

Abstract
LFA-1/ICAM-1 interaction plays an important role in the formation of the immunological synapse between T cells and antigen-presenting cells (APC). Blocking of LFA-1/ICAM-1 interactions has been shown to suppress the progression of autoimmune diseases. cIBR peptide (cyclo(1,12)PenPRGGSVLVTGC) inhibits ICAM-1/LFA-1 interaction by binding to the I-domain of LFA-1. To increase the bioactivity of cIBR peptide, we systemically modified the structure of the peptide by (a) replacing the Pen residue at the N-terminus with Cys, (b) cyclization using amide bond formation between Lys-Glu side chains, and (c) reducing the peptide size by eliminating the C-terminal residue. We found that the activity of cIBR peptide was not affected by replacing Phe with Cys. Peptide cyclization by forming the Lys-Glu amide bond also increased the activity of cIBR peptide, presumably due to the resistance of the amide bond to the reducing nature of glutathione in plasma. We also found that a reduced derivative of cIBR with eight residues (cyclo(1,8)CPRGGSVC) has a bioactivity similar to that of the larger cIBR peptides. Our findings suggest that, by systemically modifying the structure of cIBR peptide, the biological activity of these derivatives can be optimized for future use to inhibit T-cell adhesion in in vivo models of autoimmune diseases.
Introduction
T-cell activation transpires when resting T cells interact with antigen-presenting cells (APC) accompanied by the formation of the “immunological synapse” with a “bull’s eye”-like appearance at the T cell-APC interface. The immunological synapse is generated by Signal-1 and Signal-2 (1). Signal-1 is derived from the interaction between T-cell receptor (TCR) and antigen-major histocompatibility complex (Ag-MHC), which produces the central zone supramolecular activation complex (cSMAC) at the center of the bull’s eye. Signal-2 derives from a costimulatory signal that is derived from a pair of molecules, i.e., the interactions between lymphocyte function-associated antigen-1 (LFA-1) and intercellular adhesion molecule-1 (ICAM-1) that generates the peripheral zone supramolecular activation complex (pSMAC) at the outer ring of the bull’s-eye. Prior to establishing the immunological synapse, Signal-1 is found at the outer ring and Signal-2 is at the inner ring. During early events of the immune synapse formation, LFA-1/ICAM-1 (Signal-2) provides a support for protrusive cytoskeletal networks that force the outermost ring of the T-cell membranes to move closer to APC, which enables TCR to bind to Ag-MHC (Signal-1). Then, these two signals translocate to switch position to form the immunological synapse (1). Therefore, the disruption of Signal-2 formation (i.e., LFA-1/ICAM-1 interaction) could disrupt the immunological formation to suppress T-cell activation and proliferation. The LFA-1-deficient transgenic animals show that the LFA-1/ICAM-1 interaction significantly contributes to T-cell activation, and this role of LFA-1/ICAM-1 interaction cannot be overcome entirely by increasing Ag-MHC density or adding exogenous IL-2 (2).
LFA-1 is a member of the integrin family that binds to several different ligands, including intercellular adhesion molecule-1 (ICAM-1) (3,4), ICAM-2 (5), ICAM-3 (6), ICAM-4 (7), ICAM-5 (8), and junctional adhesion molecule-A (JAM-A) (9). LFA-1 consists of an α-subunit (CD11a) and β-subunit (CD18) connected by physical interactions at the insert (I) domain on the “top” of CD11a (10). The ICAM-1 binds LFA-1 via the I-domain of CD11a, and this binding is primed by CD18. In leukocyte adhesion deficiency disease (LAD), a mutation in the sequence of CD18 makes LFA-1 nonfunctional and prevents adhesion of leukocytes (11,12).
LFA-1 is an attractive therapeutic target for developing large and small molecules to treat leukocyte-related diseases. Inhibitors of ICAM-1/LFA-1 interactions have been developed to prevent allograft rejection (13,14) and pregnancy rejection under stress (15) as well as to treat psoriasis (16,17), autoimmune uveitis (18), multiple sclerosis (19–22), lupus (23), and inflammatory arthritis (24). For example, Efalizumab (Raptiva™) is an anti-LFA-1 drug approved by the FDA for the treatment of psoriasis (25). The small molecules (i.e., XVA143 (26), LFA703 (27), and BIRT-377 (28)), peptides (29,30), and peptidomimetics (31) have been successfully designed. However, many of these small molecules have not yet successfully reached the clinical application. This may be due to a lack of understanding of mechanism of actions of these molecules in vivo. Our group found that a cyclic peptide (cIBR, cyclo-1,12-PenPRGGSVLVTGC) derived from domain-1 of ICAM-1 binds to the I-domain of LFA-1 and inhibits LFA-1/ICAM-1-mediated T-cell homotypic and heterotypic adhesion (29,30). Besides binding to the surface of LFA-1 on T cells, cIBR peptide also undergoes receptor-mediated endocytosis, and has been utilized to target drugs to LFA-1-expressing cells (i.e., leukocytes) (29). cIBR peptide binds to the metal ion-dependent adhesion site (MIDAS) and I-domain allosteric site (IDAS) on the surface of MOLT-3 cells as well as the isolated LFA-1 receptor (30). However, NMR spectroscopy studies suggest that cIBR binds only to the IDAS site in the I-domain (32); thus, it is possible that the cIBR binding to the MIDAS can only occur in the intact LFA-1 receptor.
