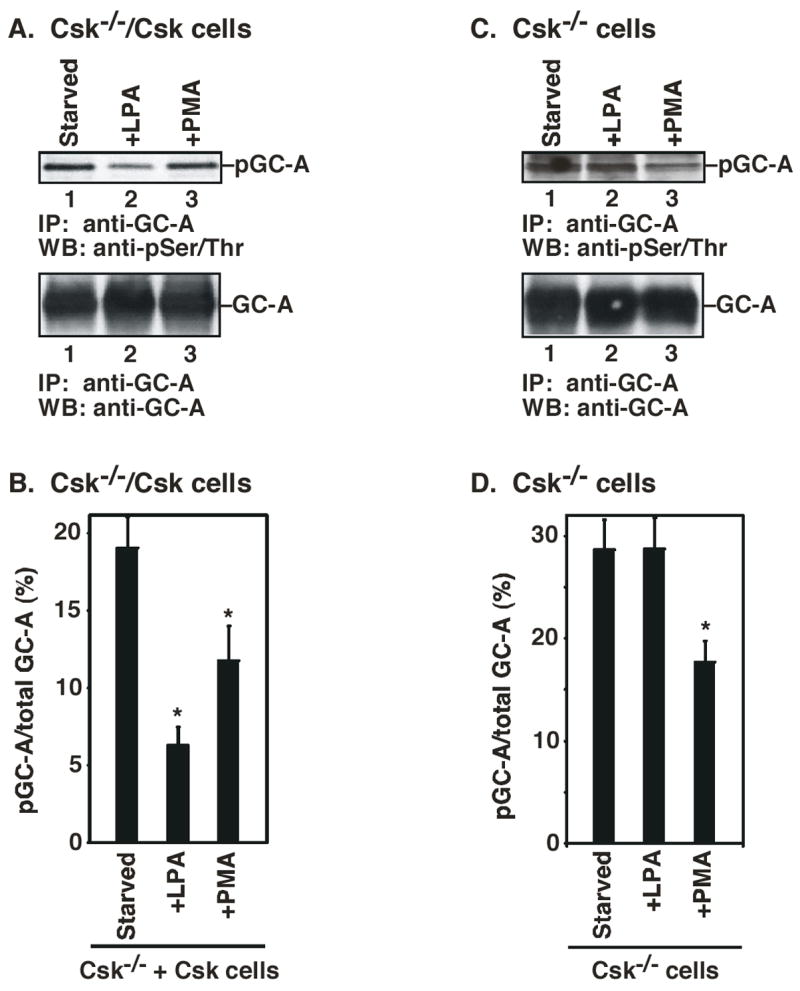
LPA-induced decrease of Ser/Thr phosphorylation of GC-A depends on Csk. A. Csk−/−/Csk cells were serum-starved. Cells were then treated with LPA or PMA. Cell lysates were immunoprecipitated with anti-GC-A antibody. After SDS-PAGE, the filters were probed with anti-phospho-Ser/Thr antibody (top panel) or anti-GC-A antibody (bottom panel). B. The intensity of each band in A was quantified by an image analyzer. The percentage of phosphorylated GC-A is corrected by the amount of the total GC-A. C. Csk−/− cells were serum-starved. Cells were then treated with LPA or PMA. Cell lysates were immunoprecipitated with anti-GC-A antibody. After SDS-PAGE, the filters were probed with anti-phospho-Ser/Thr antibody (top panel) or anti-GC-A antibody (bottom panel). D. The intensity of each band in C was quantified by an image analyzer. The percentage of phosphorylated GC-A is corrected by the amount of the total GC-A. * indicates an significant difference from the untreated cells (t-test: p<0.05). Data represent the means ± S.D. of three experiments.
DISCUSSION
In this study we show that a nonreceptor tyrosine kinase is involved in the regulation of natriuretic peptide-induced activity of membrane-bound guanylyl cyclase. We showed that the marked inhibition of ANP-induced generation of cGMP by serum and LPA was completely blocked by deletion of Csk in MEF cells. The inhibitory effect of Csk was fully restored by reintroduction of Csk in Csk−/− cells. These observations demonstrate that Csk is a negative modulator of GC-A activity and that Csk activation is essential for serum and LPA attenuation of ANP-stimulated GC-A activity in MEF cells.
