

Abstract
Necroptosis is a regulated caspase-independent cell death pathway resulting in morphology reminiscent of passive non-regulated necrosis. Several diverse structure classes of necroptosis inhibitors have been reported to date, including a series of [1,2,3]thiadiazole benzylamide derivatives. However, initial evaluation of mouse liver microsome stability indicated that this series of compounds was rapidly degraded. A structure-activity relationship (SAR) study of the [1,2,3]thiadiazole benzylamide series revealed that increased mouse liver microsome stability and increased necroptosis inhibitory activity could be accomplished by replacement of the 4-cyclopropyl-[1,2,3]thiadiazole with a 5-cyano-1-methylpyrrole. In addition, the SAR and the cellular activity profiles, utilizing different cell types and necroptosis-inducing stimuli, of representative [1,2,3]thiadiazole and pyrrole derivatives were very similar suggesting that the two compound series inhibit necroptosis in the same manner.
Necroptosis is a regulated caspase-independent cell death pathway resulting in morphological features reminiscent of passive non-regulated necrosis.1, 2 Necroptosis can be induced with various stimuli (e.g. TNF-α and Fas ligand) and in a variety of cell types (e.g. monocytes, fibroblasts, lymphocytes, macrophages, epithelial cells and neurons). Furthermore, it may represent a significant contributor to and in some cases predominant mode of cellular demise under pathological conditions involving excessive cell stress, rapid energy loss and massive oxidative species generation, not conducive for highly energy-dependent processes such as apoptosis. Discovery of regulated necrotic cell death mechanisms, such as necroptosis, raises the possibility of implementing novel therapeutic intervention strategies for the treatment of diseases where necrosis is known to play a prominent role, such as organ ischemia (i.e. stroke3, myocardial infarction4 and retinal ischemia5), traumatic brain injury6, liver injury7, cancer chemo/radiation therapy-induced necrosis8, acute necrotizing pancreatitis9 and possibly some forms of neurodegeneration.10
To date several diverse structure classes of necroptosis inhibitors have been reported, including hydantoin containing indole derivatives (i.e. 1),11 tricyclic derivatives (i.e. 2),12 substituted 3H-thieno[2,3-d]pyrimidin-4-ones (i.e. 3),13 and [1,2,3]thiadiazole benzylamides (i.e. 4)14 (Figure 1). In addition, (±)-1 and its derivatives have demonstrated in vivo activity in the temporary and permanent middle cerebral artery occlusion (MCAO) model of cerebral ischemia1, in a mouse model of ischemia/reperfusion heart injury15 and in the controlled cortical impact (CCI) model of traumatic brain injury (TBI).16 Structure-activity relationship (SAR) studies leading to significant potency improvements for necrostatins have been reported 11 – 14, however, issues of bioavailability for these promising agents have not been previously addressed, except for 1.11
Figure 1.

Necrostatins
In order to evaluate the in vivo pharmacology of other necroptosis inhibitors via preferred administration routes (i.e. oral, intravenous, intraperitoneal or subcutaneous) they must possess adequate metabolic stability, in addition to in vitro potency. One efficient and cost effective method of assessing a compound’s metabolic stability is to measure its resistance to metabolism over time in the presence of liver microsomes.17 Utilizing this technique with mouse liver microsomes, compound 4, which inhibits necroptosis induced with TNF-α in FADD-deficient variant of human Jurkat T cells with an EC50 value of 0.28 µM, demonstrated poor metabolic stability with a half-life (t1/2) of 32.5 min and intrinsic clearance (CLint) of 42.6 ± 3.2 µL/min/mg protein. Herein, we describe the results of a SAR study to optimize the mouse liver microsome stability of this inhibitor series. In addition, we evaluated the cellular activity profile of an optimized inhibitor utilizing different cell types and necroptosis-inducing stimuli.