In this work, we focused on improving the biological activity and physicochemical properties of cIBR peptide by altering the cyclization method and reducing the size of the peptide (Table 1). The hope is to find a small cyclic peptide (hexa- or octapeptide) that has similar or better biological activity than cIBR peptide. This smaller peptide will be a more manageable molecule for optimization than the larger cIBR peptide. Thus, cIBR peptide was systematically modified by (a) replacing the Pen residue at the N-terminus with Cys, (b) using an alternative cyclization method such as side chain-to-side chain amide cyclization at Lys-Glu to replace the Pen-Cys disulfide bond, and (c) reducing the peptide size by eliminating the C-terminal residues. It is interesting to find that some of these modifications improve the bioactivity of the peptides compared to the parent cIBR peptide.
Table 1.
The sequences of ICAM-1-derived peptides that are used in this study
| Name | Amino-Acid Sequence | MW |
|---|---|---|
| cIBR | Cyclo(1,12)Pen-PRGGSVLVTGC-OH | 1174.6 |
| cIBR-1 | Cyclo(1,12)CPRGGSVLVTGC-OH | 1146.7 |
| cIBR-2 | Cyclo(1,12)CPrGGSVLVTGC-OH | 1146.7 |
| cIBR-3 | Cyclo(1,12)KPRGGSVLVTGE-OH | 1181.8 |
| cIBR-4 | Cyclo(1,13)CLPRGGSVLVTGC-OH | 1259.7 |
| cIBR-5 | Cyclo(1,11)CPRGGSVLVTC-OH | 1089.7 |
| cIBR-6 | Cyclo(1,10)CPRGGSVLVC-OH | 988.7 |
| cIBR-7 | Cyclo(1,8)CPRGGSVC-OH | 776.5 |
Experimental Section
Cell cultures
Molt-3 and Caco-2 cell lines were obtained from the American Type Culture Collection (Rockville, MD, USA) and maintained as previously described (33). The Molt-3 cell line was grown in suspension in RPMI-1640 medium (Sigma, St. Louis, MO, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 100 μg/L of penicillin/streptomycin. Molt-3 cells were activated for 12–18 h with 0.2 μM phorbol 12-myristate-13-acetate (PMA) (Sigma) in 75-cm2 tissue culture flasks (Falcon, Fisher, Pittsburgh, PA, USA). The Caco-2 cell-line was grown as monolayers in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma) with 25 mM glucose containing 10% FBS, 1% nonessential amino acids, 1 mM Na-pyruvate, 1% L-glutamine, and 100 μg/L of penicillin/streptomycin. Caco-2 cells were grown in 75-cm2 tissue culture flasks (Falcon) for maintenance purposes and in a 48-well cell culture cluster (Costar) for heterotypic-adhesion experiments. Before confluency was reached, Caco-2 cells were induced with 100 U/mL IFN-γ for 12–18 h to upregulate the ICAM-1 expression. Upon reaching 90% confluency (approximately 4–5 days), cells were subcultured at a 1:2 split ratio using 0.25% trypsin/0.1% EDTA. All cell lines were grown in a 95% humidified/5% CO2 atmosphere at 37 °C.
Peptides
All peptides (Table 1) were purchased from Multiple Peptide Systems (San Diego, CA). The purity of all peptides was determined by analytical HPLC (purity greater than 95%) and electrospray ionization (ESI) mass spectroscopy. The mass spectra of all peptides used in this study are available as Supplementary Material.
Heterotypic T-cell adhesion experiments
The heterotypic adhesion assay is a modification of a previously published method (33). Caco-2 cells in 48-well culture dishes were activated at confluency with 100 units/mL of IFN-γ for 16–18 h and then washed with serum-free RPMI 1640 prior to the addition of T cells. Activated Molt-3 cells were labeled with the fluorescent dye BCECF (Molecular Probes, Eugene, OR, USA) at a concentration of 50 μg of BCECF in 50 μL of dimethyl sulfoxide/30 million cells for 1 h at 37 °C. Cells were washed extensively with PBS to remove free BCECF label and were resuspended in RPMI at 1 × 106 cells/mL. Cells were then incubated with peptide at final concentrations of 1, 10, 50, 100, or 200 μM of peptide or monoclonal antibody (mAb) for 45 min at 37 °C. After incubation, 200,000 T cells were added to the Caco-2 monolayers and allowed to adhere for 1 h at 37 °C. After three washes with HEPES/PBS to remove unbound T cells, cells remaining in each well were lysed with 0.5 mL of 2% Triton X-100 in 0.2 N NaOH for 60 min at 37 °C with agitation. Aliquots of 100 μL of the lysates were transferred to black 96-well plates for reading in a micro plate fluorescence analyzer (Bio-Tek FL600). All doses were tested in triplicate or quadruplicate. Data are presented as percent inhibition of T-cell adhesion. Relative fluorescence intensity (FL) was corrected for background FL of cell monolayer only. Percent inhibition (%) by peptide or mAb was calculated from the relative fluorescence as follows:
Molecular docking of peptides to the I-domain of LFA-1 receptor
To correlate the effect of structural modification on the biological activity of the cyclic peptides, computer simulations on binding properties of cIBR, cIBR-1, cIBR-3, and cIBR-7 peptides to the I-domain of LFA-1 receptor were carried out. In the simulation experiments, we used all available X-ray crystal structures of the I-domain of LFA-21 receptor from the Protein Data Bank (i.e., 1ZOP, 1CQP, 1XDD, 1XDG, 1XUO, 2O7N, 2ICA). These structures were minimized and superimposed based on similarity of the Cα atoms. Subsequently, the peptides were docked to every crystal structure of the I-domain of LFA-1 receptor. The initial three-dimensional structures of cIBR analogs were generated using the results of our previous study (32), where both NMR and molecular modeling approaches were used to characterize the binding site and structures of the cIBR and cIBC peptide complexes with the I-domain. We have first performed an exhaustive conformational analysis of the peptide analogs in solution and generated low-energy solution conformations of these peptides using Monte Carlo conformational search with an energy window of 6.0 kcal/mol and up to 1000 Monte Carlo steps. Energy minimization of the structure was performed using AMBER force field with the Generalized Born/Solvent Accessible Surface Area (GB/SA) solvation term. These conformations have been then utilized in docking simulations with LFA-1 receptor, where flexible peptide docking was conducted with the receptor structure to produce the energetically favorable bound peptide conformations. During peptide docking simulations with the I-domain, the protein structure was held fixed in its minimized conformation, while both rigid body degrees of freedom and the peptide rotatable angles were treated as independent variables. Docking simulations were performed using an evolutionary search algorithm and the all-atom knowledge-based energy function, which includes (a) the intramolecular energy terms for the peptide given by torsional and non-bonded functions and (b) the intermolecular ligand-protein steric and hydrogen bond interaction terms calculated from a piecewise linear potential (PLP) summed over all protein and ligand heavy atoms (34). The determined low-energy peptide bound conformations were refined using molecular dynamics simulations of the peptide complexes with the receptor and used for in the binding free energy computations with the molecular mechanics AMBER force field and the GB/SA solvation model. The details and applications of the MM/GBSA model have been extensively documented in our earlier studies (35–37).