Using a newly developed chemical rescue approach, we also demonstrated that the catalytic activity of Csk is necessary for its negative modulatory effect on GC-A. It has previously been shown that the catalytic activity of an inactive Csk mutant, CskR318A, can be rescued by chemical compounds such as imidazole in vitro and in vivo (30, 46). Csk−/− MEF cells transfected with the inactive CskR318A failed to show the characteristic inhibitory action of serum and LPA on ANP-stimulated GC-A activity. This inhibitory action was rescued upon treatment of the cells with imidazole, demonstrating that the rescued catalytic activity of CskR318A is critical to the inhibitory effect of Csk.
It has been reported that PKC, possibly working through a phosphatase to dephosphorylate GC-A, inhibits GC-A activity (20). However, we observed no effect of a general PKC inhibitor, Calphostin C, on Csk-mediated inhibition of GC-A activity. Nevertheless, our data suggests that the mechanism of inhibition of GC-A by Csk involves changes on GC-A modification. Indeed, we have shown that, in the absence of Csk, GC-A is highly Ser/Thr phosphorylated. It is known that GC-A, under basal conditions, is phosphorylated on six known Ser/Thr sites within its kinase homology domain (17, 47). Single mutation of any one of these phosphorylation sites to alanine decreases receptor activity. Mutation of all these phosphorylation sites to alanine yields a receptor that is unresponsive to the ligand ANP (17). Desensitization of GC-A is correlated with a decrease in Ser/Thr phosphorylation (48). Our finding reveals that Csk-mediated LPA inhibition of GC-A might also involve a decrease of Ser/Thr phosphorylation of GC-A. It would be interesting to investigate whether Csk is involved in GC-A homologous desensitization after ANP treatment. Given that re-expression of Csk in Csk−/− cells resulted in about 3-fold lower ANP-stimulated cGMP levels compared to that observed in vector-transfected cells (Fig. 2), Csk might be involved in GC-A homologous desensitization. On the other hand, Csk could lead to the de-phosphorylation of GC-A and thus reduced the ANP-induced cGMP accumulation without being directly in the GC-A homologous desensitization after ANP treatment. Since both PKC and Csk could lead to GC-A desensitization though via different pathways, PKC and Csk could employ the same or different phosphatases to inhibit GC-A. The underlying biochemical mechanism is the same for both PKC and Csk-mediated desensitization, that is the dephosphorylation of GC-A.
Currently it is not clear which kinase(s) or phosphatase(s) is responsible for the phosphorylation and dephosphorylation of GC-A. Interestingly, in a yeast two-hybrid screen, protein phosphatase 5 (PP5) was shown to interact with the kinase homology domain of GC-A (49). Although this interaction has not been reported in mammalian cells, PP5 has also been reported to interact with Gα12 and Gα13 which mediate the signaling from the G protein-coupled LPA receptors (50). Further investigation is needed to examine whether PP5 indeed mediates LPA initiated inhibition of GC-A.
ANP, via activation of GC-A receptors, inhibits cardiomyocyte growth, promotes relaxation of preconstricted vasculature, and is a potent anti-hypertensive and anti-hypertrophic agent. On the other hand, over-activation of several nonreceptor tyrosine kinases has been implicated in abnormal cellular growth, vasoconstriction, and development of cardiovascular diseases such as hypertension and cardiac hypertrophy (35–37). Taking these findings in conjunction with the present data, we postulate that the effects of Csk and possibly other tyrosine kinases on contraction, vascular tone, activation of the MAPK pathway, growth and hypertrophy may be mediated in part by their inhibition of the powerful vasodilatory, antiproliferative and antihypertrophic effects of natriuretic peptides.
Acknowledgments
We thank members of our laboratory for discussion and for critical reading of the manuscript.