3-Alkyl pyrrole derivatives were prepared according to the procedure outlined in Scheme 1.18 Glycine ethyl ester, 5, was treated with p-toluenesulfonyl chloride (Ts-Cl) to give 6, which upon treatment with 4-diethylaminobutan-2-one in the presence of t-BuOK gave 7. Dehydration with POCl3 yielded the dihydropyrrole derivative 8. Elimination in the presence of sodium ethoxide generated pyrrole derivative 9. The pyrrole nitrogen was deprotonated with sodium hydride and alkylated to give 10. The ester was hydrolyzed with aqueous KOH in MeOH and then the corresponding acid 11 was converted to amide 12 using EDCI.
Scheme 1.

(a) Py, TsCl, CH2Cl2, rt, 6 h; (b) 4-diethylaminobutan-2-one, t-BuOK, t-BuOH, THF, rt, 2 d; (c) POCl3, Py, rt, 18 h, (45%, over three steps); (d) NaOEt, EtOH rt, 5 h, (82%); (e) NaH, DMF, R1I, rt, 12 h, (60–90%); (f) KOH, MeOH, H2O, 50 °C, 12 h,; (g) EDCI, HOBt, ArCH2NH2, DMF, rt, 12 h, (50–85%, over two steps).
1-Alkyl pyrrole derivatives were prepared according to the procedure outlined in Scheme 2. Methyl 2-pyrrolecarboxylate, 13, was deprotonated using sodium hydride and then alkylated to give 14. The ester was hydrolyzed to give acid 15, which was coupled to a 2-chloro-6-fluorobenzylamine utilizing EDCI to give amide 16. Regioselective bromination with NBS gave 17. Finally, conversion of the aryl bromide to a nitrile was accomplished utilizing a palladium-mediated coupling with zinc cyanide to give 18 in excellent yield.19
Scheme 2.

(a) NaH, DMF, R1I, rt, 12 h, (80–95%); (b) KOH, MeOH, H2O, rt, 8 h; (c) EDCI, HOBt, ArCH(R)NH2, DMF, rt, 12 h, (50–85%, two steps); (d) NBS, MeOH, THF, rt, 6 h, (75%); (e) Zn(CN)2, Pd(PPh3)4, DMF, 95 °C, 6 h, (85%).
Alternatively, cyano- and halo-substituted pyrrole derivatives were prepared according to the procedure outlined in Scheme 3. 1-Alkylpyrroles 14 were allowed to react with chlorosulfonyl isocyanate to give two readily separable regioisomeric cyanopyrrole derivatives 19 and 20 (1:4).20 Each was converted the corresponding acid and then coupled with 2-chloro-6-fluorobenzylamine to give 21 and 22, respectively. Methyl 2-pyrrolecarboxylate, 13, was also regioselectively chlorinated with t-butyl hypochlorite to give 23.21 N-alkylation gave 24 and subsequent ester hydrolysis yielded 25, which was coupled with 2-chloro-6-fluorobenzylamine to give 26.
Scheme 3.

(a) ClSO2NCO, CH3CN, rt, 2.5 h (82%); (b); (c) EDCI, HOBt, ArCH(R)NH2, DMF, rt, 12 h, (50–85%, over two steps); (d) t-Butyl hypochlorite, CCl4, rt, 2 d, (50%); (e) NaH, DMF, MeI, rt, 12 h, (80–95%); (f) KOH, MeOH, H2O, rt, 6 h (used without purification).
Finally, a sulfonamide pyrrole derivative was prepared according to the procedure outlined in Scheme 4. N-methylpyrrole sulfonyl chloride derivative 27 was allowed to react with 2-chloro-6-fluorobenzylamine to give 28. This material was treated with ammonia gas in the presence of trimethylaluminum to give a mixture of amide 29 and nitrile 30.22 The isolated amide 29 could be converted to nitrile 30 in the presence of phosphorus oxychloride.23
Scheme 4.

(a) Py, CH2Cl2, rt, 6 h, (95%); (b) 2.0 M AlMe3 in hexane, NH3 gas, 125 °C, PhCl, 2 d; (c) POCl3, Et3N, CHCl3, rt, 2 h (80% over two steps).