Molecular dynamics of peptides to the I-domain of LFA-1 receptor
The low-energy structures of the complexes between the peptide and the I-domain were refined in molecular dynamics simulations for 5 ns using a parallel version of the NAMD 2.6 program (38). An NPT ensemble and periodic boundary conditions were used on the systems. The electrostatic term was described by using the particle mesh Ewald algorithm. The non-bonded cutoff, switching distance, and non-bonded pair-list distance were set to 9, 8, and 10.5 Angstroms, respectively. The system was maintained at constant pressure and temperature using an isotropic Langevin barostat and a Langevin thermostat. One thousand steps of conjugate gradient algorithm were used to minimize each system with the default NAMD restraints to the protein backbone atoms, followed by 1000 steps without restraints. The harmonic restraints allow a harmonic-restraining force to be applied to any set of atoms in the simulation. The harmonic restraint energy function is multiplied by this parameter, making it possible to gradually turn off constraints during refinement. For studied systems, each system was warmed up to 340 K for 40 ps with the restraints and equilibrated for 100 ps, followed by the gradual release of restraints, and finishing with no restraints at 300 K.
Results and Discussion
The effect of cyclization methods on the biological activity
The original idea of using Pen amino acid at the N-terminus of cIBR (Table 1) was to induce conformational restriction due to the presence of a gem-dimethyl group at the beta-carbon as in receptor-selective cyclic opioid peptides (39). To test whether the Pen residue is necessary for the activity of cIBR peptide, cIBR-1 peptide was synthesized by replacing the Pen1 residue with the Cys1 residue. It is interesting to find that the activity of cIBR-1 peptide in inhibiting T-cell adhesion to Caco-2 cell monolayers is similar to that of cIBR peptide (Figure 1). Because the PRGG sequence in cIBR peptide is one of the important sequences (40), the biological activity results suggest that replacing Pen1 with Cys1 does not influence the conformational recognition of the PRGG sequence in cIBR and cIBR-1 peptides. The low influence of Pen on the activity may be due to the high propensity of PRGG sequence to form a stable β-turn structure as shown in the X-ray structure of ICAM-1 (41). The binding studies of cIBR to the I-domain using NMR indicate that Glu301 cross-peak is affected by the peptide; the molecular dockig experiments suggest that the Arg3 residue is involved in salt bridge formation with the side chain of Glu301 of the I-domain. Thus, the role of the Arg3 was evaluated by replacing this residue in cIBR-1 with D-Arg to give cIBR-2 peptide. This replacement improves the activity of cIBR-2 compared to cIBR-1, suggesting that changing the configuration of the Arg3 may optimize the salt-bridge formation.
Figure 1.

The effect of mutation and cyclization method on the activity of cIBR derivatives in inhibiting ICAM-1/LFA-1-mediated T-cell adhesion to Caco-2 cell monolayers in concentration-dependent manner. Mutation of Pen1 to Cys1 (cIBR-1) does not affect the activity and replacing L-Arg-3 with D-Arg-3 improves the activity of the peptide (cIBR-2). Amide bond cyclization via the side chains in cIBR-3 enhances the activity of the peptide.
Next, an alternative method to form cyclic peptides was explored using side chain-to-side chain cyclization via a peptide bond. The Pen1 and Cys12 residues in cIBR peptide were replaced with Lys1 and Glu12 in cIBR-3 peptide, respectively, and a peptide bond was formed from the amino group of Lys1 to the carboxyl group of Glu12. The advantage of this amide bond cyclization is that it is stable to the reducing nature of glutathione in systemic circulation as in plasma, which is one of the potential problems for a disulfide bond-containing cyclic peptide (i.e., cIBR). It is interesting to discover that cIBR-3 has higher biological activity than that of the parent cIBR peptide at various concentrations (Figure 2). These results suggest that a combination of peptide bond cyclization and D-Arg3 replacement may be one way to enhance the activity of the peptide. Currently, we are investigating the effect of different cyclization methods on the conformation of PRGG sequence by NMR and molecular dynamics simulations; the biological activities of the peptides are being evaluated in protein-binding and T-cell adhesion assays.
Figure 2.