ABBREVIATIONS
- NP
natriuretic peptide
- GC
guanylyl cyclase
- LPA
lysophosphatidic acid
- ANP
atrial natriuretic peptide
- BNP
brain natriuretic peptide
- CNP
C-type natriuretic peptide
- IBMX
3-isobutyl-methylxanthine
- MEF
mouse embryonic fibroblast
- PMA
phorbol myristate acetate
Footnotes
Supported by grants from the NIH (AG014563 and GM056904).
References
- 1.Silberbach M, Roberts CT., Jr Natriuretic peptide signalling: molecular and cellular pathways to growth regulation. Cell Signal. 2001;13:221–31. doi: 10.1016/s0898-6568(01)00139-5. [DOI] [PubMed] [Google Scholar]
- 2.Wedel B, Garbers D. The guanylyl cyclase family at Y2K. Annu Rev Physiol. 2001;63:215–33. doi: 10.1146/annurev.physiol.63.1.215. [DOI] [PubMed] [Google Scholar]
- 3.Kuhn M. Structure, regulation, and function of mammalian membrane guanylyl cyclase receptors, with a focus on guanylyl cyclase-A. Circ Res. 2003;93:700–9. doi: 10.1161/01.RES.0000094745.28948.4D. [DOI] [PubMed] [Google Scholar]
- 4.Chrisman TD, Schulz S, Potter LR, Garbers DL. Seminal plasma factors that cause large elevations in cellular cyclic GMP are C-type natriuretic peptides. J Biol Chem. 1993;268:3698–703. [PubMed] [Google Scholar]
- 5.Atlas SA, Kleinert HD, Camargo MJ, Januszewicz A, Sealey JE, Laragh JH, Schilling JW, Lewicki JA, Johnson LK, Maack T. Purification, sequencing and synthesis of natriuretic and vasoactive rat atrial peptide. Nature. 1984;309:717–9. doi: 10.1038/309717a0. [DOI] [PubMed] [Google Scholar]
- 6.Maack T. Receptors of atrial natriuretic factor. Annu Rev Physiol. 1992;54:11–27. doi: 10.1146/annurev.ph.54.030192.000303. [DOI] [PubMed] [Google Scholar]
- 7.Chinkers M, Garbers DL, Chang MS, Lowe DG, Chin HM, Goeddel DV, Schulz S. A membrane form of guanylate cyclase is an atrial natriuretic peptide receptor. Nature. 1989;338:78–83. doi: 10.1038/338078a0. [DOI] [PubMed] [Google Scholar]
- 8.Maack T. Role of atrial natriuretic factor in volume control. Kidney Int. 1996;49:1732–7. doi: 10.1038/ki.1996.257. [DOI] [PubMed] [Google Scholar]
- 9.Cameron VA, Rademaker MT, Ellmers LJ, Espiner EA, Nicholls MG, Richards AM. Atrial (ANP) and brain natriuretic peptide (BNP) expression after myocardial infarction in sheep: ANP is synthesized by fibroblasts infiltrating the infarct. Endocrinology. 2000;141:4690–7. doi: 10.1210/endo.141.12.7847. [DOI] [PubMed] [Google Scholar]
- 10.Calderone A, Thaik CM, Takahashi N, Chang DL, Colucci WS. Nitric oxide, atrial natriuretic peptide, and cyclic GMP inhibit the growth-promoting effects of norepinephrine in cardiac myocytes and fibroblasts. J Clin Invest. 1998;101:812–8. doi: 10.1172/JCI119883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horio T, Nishikimi T, Yoshihara F, Matsuo H, Takishita S, Kangawa K. Inhibitory regulation of hypertrophy by endogenous atrial natriuretic peptide in cultured cardiac myocytes. Hypertension. 2000;35:19–24. doi: 10.1161/01.hyp.35.1.19. [DOI] [PubMed] [Google Scholar]