Evaluation of compounds 26, 30 and 31 – 50 (Table 1) for necroptosis inhibitory activity was performed using a FADD-deficient variant of human Jurkat T cells treated with TNF-α as previously described.1, 12 Utilizing these conditions the cells efficiently underwent necroptosis, which was completely and selectively inhibited by 1 (EC50 = 0.050 µM). For EC50 value determinations, cells were treated with 10 ng/mL of human TNF-α in the presence of increasing concentration of test compounds for 24 h followed by ATP-based viability assessment. Microsome stability was determined in pooled mouse liver microsomes.24 The results of the biological studies are shown in Table 2.
Table 1.
Pyrrrole amide derivatives prepared for inhibition of necroptosis in FADD-deficient Jurkat T cells treated with TNF-α and mouse microsome stability testing.
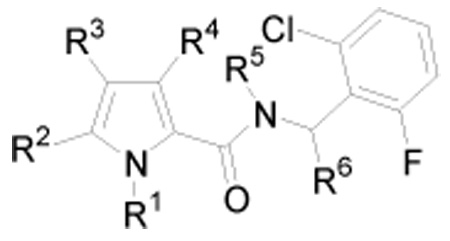 | ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | R5 | R6 |
| 26 | Me | Cl | H | H | H | H |
| 31 | H | H | H | H | H | H |
| 32 | H | H | H | Me | H | H |
| 33 | Me | H | H | Me | H | H |
| 34 | i-Pr | H | H | Me | H | H |
| 35 | Me | H | H | H | H | H |
| 36 | i-Pr | H | H | H | H | H |
| 37 | Bn | H | H | H | H | H |
| 38 | CH(Me)Ph | H | H | H | H | H |
| 39 | Me | H | H | H | Me | H |
| 40 | i-Pr | H | H | H | H | (±)-Me |
| 41 | i-Pr | H | H | H | H | (S)-Me |
| 42 | i-Pr | H | H | H | H | (R)-Me |
| 43 | i-Pr | H | H | H | H | (±)-n-Bu |
| 44 | Et | Br | H | H | H | H |
| 45 | Me | CN | H | H | H | H |
| 46 | Et | CN | H | H | H | H |
| 47 | i-Pr | CN | H | H | H | H |
| 48 | Me | CN | H | H | H | (S)-Me |
| 49 | Me | H | CN | H | H | H |
| 50 | i-Pr | H | CN | H | H | H |
Table 2.
EC50 determinations for necroptosis inhibition in FADD-deficient Jurkat T cells treated with TNF-α and mouse microsome stability values.
| Compound | EC50 (µM)a | t1/2 (min) | CLint (µL/min/mg protein) |
|---|---|---|---|
| 4 | 0.28 | 32.5 | 42.6 ± 3.2 |
| 26 | 0.74 | 15.4 | 89.8 ± 3.0 |
| 30 | > 20 | --- | --- |
| 31 | > 20 | --- | --- |
| 32 | 4.9 | --- | --- |
| 33 | > 20 | --- | --- |
| 34 | 7.8 | --- | --- |
| 35 | 0.90 | 17.2 | 80.6 ± 4.4 |
| 36 | 0.44 | --- | --- |
| 37 | > 20 | --- | --- |
| 38 | > 20 | --- | --- |
| 39 | > 20 | --- | --- |
| 40 | 2.1 | --- | --- |
| 41 | 0.52 | --- | --- |
| 42 | > 20 | --- | --- |
| 43 | > 20 | --- | --- |
| 44 | 2.4 | --- | --- |
| 45 | 0.34 | 42.3 | 32.8 ± 2.2 |
| 46 | 1.9 | --- | --- |
| 47 | 1.4 | --- | --- |
| 48 | 0.092 | 236 | 5.9 ± 2.5 |
| 49 | > 20 | --- | --- |
| 50 | > 20 | --- | --- |
Standard deviation < 10%.