The effect of adding Leu to the N-terminus of PRG sequence and size reduction of the cyclic peptide on the biological activity of cIBR derivatives. Adding Leu residue to the N-terminus of PRG sequence in cIBR-4 enhances the activity of the peptide compared to cIBR-1. Size reduction of the cIBR-1 peptide to cIBR-7 maintains the activity of the peptide.
The effect of cyclic peptide size on biological activity
The flanking residues to the PRGG sequence and the size of the cyclic peptide may influence the activity of the peptide. To test whether adding a residue to the N-terminal region of cIBR-1 peptide influences the activity, cIBR-4 peptide was synthesized with addition of a Leu residue to the N-terminus of PRGG sequence. It is interesting to find that cIBR-4 has better activity than cIBR-1 (Figure 2), suggesting that the hydrophobic side chain of the Leu residue may be involved in binding to a hydrophobic pocket of the I-domain.
The effect of reducing the size of cyclic peptide on biological activity was investigated by eliminating the amino acid from the C-terminus of cIBR-1 but maintaining the PRGG sequence (40). Cyclic peptides (cIBR-5, cIBR-6, and cIBR-7, Table 1) were synthesized and their inhibitory activities were evaluated in T-cell adhesion assay. Eliminating Gly11 from cIBR-1 to give cIBR-5 dramatically reduces the activity of the peptide (Figure 2). Next, the Thr10 in cIBR-5 was removed to give cIBR-6 with activity similar to that of the parent cIBR-1. The results suggest that the backbone ring size has an effect on the conformation of the peptide and peptide activity. Finally, two C-terminal residues were removed from cIBR-6 to give cIBR-7, which has better activity than cIBR-1 and cIBR-6. At high concentration (100–200 μM), cIBR-7 has activity comparable to that of cIBR-3 and cIBR-4. These results suggest that the smaller backbone ring size of cIBR-7 induces conformational restriction in the PRGG sequence and improves the selectivity of this peptide.
Molecular modeling of binding between peptide and I-domain