- 12.Holtwick R, van Eickels M, Skryabin BV, Baba HA, Bubikat A, Begrow F, Schneider MD, Garbers DL, Kuhn M. Pressure-independent cardiac hypertrophy in mice with cardiomyocyte-restricted inactivation of the atrial natriuretic peptide receptor guanylyl cyclase-A. J Clin Invest. 2003;111:1399–407. doi: 10.1172/JCI17061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang D, Oparil S, Feng JA, Li P, Perry G, Chen LB, Dai M, John SW, Chen YF. Effects of pressure overload on extracellular matrix expression in the heart of the atrial natriuretic peptide-null mouse. Hypertension. 2003;42:88–95. doi: 10.1161/01.HYP.0000074905.22908.A6. [DOI] [PubMed] [Google Scholar]
- 14.John SW, Krege JH, Oliver PM, Hagaman JR, Hodgin JB, Pang SC, Flynn TG, Smithies O. Genetic decreases in atrial natriuretic peptide and salt-sensitive hypertension. Science. 1995;267:679–81. doi: 10.1126/science.7839143. [DOI] [PubMed] [Google Scholar]
- 15.Oliver PM, Fox JE, Kim R, Rockman HA, Kim HS, Reddick RL, Pandey KN, Milgram SL, Smithies O, Maeda N. Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A. Proc Natl Acad Sci U S A. 1997;94:14730–5. doi: 10.1073/pnas.94.26.14730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenkranz AC, Woods RL, Dusting GJ, Ritchie RH. Antihypertrophic actions of the natriuretic peptides in adult rat cardiomyocytes: importance of cyclic GMP. Cardiovasc Res. 2003;57:515–22. doi: 10.1016/s0008-6363(02)00667-3. [DOI] [PubMed] [Google Scholar]
- 17.Potter LR, Hunter T. Phosphorylation of the kinase homology domain is essential for activation of the A-type natriuretic peptide receptor. Mol Cell Biol. 1998;18:2164–72. doi: 10.1128/mcb.18.4.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duda T, Yadav P, Jankowska A, Venkataraman V, Sharma RK. Three dimensional atomic model and experimental validation for the ATP-Regulated Module (ARM) of the atrial natriuretic factor receptor guanylate cyclase. Mol Cell Biochem. 2001;217:165–72. doi: 10.1023/a:1007236917061. [DOI] [PubMed] [Google Scholar]
- 19.Potter LR, Hunter T. Identification and characterization of the phosphorylation sites of the guanylyl cyclase-linked natriuretic peptide receptors A and B. Methods. 1999;19:506–20. doi: 10.1006/meth.1999.0893. [DOI] [PubMed] [Google Scholar]
- 20.Bryan PM, Potter LR. The atrial natriuretic peptide receptor (NPR-A/GC-A) is dephosphorylated by distinct microcystin-sensitive and magnesium-dependent protein phosphatases. J Biol Chem. 2002;277:16041–7. doi: 10.1074/jbc.M110626200. [DOI] [PubMed] [Google Scholar]
- 21.Chrisman TD, Garbers DL. Reciprocal antagonism coordinates C-type natriuretic peptide and mitogen-signaling pathways in fibroblasts. J Biol Chem. 1999;274:4293–9. doi: 10.1074/jbc.274.7.4293. [DOI] [PubMed] [Google Scholar]
- 22.Abbey SE, Potter LR. Lysophosphatidic acid inhibits C-type natriuretic peptide activation of guanylyl cyclase-B. Endocrinology. 2003;144:240–6. doi: 10.1210/en.2002-220702. [DOI] [PubMed] [Google Scholar]
- 23.Nada S, Okada M, MacAuley A, Cooper JA, Nakagawa H. Cloning of a complementary DNA for a protein-tyrosine kinase that specifically phosphorylates a negative regulatory site of p60c-src. Nature. 1991;351:69–72. doi: 10.1038/351069a0. [DOI] [PubMed] [Google Scholar]