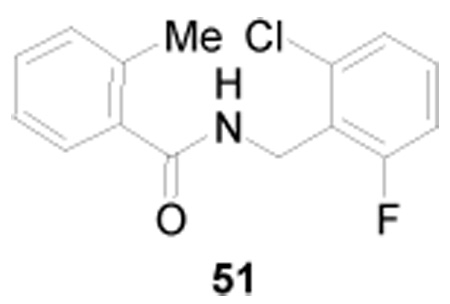
The heterocyclic ring in 4 was presumed to be most likely responsible for the compound’s lack of metabolic stability. To test this hypothesis, a related derivative that replaced the 4-alkyl-[1,2,3]thiadiazole with a 2-methylphenyl (i.e. 51) was prepared. This compound was devoid of necroptosis inhibitory activity, but demonstrated a significant improvement in mouse microsome stability with a t1/2 of 194 min and CLint of 7.13 ± 7.3 µL/min/mg protein. Based on these findings other surrogates for the [1,2,3]thiadiazole were sought that would retain necroptosis inhibitory activity.
Replacement of the 4-cyclopropyl-[1,2,3]thiadiazole with a pyrrole (31), a 3-alkylpyrrole (32) or 1,3-dialkylpyrroles (33 and 34) resulted in significant decreases in necroptosis inhibitory activity compared with 4. However, replacement with 1-methyl- or 1-isopropylpyrrole (35 and 36) resulted in a less significant decrease in activity suggesting that N-alkylpyrroles may present a possible replacement. However, 35 demonstrated poor mouse microsome stability presumably due to the electron-rich nature of the pyrrole ring. Increasing the size of the alkyl group (37 and 38) resulted in further decreases in necroptosis inhibitory activity. In addition, a tertiary amide (39) was not tolerated. This result was consistent with the SAR previously established for the [1,2,3]thiadiazole series.14 Introduction of a methyl group on the benzylic position of the amide (40) resulted in a decrease in necroptosis inhibitory activity. However, again consistent with the SAR for the [1,2,3]thiadiazole series, all of the activity resided with the (S)-enantiomer (41 vs. 42) and large substituents on the benzylic carbon (43) were not tolerated. Addition of electron-withdrawing groups to the 5-position of the pyrrole that would render it less electron rich, were tolerated (26 and 44), with nitrile (45) being best. In the case of 26 mouse microsome stability was worst compared with 4. However, 45 showed a slight improvement in stability. Interestingly, introduction of the nitrile to the 4-position of the pyrrole (49 and 50) eliminated necroptosis inhibitory activity. Increasing the size of the alkyl group on the 1-poistion of the pyrrole (46 and 47) was also detrimental to necroptosis inhibitory activity. However, introduction of a (S)-methyl substituent to the benzylic amide position of the 5-cyano-1-methylpyrrole (48)25 resulted in a significant increase in both necroptosis inhibitory activity (EC50 = 92 nM) and mouse microsome stability (t1/2 = 236 min and in CLint of 5.9 ± 2.5 µL/min/mg protein). Finally, sulfonamide 30 was found to be inactive as a necroptosis inhibitor further demonstrating the importance of the secondary amide functionality.
Although the precise mechanism of necroptosis inhibition by the [1,2,3]thiadiazole series is not currently known, it has shown an unique inhibitory profile compared to (±)-1 and 2 when evaluated in different cell types and utilizing different necroptosis-inducing stimuli.12, 14 Therefore, a similar analysis was conducted comparing 48 with (±)-1 and 4 (Figure 2). The cellular activity profiles for the [1,2,3]thiadiazole and pyrrole inhibitors were very similar suggesting that the two compound series inhibit necroptosis in the same manner and that the 5-cyano-1-methylpyrrole in 48 acts as a bioisostere for the 4-cyclopropyl-[1,2,3]thiadiazole in 4.26 Furthermore, both compounds demonstrated a very different profile compared to (±)-1 suggesting that they posses a distinct mode of necroptosis inhibition.
Figure 2.

Cell type/stimulus specific activities of necrostatins. FADD-deficient Jurkat, L929 and mouse adult lung fibroblast cells were treated for 24 hr with 10 ng/mL human TNF-α and/or 100 µM zVAD.fmk as indicated in the presence of 30 µM of necrostatin (±)-1, 4 or 48. Cell viability was determined using an ATP-based assessment method. Values were normalized to cells treated with necrostatins in the absence of necroptotic stimulus, which were set as 100% viability. Error bars reflect standard deviation values (N = 2).