To explore the relationship between peptide structure and peptide-binding properties to the I-domain, computational docking and molecular dynamics simulations were first carried out by comparing the binding properties of cIBR and cIBR-1. Our previous NMR and docking experiments indicated that cIBR peptides binds to the L-site of the I-domain (32). However, cIBR peptide binding studies using an anti-I-domain antibodies indicated that cIBR could also bind to the MIDAS of I-domain (29). In this study, we compared the docking properties of cIBR and cIBR-1 at the L-site hydrophobic pocket of I-domain protein and both molecules have similar binding energies, i.e., −7.4 kcal/mol for cIBR and −7.87 kcal/mol cIBR-1. The binding orientations of cIBR and cIBR-1 on the L-site are also similar, which explains the similarity in their biological activities (Figure 3A). Both peptides bind to the L-site of the I-domain by protruding their Gly5 to Gly11 residues into the hydrophobic pocket, forming a number of favorable hydrophobic contacts. A hydrogen-bonding network was observed between the peptide and the I-domain, including a hydrogen bond (1) between the hydroxyl of Thr10 of the peptide and the side chain of Tyr166 of the I-domain, (2) between the carbonyl oxygen of Thr10 of the peptide and Tyr307 of the protein, and (3) between the NH of Gly5 and carbonyl of the Glu301 of the I-domain. Finally, the side chain of Arg3 in the cIBR peptide forms a stable salt bridge to the side chain of Glu301 of the I-domain. The molecular modeling suggests that the salt bridge hinders the movement of α-helix-7 of the I-domain to open the L-site that is necessary for binding with ICAM-1 on the cell surface (Figure 3B).
Figure 3.
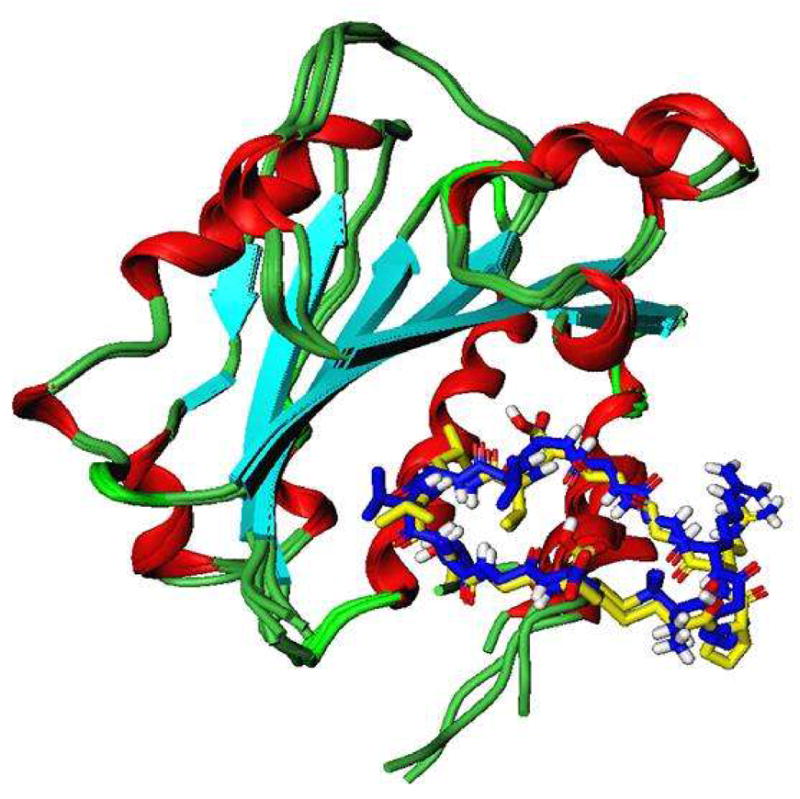
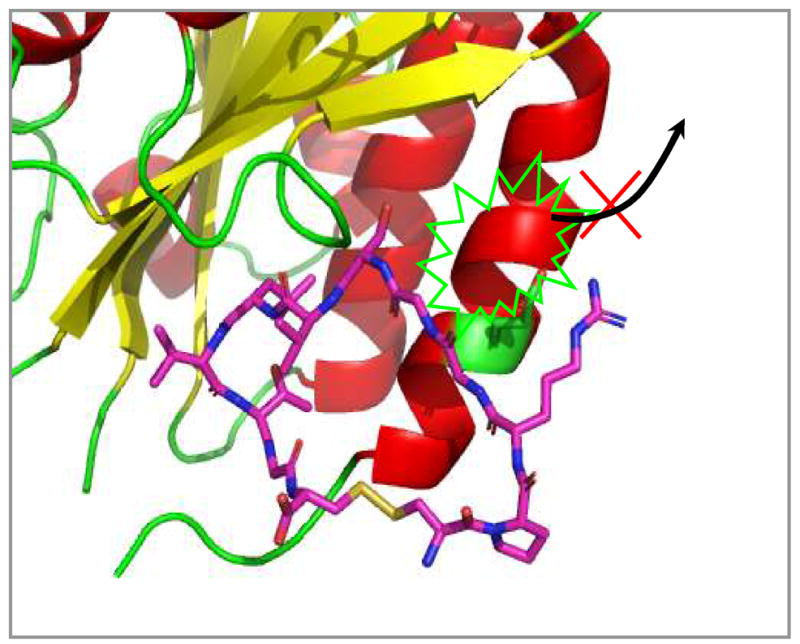
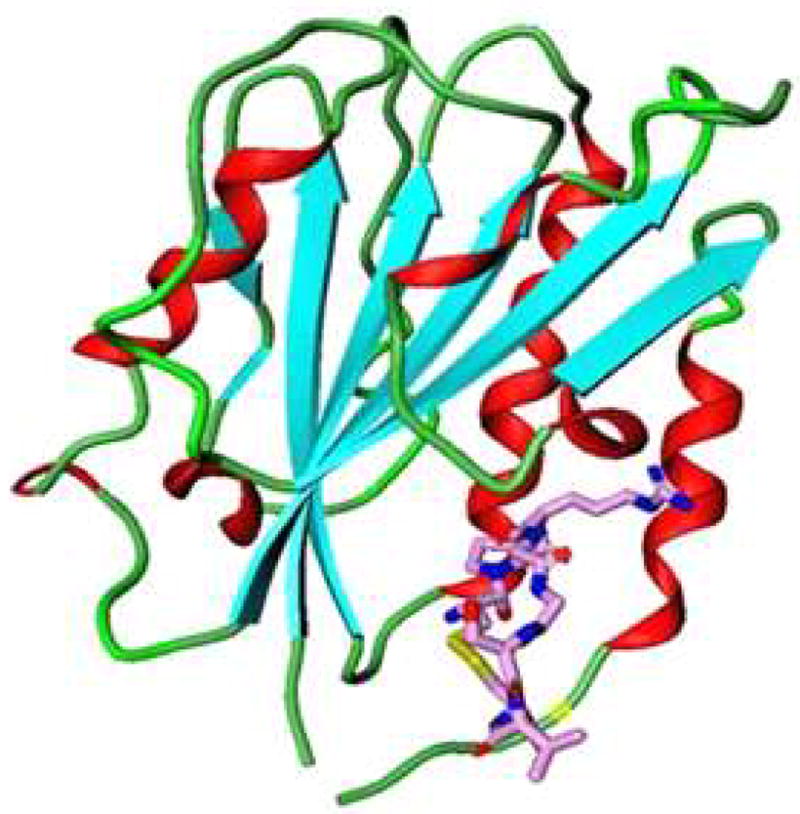
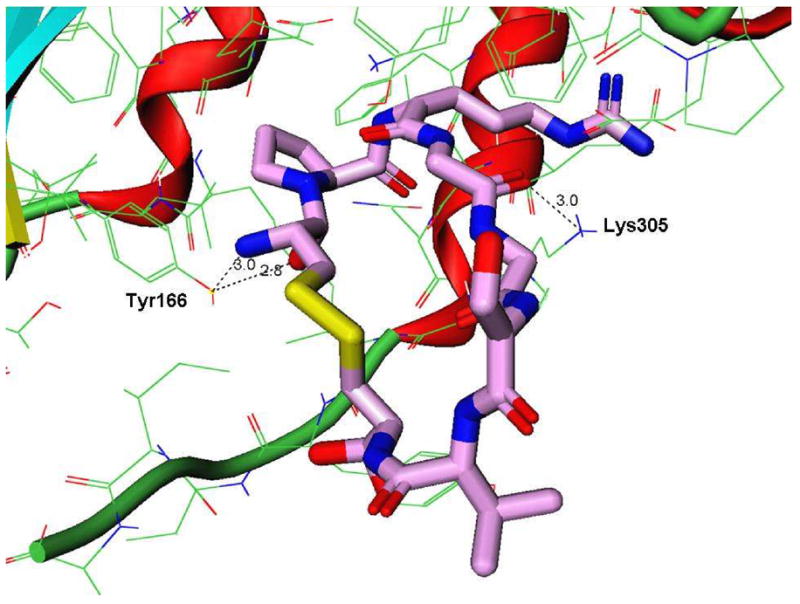
The docking structures of cIBR and its derivatives to the I-domain. (A) Comparison the predictive binding mode between cIBR (blue) and cIBR-1 (yellow) upon docking to the I-domain. Both peptides bind to the L-site of the I-domain in a similar fashion. (B) The proposed interaction of Arg3 of cIBR peptide and Glu301 on the α-helix-7 on the I-domain that prevent the movement of this helical segment to open the L-site for I-domain binding to ICAM-1. (C) Binding property of cIBR-7 to the L-site of the I-domain with the Arg-3 directly located in the L-site pocket. (D) The hydrogen bonding network between Arg3 on cIBR-7 and the L-site of the I-domain.
