

Abstract
Pancreatic adenocarcinoma is characterized by a dense background of tumor associated stroma originating from abundant pancreatic stellate cells. The aim of this study was to determine the effect of human pancreatic stellate cells (HPSC) on pancreatic tumor progression. HPSCs were isolated from resected pancreatic adenocarcinoma samples and immortalized with telomerase and SV40 large T antigen. Effects of HPSC conditioned medium (HPSC-CM) on in vitro proliferation, migration, invasion, soft-agar colony formation, and survival in the presence of gemcitabine or radiation therapy were measured in two pancreatic cancer cell lines. The effects of HPSCs on tumors were examined in an orthotopic murine model of pancreatic cancer by co-injecting them with cancer cells and analyzing growth and metastasis. HPSC-CM dose-dependently increased BxPC3 and Panc1 tumor cell proliferation, migration, invasion, and colony formation. Furthermore, gemcitabine and radiation therapy were less effective in tumor cells treated with HPSC-CM. HPSC-CM activated the mitogen-activated protein kinase and Akt pathways in tumor cells. Co-injection of tumor cells with HPSCs in an orthotopic model resulted in increased primary tumor incidence, size, and metastasis, which corresponded with the proportion of HPSCs. HPSCs produce soluble factors that stimulate signaling pathways related to proliferation and survival of pancreatic cancer cells, and the presence of HPSCs in tumors increases the growth and metastasis of these cells. These data indicate that stellate cells have an important role in supporting and promoting pancreatic cancer. Identification of HPSC-derived factors may lead to novel stroma-targeted therapies for pancreatic cancer.
Introduction
Pancreatic ductal adenocarcinoma is characterized by a highly malignant phenotype resistant to currently available systemic therapies; incidence rates are nearly equal to mortality rates (1). The extremely dense desmoplastic infiltration that is characteristic of pancreatic adenocarcinoma is unique among solid tumors, and may impede effective systemic treatments on a molecular level. Importantly, in many tumor types, the local microenvironment is thought to be an active participant in the process of cancer initiation, progression, and metastasis (2, 3). In many solid tumors, the stroma is increasingly recognized to be important in promoting tumor proliferation, invasion, metastasis, and chemoresistance (2, 4). However, despite the abundant desmoplastic reaction that is a hallmark of pancreatic adenocarcinoma, the role of tumor-associated stroma in pancreatic cancer is not well described.
Pancreatic stellate cells are myofibroblast-like cells that were originally identified as the source of the fibrosis in chronic pancreatitis (5) and are now thought to be responsible for the dense stroma associated with pancreatic adenocarcinoma (6). Pancreatic stellate cells are similar to hepatic stellate cells, which are important effector cells in hepatic fibrosis (7–9). Both pancreatic and hepatic stellate cells stain positive for vimentin, desmin, and α smooth muscle actin (αSMA) and contain vitamin A–storing lipid droplets in the cytoplasm, indicating that they are neither fibroblasts nor smooth muscle cells (5). The precise role of pancreatic stellate cells in tumor-stroma interactions in pancreatic cancer has not been well defined, in part because of the difficulty in obtaining primary HPSCs in sufficient quantities for study. Once isolated, HPSCs can only be passaged for a limited number of times before undergoing senescence. Most studies using pancreatic stellate cells have used cells isolated from rats (10, 11), with relatively few studies using primary HPSCs from pancreatic cancer (12, 13). Detailed characterization of the role of pancreatic stellate cells in human pancreatic cancer progression would provide a set of potential targets for stromal-directed therapy. In addition, pancreatic cancer–associated stromal factors may also serve as tumor biomarkers and possible targets for cancer prevention.
The purpose of this study was to investigate the effects of primary HPSCs on pancreatic tumor progression and response to therapy in vitro and in an orthotopic model of pancreatic cancer. For this purpose, we isolated human adenocarcinoma-associated pancreatic stellate cells from resected surgical specimens. These cells were immortalized and examined for their influence on pancreatic cancer cells in vitro and in vivo. We found that the stellate cells had several effects that promoted the progression of pancreatic adenocarcinoma.
Materials and Methods
Cell culture
BxPC3 and Panc1 pancreatic tumor cells were obtained from American Type Culture Collection. Tumor cells were cultured in DMEM (Invitrogen) containing 10% FCS with 2 mmol/L glutamine and penicillin/streptomycin (both from Invitrogen) at 37°C in a humidified atmosphere of 5% CO2.