In conclusion, 5-cyano-1-methylpyrrole was found to be an excellent surrogate for the 4-cyclopropyl-[1,2,3]thiadiazole in 4 resulting in 48, which demonstrated significantly increased necroptosis inhibitory activity and has improved mouse microsome stability. The SAR of the pyrrole derivatives for necroptosis inhibition paralleled the [1,2,3]thiadiazoles in many respects, including the requirement for a secondary amide and when a benzylic methyl was present all of the necroptosis activity resided with the (S)-enantiomer. Finally, cellular activity profiles of the 5-cyano-1-methylpyrrole 48 and the [1,2,3]thiadiazole 4 utilizing different cell types and necroptosis-inducing stimuli were very similar suggesting that the two compound series inhibit necroptosis in the same manner and are distinct from 1 and 2. Studies are currently underway to further evaluate 5-cyano-1-methylpyrrole necroptosis inhibitors in animal models of disease where necroptosis is likely to play a substantial role (i.e. liver injury) and to investigate the mechanism of necroptosis inhibition by these derivatives.
Acknowledgements
XT and GDC thank the Harvard NeuroDiscovery Center (HNC) for financial support. AD and JY thank the National Institute on Aging, National Institute of General Medical Sciences and American Health Assistance Foundation for financial support. XT, GDC and JY thank the National Institute of Neurological Disorders and Stroke (NINDS) for financial support. AD is a recipient of an NIH Mentored Scientist Development Award from the National Institute on Aging (NIA) and the Smith Family New Investigator Award from the Massachusetts Medical Foundation. We also thank Roz Southall (Cyprotex, Inc.) for assistance with the microsome stability experiments. The SV40-transformed adult mouse lung fibroblasts were a generous gift of Dr. Philip Tsichlis (Tufts University).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison T, Moskowitz M, Yuan J. Nat. Chem. Biol. 2005;1:112. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 2.For literature related to caspase-independent cell death see: Kitanaka C, Kuchino Y. Cell Death Differ. 1999;6:508. doi: 10.1038/sj.cdd.4400526.Fiers W, Beyaert R, Declercq W, Vandenabeele P. Oncogene. 1999;18:7719. doi: 10.1038/sj.onc.1203249.Borner C, Monney L. Cell. Death Differ. 1999;6:497. doi: 10.1038/sj.cdd.4400525.Edinger AL, Thompson CB. Curr. Opin. Cell Biol. 2004;16:663. doi: 10.1016/j.ceb.2004.09.011.Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Science. 2004;304:1500. doi: 10.1126/science.1096645.Chipuk JE, Green DR. Nat. Rev. Mol. Cell Biol. 2005;6:268. doi: 10.1038/nrm1573.Bröker LE, Kruyt FAE, Giaccone G. Clin. Cancer Res. 2005;11:3155. doi: 10.1158/1078-0432.CCR-04-2223.Fink SL, Cookson BT. Infect. Immun. 2005;73:1907. doi: 10.1128/IAI.73.4.1907-1916.2005.Kroemer G, Martin SJ. Nat. Med. 2005;11:725. doi: 10.1038/nm1263.Vandenabeele P, Vanden Berghe T, Festjens N. Sci. STKE. 2006;358:pe44. doi: 10.1126/stke.3582006pe44.Martinet W, Schrijvers DM, Herman AG, De Meyer GR. Autophagy. 2006;2:312. doi: 10.4161/auto.2966.