Reducing the size of cyclic peptides and changing the method of cyclization have been shown to improve the biological activity of cIBR peptide derivatives. To describe the structure-activity relationships of cIBR derivatives, docking properties and the energetics of docking of cIBR, cIBR-1, cIBR-3, and cIBR-7 to the I-domain were compared. Unlike cIBR and cIBR-1, the Arg3 residue in cIBR-7 sits in the binding pocket of the L-site with a network of hydrogen to the side chains of the Tyr166 and Lys305 residues (Figure 3D). As suggested previously, the Arg residue may prevent the movement of the a7-helix for activating the I-domain to bind LFA-1. The binding energies of these four peptides were compared, and the rank energy is in the following order cIBR > cIBR-1 > cIBR-7 > cIBR-3 (Figure 4). This rank is congruent with the biological activities of these peptides. The high activities of cIBR-3 and cIBR-7 could be due to the conformation of the peptide imposed by the cyclization method. To evaluate the effect of cyclization method and conformation, we are currently studying the solution structures of all of these peptides in the absence and presence of the I-domain using NMR.
Figure 4.

The correlation between computed free energy of binding and % inhibition of T-cell adhesion at 200 μM of peptide of cIBR, cIBR-1, cIBR-3, and cIBR-7. The results show a good correlation between the activity of the peptide and the binding energy.
Conclusions
This study indicates that there are several factors that influence the biological activity of cIBR peptide and its derivatives. First, the Pen1 residue can be replaced with a Cys1 residue as in cIBR-1, and the cyclization using the amide bond via side chains of Lys1 and Glu12 enhances the activity of cIBR-3 peptide. Addition of Leu residue and replacement of L-Arg-3 with D-Arg-3 also improves the activity of cIBR derivatives. Finally, a smaller cIBR-7 peptide has better activity than the parent cIBR peptide. In the future, we will modify cIBR-7 peptide by adding a Leu residue at the N-terminus and replacing the L-Arg residue with the D-Arg residue. Finally, we will investigate several different types of the amide bond cyclization (i.e., backbone-to-backbone, backbone-to-side chain, and side chain-to-side chain) to make cIBR-7 derivatives for further improvement of the biological activity.
Supplementary Material
The ESI mass spectra of eight peptides used in this study.
Acknowledgments
This work is supported in part by the National Institutes of Health (R01-AI-063002). We gratefully acknowledge Nancy Harmony for her excellent assistance in proofreading the manuscript.
References
- 1.Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, et al. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- 2.Shier P, Ngo K, Fung-Leung WP. Defective CD8+ T cell activation and cytolytic function in the absence of LFA-1 cannot be restored by increased TCR signaling. J Immunol. 1999;163:4826–4832. [PubMed] [Google Scholar]
- 3.Rothlein R, Dustin ML, Marlin SD, Springer TA. A human intercellular adhesion molecule (ICAM-1) distinct from LFA-1. J Immunol. 1986;137:1270–1274. [PubMed] [Google Scholar]
- 4.Marlin SD, Springer TA. Purified intercellular adhesion molecule-1 (ICAM-1) is a ligand for lymphocyte function-associated antigen 1 (LFA-1) Cell. 1987;51:813–819. doi: 10.1016/0092-8674(87)90104-8. [DOI] [PubMed] [Google Scholar]
- 5.Edwards CP, Fisher KL, Presta LG, Bodary SC. Mapping the intercellular adhesion molecule-1 and -2 binding site on the inserted domain of leukocyte function-associated antigen-1. J Biol Chem. 1998;273:28937–28944. doi: 10.1074/jbc.273.44.28937. [DOI] [PubMed] [Google Scholar]
- 6.Song G, Yang Y, Liu JH, Casasnovas JM, Shimaoka M, Springer TA, et al. An atomic resolution view of ICAM recognition in a complex between the binding domains of ICAM-3 and integrin aLβ2. Proc Natl Acad Sci U S A. 2005;102:3366–3371. doi: 10.1073/pnas.0500200102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hermand P, Huet M, Callebaut I, Gane P, Ihanus E, Gahmberg CG, et al. Binding sites of leukocyte β 2 integrins (LFA-1, Mac-1) on the human ICAM-4/LW blood group protein. J Biol Chem. 2000;275:26002–26010. doi: 10.1074/jbc.M002823200. [DOI] [PubMed] [Google Scholar]
- 8.Tian L, Kilgannon P, Yoshihara Y, Mori K, Gallatin WM, Carpen O, et al. Binding of T lymphocytes to hippocampal neurons through ICAM-5 (telencephalin) and characterization of its interaction with the leukocyte integrin CD11a/CD18. Eur J Immunol. 2000;30:810–818. doi: 10.1002/1521-4141(200003)30:3<810::AID-IMMU810>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 9.Ostermann G, Weber KS, Zernecke A, Schroder A, Weber C. JAM-1 is a ligand of the β(2) integrin LFA-1 involved in transendothelial migration of leukocytes. Nat Immunol. 2002;3:151–158. doi: 10.1038/ni755. [DOI] [PubMed] [Google Scholar]