Human pancreatic stellate cells (HPSC) were prepared by the outgrowth method (12). Fresh tissue was obtained from residual pancreatic adenocarcinoma specimens from patients undergoing primary surgical resection at The University of Texas M. D. Anderson Cancer Center. All human samples were obtained in accordance with the policies and practices of the Institutional Review Board of The University of Texas M. D. Anderson Cancer Center. Briefly, tumor samples were minced and seeded in six-well plates containing 15% FCS/DMEM, l-glutamine (2 mmol/L), penicillin/streptomycin, and amphotericin. After ∼5 days, cells were able to grow out from the tissue clumps. Medium was changed every 3 days. All cells were maintained at 37°C in a humidified atmosphere of 5% CO2. When HPSCs grew to confluence, cells were trypsinized and passaged 1:3.
Cell purity was determined by immunohistochemistry for αSMA, vimentin, and desmin, as well as morphology (spindle-shaped cells with cytoplasmic extensions) and positive staining with Oil Red O (lipid inclusions were visualized). Antibodies used were antihuman αSMA (clone 1A4), anti-vimentin (clone V9), and antihuman desmin (clone D33; all from DAKO). For the assays using nonimmortalized primary HPSCs, passage numbers 5 to 12 were used.
HPSCs were immortalized using a HPSC line derived from the pancreatic tumor of a patient who had received no prior therapy before surgery. Lentiviral vectors with human telomerase (hTERT) or SV40 large T antigen (TAg) were produced by transfection of 293 cells. Plasmids containing TAg (pHIV7-CNPO-TAg) and hTERT (pHIV7-CNPO-hTERT) have been described in detail (14) and were kindly provided by Dr. Jiing-Kuan Yee (City of Hope National Medical Center, Duarte, CA). After 48 h, viral supernatant was collected and added to HPSCs. HPSCs carrying the hTERT or SV40-T were selected in 1 to 3 mg/mL G418 for 3 weeks (Invitrogen). The presence of SV40-T was confirmed by Western blotting (Santa Cruz Biotech). Expression of αSMA and vimentin was confirmed as described above (Fig. 1). To quantify growth rate, cells were grown in six-well plates and counted every 2 days. Doubling time was calculated as (0.301 × Δt) / (log10 N/N0), where Δt represents the difference in time, measured in hours, and N0 and N represent the number of cells at the initial and end time points.
Figure 1.
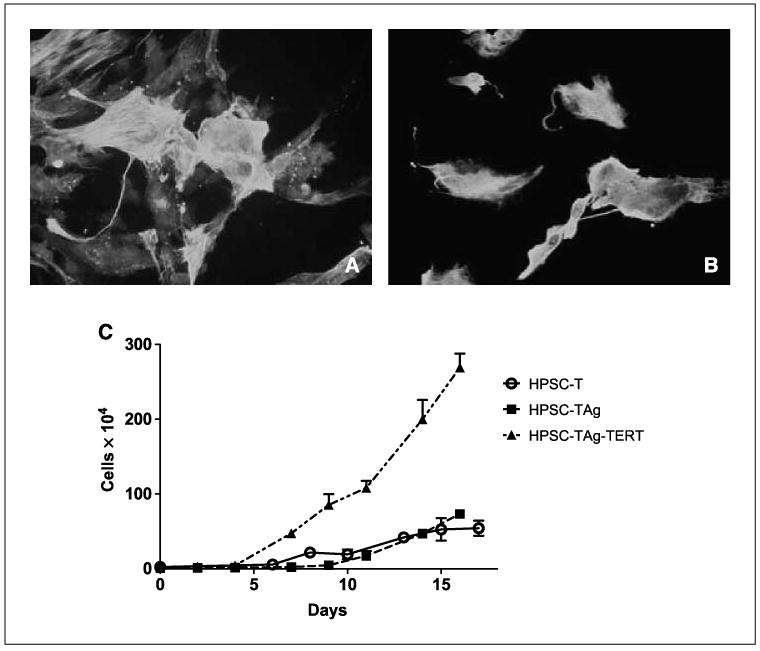
Immortalization of HPSCs. Primary HPSCs were exposed to lentiviral vectors carrying hTERT and SV40-TAg. Immunohistochemical staining of HPSCs transduced with hTERT and SV40-TAg for αSMA (A) and vimentin (B). C, growth curve for primary HPSCs, HPSCs transduced with SV40-TAg, and HPSCs transduced with both hTERT and SV40-TAg. HPSCs carrying hTERT and Tag show no evidence of senescence at >100 passages.
Preparation of conditioned media
HPSCs, both primary and immortalized, were grown to 70% to 80% confluence in 20-cm2 dishes in DMEM/10% FCS. The media were changed to serum-free DMEM and cells were cultured for 48 h. Media were collected, centrifuged at 1,000 × g for 10 min, and the supernatant was concentrated with Centricon YM3 filters (Millipore). Protein content of the concentrated HPSC conditioned media (HPSC-CM) was determined by Bradford assay (Bio-Rad Laboratories) and aliquots were stored at −80°C until use. For all functional assays using HPSC-CM, conditioned media from immortalized and nonimmortalized HPSCs were used and produced similar results.