- 24.Miller MA, Malik IA, Shenk MA, Steele RE. The Src/Csk regulatory circuit arose early in metazoan evolution. Oncogene. 2000;19:3925–30. doi: 10.1038/sj.onc.1203714. [DOI] [PubMed] [Google Scholar]
- 25.Imamoto A, Soriano P. Disruption of the csk gene, encoding a negative regulator of Src family tyrosine kinases, leads to neural tube defects and embryonic lethality in mice. Cell. 1993;73:1117–24. doi: 10.1016/0092-8674(93)90641-3. [DOI] [PubMed] [Google Scholar]
- 26.Nada S, Yagi T, Takeda H, Tokunaga T, Nakagawa H, Ikawa Y, Okada M, Aizawa S. Constitutive activation of Src family kinases in mouse embryos that lack Csk. Cell. 1993;73:1125–35. doi: 10.1016/0092-8674(93)90642-4. [DOI] [PubMed] [Google Scholar]
- 27.Thomas SM, Soriano P, Imamoto A. Specific and redundant roles of Src and Fyn in organizing the cytoskeleton. Nature. 1995;376:267–71. doi: 10.1038/376267a0. [DOI] [PubMed] [Google Scholar]
- 28.Gross JA, Appleby MW, Chien S, Nada S, Bartelmez SH, Okada M, Aizawa S, Perlmutter RM. Control of lymphopoiesis by p50csk, a regulatory protein tyrosine kinase. J Exp Med. 1995;181:463–73. doi: 10.1084/jem.181.2.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wan Y, Bence K, Hata A, Kurosaki T, Veillette A, Huang XY. Genetic evidence for a tyrosine kinase cascade preceding the mitogen- activated protein kinase cascade in vertebrate G protein signaling. J Biol Chem. 1997;272:17209–15. doi: 10.1074/jbc.272.27.17209. [DOI] [PubMed] [Google Scholar]
- 30.Lowry WE, Huang J, Ma YC, Ali S, Wang D, Williams DM, Okada M, Cole PA, Huang XY. Csk, a critical link of g protein signals to actin cytoskeletal reorganization. Dev Cell. 2002;2:733–44. doi: 10.1016/s1534-5807(02)00175-2. [DOI] [PubMed] [Google Scholar]
- 31.Wan Y, Kurosaki T, Huang XY. Tyrosine kinases in activation of the MAP kinase cascade by G-protein- coupled receptors. Nature. 1996;380:541–4. doi: 10.1038/380541a0. [DOI] [PubMed] [Google Scholar]
- 32.Bence K, Ma W, Kozasa T, Huang XY. Direct stimulation of Bruton’s tyrosine kinase by G(q)-protein alpha- subunit. Nature. 1997;389:296–9. doi: 10.1038/38520. [DOI] [PubMed] [Google Scholar]
- 33.Jiang Y, Ma W, Wan Y, Kozasa T, Hattori S, Huang XY. The G protein G alpha12 stimulates Bruton’s tyrosine kinase and a rasGAP through a conserved PH/BM domain. Nature. 1998;395:808–13. doi: 10.1038/27454. [DOI] [PubMed] [Google Scholar]
- 34.Ma YC, Huang J, Ali S, Lowry W, Huang XY. Src tyrosine kinase is a novel direct effector of G proteins. Cell. 2000;102:635–46. doi: 10.1016/s0092-8674(00)00086-6. [DOI] [PubMed] [Google Scholar]
- 35.Taylor JM, Rovin JD, Parsons JT. A role for focal adhesion kinase in phenylephrine-induced hypertrophy of rat ventricular cardiomyocytes. J Biol Chem. 2000;275:19250–7. doi: 10.1074/jbc.M909099199. [DOI] [PubMed] [Google Scholar]
- 36.He Q, LaPointe MC. Src and Rac mediate endothelin-1 and lysophosphatidic acid stimulation of the human brain natriuretic peptide promoter. Hypertension. 2001;37:478–84. doi: 10.1161/01.hyp.37.2.478. [DOI] [PubMed] [Google Scholar]