- 10.Qu A, Leahy DJ. Crystal structure of the I-domain from the CD11a/CD18 (LFA-1, αLβ2) integrin. Proc Natl Acad Sci U S A. 1995;92:10277–10281. doi: 10.1073/pnas.92.22.10277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hogg N, Stewart MP, Scarth SL, Newton R, Shaw JM, Law SK, et al. A novel leukocyte adhesion deficiency caused by expressed but nonfunctional beta2 integrins Mac-1 and LFA-1. J Clin Invest. 1999;103:97–106. doi: 10.1172/JCI3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hogg N, Henderson R, Leitinger B, McDowall A, Porter J, Stanley P. Mechanisms contributing to the activity of integrins on leukocytes. Immunol Rev. 2002;186:164–171. doi: 10.1034/j.1600-065x.2002.18614.x. [DOI] [PubMed] [Google Scholar]
- 13.Soleimani B, Wieczorek G, Katopodis A, Zenke G, George AJ, Hornick PI, et al. Anti-LFA-1 monotherapy prevents neointimal formation in a murine model of transplant intimal hyperplasia. J Heart Lung Transplant. 2007;26:724–731. doi: 10.1016/j.healun.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 14.Go S, Fleischmann A, Lantz O, Cretolle C, Brousse N, Cerf-Bensussan N, et al. Anti-LFA-1 antibody postpones T-cell receptor triggering while preserving generation of regulatory T cells in T-cell receptor anti-HY transgenic mice. Transplantation. 2006;82:119–126. doi: 10.1097/01.tp.0000225804.85830.de. [DOI] [PubMed] [Google Scholar]
- 15.Blois S, Tometten M, Kandil J, Hagen E, Klapp BF, Margni RA, et al. Intercellular adhesion molecule-1/LFA-1 cross talk is a proximate mediator capable of disrupting immune integration and tolerance mechanism at the feto-maternal interface in murine pregnancies. J Immunol. 2005;174:1820–1829. doi: 10.4049/jimmunol.174.4.1820. [DOI] [PubMed] [Google Scholar]
- 16.Gottlieb AB, Krueger JG, Wittkowski K, Dedrick R, Walicke PA, Garovoy M. Psoriasis as a model for T-cell-mediated disease: immunobiologic and clinical effects of treatment with multiple doses of efalizumab, an anti-CD11a antibody. Arch Dermatol. 2002;138:591–600. doi: 10.1001/archderm.138.5.591. [DOI] [PubMed] [Google Scholar]
- 17.Papp K, Bissonnette R, Krueger JG, Carey W, Gratton D, Gulliver WP, et al. The treatment of moderate to severe psoriasis with a new anti-CD11a monoclonal antibody. J Am Acad Dermatol. 2001;45:665–674. doi: 10.1067/mjd.2001.117850. [DOI] [PubMed] [Google Scholar]
- 18.Ke Y, Sun D, Zhang P, Jiang G, Kaplan HJ, Shao H. Suppression of established experimental autoimmune uveitis by anti-LFA-1alpha Ab. Invest Ophthalmol Vis Sci. 2007;48:2667–2675. doi: 10.1167/iovs.06-1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Willenborg DO, Staykova MA, Miyasaka M. Short term treatment with soluble neuroantigen and anti-CD11a (LFA-1) protects rats against autoimmune encephalomyelitis: treatment abrogates autoimmune disease but not autoimmunity. J Immunol. 1996;157:1973–1980. [PubMed] [Google Scholar]
- 20.Kobayashi Y, Kawai K, Honda H, Tomida S, Niimi N, Tamatani T, et al. Antibodies against leukocyte function-associated antigen-1 and against intercellular adhesion molecule-1 together suppress the progression of experimental allergic encephalomyelitis. Cell Immunol. 1995;164:295–305. doi: 10.1006/cimm.1995.1173. [DOI] [PubMed] [Google Scholar]
- 21.Gordon EJ, Myers KJ, Dougherty JP, Rosen H, Ron Y. Both anti-CD11a (LFA-1) and anti-CD11b (MAC-1) therapy delay the onset and diminish the severity of experimental autoimmune encephalomyelitis. J Neuroimmunol. 1995;62:153–160. doi: 10.1016/0165-5728(95)00120-2. [DOI] [PubMed] [Google Scholar]
- 22.Kawai K, Kobayashi Y, Shiratori M, Sobue G, Tamatani T, Miyasaka M, et al. Intrathecal administration of antibodies against LFA-1 and against ICAM-1 suppresses experimental allergic encephalomyelitis in rats. Cell Immunol. 1996;171:262–268. doi: 10.1006/cimm.1996.0202. [DOI] [PubMed] [Google Scholar]
- 23.Connolly MK, Kitchens EA, Chan B, Jardieu P, Wofsy D. Treatment of murine lupus with monoclonal antibodies to lymphocyte function-associated antigen-1: dose-dependent inhibition of autoantibody production and blockade of the immune response to therapy. Clin Immunol Immunopathol. 1994;72:198–203. doi: 10.1006/clin.1994.1130. [DOI] [PubMed] [Google Scholar]
- 24.Kakimoto K, Nakamura T, Ishii K, Takashi T, Iigou H, Yagita H, et al. The effect of anti-adhesion molecule antibody on the development of collagen-induced arthritis. Cell Immunol. 1992;142:326–337. doi: 10.1016/0008-8749(92)90294-y. [DOI] [PubMed] [Google Scholar]