Proliferation assay
BxPC3 and Panc1 cells were seeded at 3,000 per well in 96-well plates and cultured in DMEM/10% FCS. Medium was changed to serum-free DMEM for overnight incubation. Concentrated HPSC-CM was added to cells at varying concentrations (0.1, 0.25, 0.5, 1.0 μg/μL); serum-free DMEM was added to control wells. Cell growth was analyzed at 24, 48, and 72 h with the MTS reagent (Promega) added 1 h before taking the spectrophotometric reading, according to the manufacturer's directions.
Invasion assays
For studies of cell invasiveness, BioCoat Matrigel-coated invasion chambers (BD Biosciences) were used. Briefly, 2 × 104 BxPC3 or Panc1 cells in 500-μL serum-free medium were added to the upper chamber. Medium containing 10% FCS or concentrated HPSC-CM (0.1, 0.5, and 1.0 μg/μL) was added into the lower chamber. Serum-free medium was added to the lower chamber of control wells. The cells were allowed to invade the Matrigel for 48 h at 37°C in a 5% CO2 atmosphere. The noninvading cells on the upper surface of the membrane were removed with a cotton swab. Cells were fixed in methanol, stained with hematoxylin, and air-dried. Invading cells in three adjacent microscope fields for each membrane were counted at ×20 magnification.
Soft-agar colony formation
BxPC3 or Panc1 cells were plated in 24-well plates (104 per well) with 0.3% agar on a 0.6% agar (Noble agar, Becton Dickinson) underlayer containing HPSC-CM, serum-free medium, or DMEM containing 10% FCS. HPSC-CM was replenished daily on the top layer. Cells were cultured for 15 days and colonies were counted in 10 random fields per well.
Western blot analysis
To evaluate activation of the mitogen-activated protein kinase (MAPK) and Akt pathways in the presence of HPSC-CM, BxPC3 cells were grown in six-well plates in 10% DMEM to 70% confluency, serum starved overnight, and treated with HPSC-CM (1 μg/μL) for 0 to 120 min. Cells were washed twice with PBS and lysed. Cell lysates were prepared and separated by SDS-PAGE and transferred to nitrocellulose. Membranes were blocked for 1 h at room temperature in 5% nonfat milk in 0.1% Tween, washed, and incubated overnight at 4°C with one of the following antibodies: total p44/42, phospho p44/42 (Thr202/Tyr204), Akt, or phospho Akt (Ser473), all from Cell Signaling Technology. Immunodetection was done with the corresponding secondary Alexa Fluor 680–conjugated antibodies. Fluorescence was detected with the Odyssey fluorescent imager (Li-Cor Biosciences).
Response to chemotherapy and radiation treatment
BxPC3 cells were seeded in eight-well chamber slides (1 × 105 per well) in DMEM with 10% FCS. After incubation overnight, cells were washed and medium was changed to DMEM + 10% FCS, with or without HPSC-CM (1 μg/μL). Cells were treated with either 100 μmol/L gemcitabine (Eli Lilly) or vehicle alone. After 48 h, apoptotic cells were analyzed with the terminal deoxyribonucleotidyl transferase–mediated dUTP nick end labeling (TUNEL) assay (ApopTag, Chemicon) and nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Cells were counted using Simple PCI software (Compix, Inc.). The ratio of apoptotic cells was calculated as (number of cells positive for TUNEL / number of nuclei stained with DAPI) × 100. Experiments were done in duplicate and 10 random fields were counted for each condition.
The effect of HPSC-CM on tumor cell response to radiation was done in a similar fashion. After addition of HPSC-CM (or DMEM with 10% FCS for control wells), cells were exposed to either 0 or 100 Gy of ionizing radiation. After 24-h incubation, caspase activation was detected using Magic Red kit (Immunochemistry Technologies, Inc.) and cell nuclei were stained with DAPI. The percentage of cells undergoing apoptosis and the total number of nuclei were counted using Simple PCI software (Compix, Inc.). Experiments were done in duplicate and 10 random fields were counted for each condition.
Orthotopic model of pancreatic cancer
All animal experiments were reviewed and approved by the University of Texas M. D. Anderson Institutional Animal Care and Use Committee. An orthotopic nude mouse model of pancreatic cancer using BxPC3 pancreatic tumor cells labeled with firefly luciferase (BxPC3-FL) has previously been described by this lab (15). All mice were divided into groups receiving intrapancreatic injections of (a) BxPC3-FL alone, either 0.5 × 106 or 1 × 106 per mouse; (b) BxPC3-FL with immortalized HPSCs, in varying tumor-to-stroma ratios (1:1, 1:1, or 1:5); or (c) immortalized HPSCs alone (0.5 × 106 or 1 × 106). All cell suspensions, including the mixtures of BxPC3 and HPSCs, were injected in a 50-μL volume of HBSS. Bioluminescent imaging was done weekly to follow the luciferase signal from BxPC3 cells. Mice were sacrificed and tumors were harvested and measured.