- 37.Kovacic-Milivojevic B, Roediger F, Almeida EA, Damsky CH, Gardner DG, Ilic D. Focal adhesion kinase and p130Cas mediate both sarcomeric organization and activation of genes associated with cardiac myocyte hypertrophy. Mol Biol Cell. 2001;12:2290–307. doi: 10.1091/mbc.12.8.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cvejic S, Zhu Z, Felice SJ, Berman Y, Huang XY. The endogenous ligand Stunted of the GPCR Methuselah extends lifespan in Drosophila. Nat Cell Biol. 2004;6:540–6. doi: 10.1038/ncb1133. [DOI] [PubMed] [Google Scholar]
- 39.Koh GY, Nussenzveig DR, Okolicany J, Price DA, Maack T. Dynamics of atrial natriuretic factor-guanylate cyclase receptors and receptor-ligand complexes in cultured glomerular mesangial and renomedullary interstitial cells. J Biol Chem. 1992;267:11987–94. [PubMed] [Google Scholar]
- 40.Vieira MA, Gao M, Nikonova LN, Maack T. Molecular and cellular physiology of the dissociation of atrial natriuretic peptide from guanylyl cyclase a receptors. J Biol Chem. 2001;276:36438–45. doi: 10.1074/jbc.M102208200. [DOI] [PubMed] [Google Scholar]
- 41.Wang D, Huang XY, Cole PA. Molecular determinants for Csk-catalyzed tyrosine phosphorylation of the Src tail. Biochemistry. 2001;40:2004–10. doi: 10.1021/bi002342n. [DOI] [PubMed] [Google Scholar]
- 42.Tang H, Zhao ZJ, Huang XY, Landon EJ, Inagami T. Fyn kinase-directed activation of SH2 domain-containing protein-tyrosine phosphatase SHP-2 by Gi protein-coupled receptors in Madin-Darby canine kidney cells. J Biol Chem. 1999;274:12401–7. doi: 10.1074/jbc.274.18.12401. [DOI] [PubMed] [Google Scholar]
- 43.Ridley AJ, Hall A. Signal transduction pathways regulating Rho-mediated stress fibre formation: requirement for a tyrosine kinase. Embo J. 1994;13:2600–10. doi: 10.1002/j.1460-2075.1994.tb06550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fukushima N, Ishii I, Contos JJ, Weiner JA, Chun J. Lysophospholipid receptors. Annu Rev Pharmacol Toxicol. 2001;41:507–34. doi: 10.1146/annurev.pharmtox.41.1.507. [DOI] [PubMed] [Google Scholar]
- 45.Daaka Y. Mitogenic action of LPA in prostate. Biochim Biophys Acta. 2002;1582:265–9. doi: 10.1016/s1388-1981(02)00180-4. [DOI] [PubMed] [Google Scholar]
- 46.Williams DM, Wang D, Cole PA. Chemical rescue of a mutant protein-tyrosine kinase. J Biol Chem. 2000;275:38127–30. doi: 10.1074/jbc.C000606200. [DOI] [PubMed] [Google Scholar]
- 47.Koller KJ, Lipari MT, Goeddel DV. Proper glycosylation and phosphorylation of the type A natriuretic peptide receptor are required for hormone-stimulated guanylyl cyclase activity. J Biol Chem. 1993;268:5997–6003. [PubMed] [Google Scholar]
- 48.Potter LR, Garbers DL. Dephosphorylation of the guanylyl cyclase-A receptor causes desensitization. J Biol Chem. 1992;267:14531–4. [PubMed] [Google Scholar]
- 49.Chinkers M. Targeting of a distinctive protein-serine phosphatase to the protein kinase-like domain of the atrial natriuretic peptide receptor. Proc Natl Acad Sci U S A. 1994;91:11075–9. doi: 10.1073/pnas.91.23.11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamaguchi Y, Katoh H, Mori K, Negishi M. Galpha(12) and Galpha(13) interact with Ser/Thr protein phosphatase type 5 and stimulate its phosphatase activity. Curr Biol. 2002;12:1353–8. doi: 10.1016/s0960-9822(02)01034-5. [DOI] [PubMed] [Google Scholar]