- 25.Lebwohl M, Tyring SK, Hamilton TK, Toth D, Glazer S, Tawfik NH, et al. A novel targeted T-cell modulator, efalizumab, for plaque psoriasis. N Engl J Med. 2003;349:2004–2013. doi: 10.1056/NEJMoa030002. [DOI] [PubMed] [Google Scholar]
- 26.Salas A, Shimaoka M, Kogan AN, Harwood C, von Andrian UH, Springer TA. Rolling adhesion through an extended conformation of integrin aLβ2 and relation to a I and β I-like domain interaction. Immunity. 2004;20:393–406. doi: 10.1016/s1074-7613(04)00082-2. [DOI] [PubMed] [Google Scholar]
- 27.Weitz-Schmidt G, Welzenbach K, Brinkmann V, Kamata T, Kallen J, Bruns C, et al. Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nat Med. 2001;7:687–692. doi: 10.1038/89058. [DOI] [PubMed] [Google Scholar]
- 28.Larson RS, Davis T, Bologa C, Semenuk G, Vijayan S, Li Y, et al. Dissociation of I domain and global conformational changes in LFA-1: refinement of small molecule-I domain structure-activity relationships. Biochemistry. 2005;44:4322–4331. doi: 10.1021/bi048187k. [DOI] [PubMed] [Google Scholar]
- 29.Anderson ME, Siahaan TJ. Mechanism of binding and internalization of ICAM-1-derived cyclic peptides by LFA-1 on the surface of T cells: a potential method for targeted drug delivery. Pharm Res. 2003;20:1523–1532. doi: 10.1023/a:1026188212126. [DOI] [PubMed] [Google Scholar]
- 30.Anderson ME, Tejo BA, Yakovleva T, Siahaan TJ. Characterization of binding properties of ICAM-1 peptides to LFA-1: inhibitors of T-cell adhesion. Chem Biol Drug Des. 2006;68:20–28. doi: 10.1111/j.1747-0285.2006.00407.x. [DOI] [PubMed] [Google Scholar]
- 31.Gadek TR, Burdick DJ, McDowell RS, Stanley MS, Marsters JC, Jr, Paris KJ, et al. Generation of an LFA-1 antagonist by the transfer of the ICAM-1 immunoregulatory epitope to a small molecule. Science. 2002;295:1086–1089. doi: 10.1126/science.295.5557.1086. [DOI] [PubMed] [Google Scholar]
- 32.Zimmerman T, Oyarzabal J, Sebastian ES, Majumdar S, Tejo BA, Siahaan TJ, et al. ICAM-1 peptide inhibitors of T-cell adhesion bind to the allosteric site of LFA-1. An NMR characterization. Chem Biol Drug Des. 2007;70:347–353. doi: 10.1111/j.1747-0285.2007.00566.x. [DOI] [PubMed] [Google Scholar]
- 33.Yusuf-Makagiansar H, Makagiansar IT, Siahaan TJ. Inhibition of the adherence of T-lymphocytes to epithelial cells by a cyclic peptide derived from inserted domain of lymphocyte function-associated antigen-1. Inflammation. 2001;25:203–214. doi: 10.1023/a:1011044616170. [DOI] [PubMed] [Google Scholar]
- 34.Verkhivker GM, Bouzida D, Gehlhaar DK, Rejto PA, Arthurs S, Colson AB, et al. Deciphering common failures in molecular docking of ligand-protein complexes. J Comput Aided Mol Des. 2000;14:731–751. doi: 10.1023/a:1008158231558. [DOI] [PubMed] [Google Scholar]
- 35.Verkhivker GM. Computational analysis of ligand binding dynamics at the intermolecular hot spots with the aid of simulated tempering and binding free energy calculations. J Mol Graph Model. 2004;22:335–348. doi: 10.1016/j.jmgm.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 36.Verkhivker GM. Computational proteomics of biomolecular interactions in the sequence and structure space of the tyrosine kinome: deciphering the molecular basis of the kinase inhibitors selectivity. Proteins. 2007;66:912–929. doi: 10.1002/prot.21287. [DOI] [PubMed] [Google Scholar]
- 37.Verkhivker GM. In silico profiling of tyrosine kinases binding specificity and drug resistance using Monte Carlo simulations with the ensembles of protein kinase crystal structures. Biopolymers. 2007;85:333–348. doi: 10.1002/bip.20656. [DOI] [PubMed] [Google Scholar]
- 38.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McFadyen IJ, Ho JC, Mosberg HI, Traynor JR. Modifications of the cyclic mu receptor selective tetrapeptide Tyr-c[D-Cys-Phe-D-Pen]NH2 (Et): effects on opioid receptor binding and activation. J Pept Res. 2000;55:255–261. doi: 10.1034/j.1399-3011.2000.00177.x. [DOI] [PubMed] [Google Scholar]
- 40.Anderson ME, Yakovleva T, Hu Y, Siahaan TJ. Inhibition of ICAM-1/LFA-1-mediated heterotypic T-cell adhesion to epithelial cells: design of ICAM-1 cyclic peptides. Bioorg Med Chem Lett. 2004;14:1399–1402. doi: 10.1016/j.bmcl.2003.09.100. [DOI] [PubMed] [Google Scholar]
- 41.Gursoy RN, Jois DS, Siahaan TJ. Structural recognition of an ICAM-1 peptide by its receptor on the surface of T cells: conformational studies of cyclo (1, 12)-Pen-Pro-Arg-Gly-Gly-Ser-Val-Leu-Val-Thr-Gly-Cys-OH. J Pept Res. 1999;53:422–431. doi: 10.1034/j.1399-3011.1999.00080.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The ESI mass spectra of eight peptides used in this study.