- 3.Lo EH, Dalkara T, Moskowitz MA. Nat. Rev. Neurosci. 2003;4:399. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- 4.McCully JD, Wakiyama H, Hsieh YJ, Jones M, Levitsky S. Am. J. Physiol. Heart Circ. Physiol. 2004;286:H1923. doi: 10.1152/ajpheart.00935.2003. [DOI] [PubMed] [Google Scholar]
- 5.Osborn NN, Casson RJ, Wood JP, Chidlow G, Graham M, Melena J. Prog. Retin. Eye Res. 2004;23:91. doi: 10.1016/j.preteyeres.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 6.Gennarelli TA, Graham DI. In: Textbook of Traumatic Brain Injury. Silver JM, McAllister TW, Yudofsky SC, editors. Washington DC: American Psychiatric Publishing Inc.; 2005. p. 37. [Google Scholar]
- 7.(a) Kaplowitz N. J. Hepatol. 2000;32(1 Suppl):39. doi: 10.1016/s0168-8278(00)80414-6. [DOI] [PubMed] [Google Scholar]; (b) Malhi H, Gores GJ, Lemasters JJ. Hepatology. 2006;43 2 Suppl. 1:S31. doi: 10.1002/hep.21062. [DOI] [PubMed] [Google Scholar]; (c) Ferrell LD, Theise ND, Scheuer PJ. In: Pathology of the Liver. 4th Edition. MacSween RNM, Burt AD, Portmann BC, Ishak KG, Scheuer PJ, Anthony PP, editors. London: Churchhill Livingstone; 2002. p. 314. [Google Scholar]
- 8.(a) Giglio P, Gilbert MR. Neurologist. 2003;9:180. doi: 10.1097/01.nrl.0000080951.78533.c4. [DOI] [PubMed] [Google Scholar]; (b) Ramesh G, Reeves WB. Am. J. Physiol Renal Physiol. 2003;285:F610. doi: 10.1152/ajprenal.00101.2003. [DOI] [PubMed] [Google Scholar]; (c) Miyaguchi M, Takashima H, Kubo T. J. Laryngol. Otol. 1997;111:763. doi: 10.1017/s0022215100138563. [DOI] [PubMed] [Google Scholar]
- 9.(a) Rosai J. Rosai and Ackerman’s Surgical Pathology. 9th Edition. Vol. 1. New York: Mosby; 2004. p. 1063. [Google Scholar]; (b) Wrobleski DM, Barth MM, Oyen LJ. AACN Clin. Issues. 1999;10:464. doi: 10.1097/00044067-199911000-00006. [DOI] [PubMed] [Google Scholar]
- 10.Martin LJ, Al-Abdulla NA, Brambrink AM, Kirsch JR, Sieber FE, Portera-Cailliau C. Brain Res. Bull. 1998;46:281. doi: 10.1016/s0361-9230(98)00024-0. [DOI] [PubMed] [Google Scholar]
- 11.Teng X, Degterev A, Jagtap P, Xing X, Choi S, Denu R, Yuan J, Cuny GD. Bioorg. Med. Chem. Lett. 2005;15:5039. doi: 10.1016/j.bmcl.2005.07.077. [DOI] [PubMed] [Google Scholar]
- 12.Jagtap PG, Degterev A, Choi S, Keys H, Yuan J, Cuny GD. J. Med. Chem. 2007;50:1886. doi: 10.1021/jm061016o. [DOI] [PubMed] [Google Scholar]
- 13.Wang K, Li J, Degterev A, Hsu E, Yuan J, Yuan C. Bioorg. Med. Chem. Lett. 2007;17:1455. doi: 10.1016/j.bmcl.2006.11.056. [DOI] [PubMed] [Google Scholar]
- 14.Teng X, Keys H, Jeevanandam A, Porco JA, Jr, Degterev A, Yuan J, Cuny GD. Bioorg. Med. Chem. Lett. 2007;17:6836. doi: 10.1016/j.bmcl.2007.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith CCT, Davidson SM, Lim SY, Simpkin JC, Hothersall JS, Yellon DM. Cardiovasc. Drugs Ther. 2007;21:227. doi: 10.1007/s10557-007-6035-1. [DOI] [PubMed] [Google Scholar]
- 16.You Z, Savitz SI, Yang J, Degterev A, Yuan J, Cuny GD, Moskowitz MA, Whalen MJ. J. Cereb. Blood Flow Metab. doi: 10.1038/jcbfm.2008.44. submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baranczewski P, Stańczak A, Sundberg K, Svensson R, Wallin A, Jannson J, Garberg P, Postlind H. Pharmacol. Rep. 2006;58:453. [PubMed] [Google Scholar]
- 18.Terry WG, Jackson AH, Kenner GW, Kornis G. J. Chem. Soc. 1965:4389. [Google Scholar]
- 19.Burgey CS, Robinson KA, Lyle TA, Sanderson PEJ, Lewis SD, Lucas BJ, Krueger JA, Singh R, Miller-Stein C, White RB, Wong B, Lyle EA, Williams PD, Coburn CA, Dorsey BD, Barrow JC, Stranieri MT, Holahan MA, Sitko GR, Cook JJ, McMasters DR, McDonough CM, Sanders WM, Wallace AA, Clayton FC, Bohn D, Leonard YM, Detwiler TJ, Jr, Lynch JJ, Jr, Yan Y, Chen Z, Kuo L, Gardell SJ, Shafer JA, Vacca JP. J Med. Chem. 2003;46:461. doi: 10.1021/jm020311f. [DOI] [PubMed] [Google Scholar]
- 20.Loader CE, Anderson HJ. Can. J. Chem. 1981;59:2673. [Google Scholar]
- 21.Kim S, Lu S, Cornelius LA, Lombardo LJ, Borzilleri RM, Schroeder GM, Sheng C, Rovnyak G, Crews D, Schmidt RJ, Williams DK, Bhide RS, Traeger SC, McDonnell PA, Mueller L, Sheriff S, Newitt JA, Pudzianowski AT, Yang Z, Wild R, Lee FY, Batorsky R, Ryder JS, Ortega-Nanos M, Shen H, Gottardis M, Roussell DL. Bioorg. Med. Chem. Lett. 2006;16:3937. doi: 10.1016/j.bmcl.2006.05.037. [DOI] [PubMed] [Google Scholar]
- 22.Li X, Reuman M, Russell RK. Heterocycles. 2006;70:201. [Google Scholar]
- 23.Kini GD, Robins RK, Avery TL. J. Med. Chem. 1989;32:1447. doi: 10.1021/jm00127a008. [DOI] [PubMed] [Google Scholar]
-
24.Microsome stability was determined in pooled mouse liver microsomes. Test compound (3 µM final concentration) along with 0.5 mg/mL microsome protein and 1 mM NADPH was incubated for 0, 5, 15, 30 and 60 min. Incubation of test compound and microsomes in the absence of NADPH served as a negative control. The samples were quenched with methanol and centrifuged for 20 min at 2500 rpm to precipitate proteins. Sample supernatants were analyzed (N=3) by LC/MS. The ln peak area ratio (compound peak area/internal standard peak area) was plotted against time and the slope of the line determined to give the elimination rate constant [k = (−1)(slope)]. The half life (t1/2 in minutes), and the in vitro intrinsic clearance (CLint in µL/min/mg protein) were calculated according to the following equations, where V = incubation volume in µL/mg protein:
- 25.48: 1H NMR (400 MHz, CDCl3):δ. 7.23-7.15 (m, 2H), 7.05-6.97 (m, 1H), 6.79 (d, 1H, J = 8.8 Hz), 6.73 (d, 1H, J = 4.4 Hz), 6.54 (d, 1H, J = 4.4 Hz), 5.95 – 5.84 (m, 1H), 4.03 (s, 3H), 1.59 (d, 3H, J = 7.2 Hz); 13H NMR (100 MHz, CDCl3):δ 163.0, 160.5, 159.4, 133.8, 133.7, 130.5, 129.3, 129.2, 128.5, 128.4, 126.3, 118.2, 115.2, 115.0, 113.0, 111.4, 109.8, 43.9, 35.3, 20.4; HRMS Calculated for C15H14ClFN3O [M + H]+ 306.0809; Found 306.0804.
- 26.(a) Chen X, Wang W. Ann. Reports Med. Chem. 2003;38:333. [Google Scholar]; (b) Olesen PH. Curr. Opin. Drug. Discov. Devel. 2001;4:471. [PubMed] [Google Scholar]