Statistical analysis
Statistical analysis was done using GraphPad Prism (GraphPad). Comparisons were made using two-tailed Student's t test and significant difference was defined as P < 0.05. Data are shown as mean ± SE.
Results
Primary culture and immortalization of HPSCs
Pancreatic stellate cells were isolated from residual tissue from a fresh human pancreatic adenocarcinoma surgical specimen and their identity was confirmed by immunohistochemical staining for αSMA and vimentin (data not shown). After lentiviral transfer of hTERT and SV40-TAg and selection in G418, HPSCs continued to express αSMA and vimentin (Fig. 1). The growth rate of HPSCs was highest for cells immortalized with both hTERT and SV40-TAg compared with cells transduced with SV40-TAg alone or nonimmortalized HPSCs (doubling time, 12 versus 42 versus 113 h, respectively). HPSCs immortalized with hTERT and SV40-Tag showed no evidence of senescence at >100 passages. Morphology of HPSCs immortalized with either SV40 Tag alone or both hTERT and SV40-TAg was not significantly different compared with nonimmortalized HPSCs (data not shown).
Conditioned medium from HPSCs stimulates pancreatic tumor cell proliferation, invasion, and colony formation in soft agar
The addition of conditioned medium from immortalized HPSCs (expressing SV40 Tag and hTERT) to BxPC3 cells induced an increase in tumor cell proliferation compared with serum-free medium controls (Fig. 2A). The effect was dose dependent and a significant increase in proliferation compared with serum-free medium control wells was observed at 0.5 μg/μL (301.7% of control ± 43.1%; P < 0.005). Cell proliferation with the highest dose of HPSC-CM (1 μg/μL) was equivalent to medium containing 10% FCS. Panc1 cells were more sensitive to HPSC-CM because a significant increase in proliferation was observed with amounts as low as 0.25 μg/μL in a dose-dependent fashion. Maximal effects of HPSC-CM on Panc1 cell proliferation were similar to those observed in BxPC3 cells and again were equal to the effects of 10% FCS (Fig. 2A).
Figure 2.

Effect of HPSC-CM on pancreatic tumor cell phenotype. CM from HPSCs (μg/μL) was added to BxPC3 or Panc1 pancreatic tumor cells. A, tumor cell proliferation was measured by MTS assay at 48 h. B, tumor cell invasion was evaluated at 48 h using a modified Boyden chamber assay. C, the ability of tumor cells to form colonies in soft agar was determined at 15 d after addition of HPSC-CM. D, tumor cell proliferation was measured in the presence of conditioned media from nonimmortalized HPSCs. Columns, mean of three experiments. *, P < 0.05 and **, P < 0.005 versus serum-free media (SFM) control. #, P < 0.0005 versus HPSC-CM 1.0 μg/μL.
HPSC-CM also induced a dose-dependent increase in BxPC3 and Panc1 cell migration and invasion (Fig. 2B; data on cell migration not shown). BxPC3 invasion was induced with as little as 0.1 μg/μL HPSC-CM [3.4 ± 1.78 versus 0.4 ± 0.51; P < 0.0001]. Stimulation of BxPC3 invasion increased with the dose of HPSC-CM to a maximum of 21.5 ± 6.2. HPSC-CM induced an even greater increase in cell migration of Panc1 cells than BxPC3, similar to its effects on cell proliferation. The minimum dose of 0.1 μg/μL HPSC-CM stimulated a 19-fold increase in Panc1 invasion compared with serum-free medium (P < 0.0001). The maximum dose of 1.0 μg/μL HPSC-CM resulted in Panc1 invasion at a higher level than medium containing 10% FCS (48 ± 15 versus 28.3 ± 3.4; P < 0.001).
To determine whether secreted factors from stromal fibroblasts affect anchorage-independent growth of tumor cells, soft-agar colony formation assay was done in the presence or absence of HPSC-CM. With the addition of HPSC-CM, BxPC3 cells formed more colonies in soft agar compared with serum-free medium control (Fig. 2C; 146.7 ± 7.6 for 0.5 μg/μL HPSC-CM, 178.7 ± 13.6 for 1 μg/μL HPSC-CM, and 105.3 ± 5.5 for serum-free medium; P < 0.005). Similarly, Panc1 cells formed a higher number of colonies in soft agar with the addition of HPSC-CM in a dose-dependent manner (144 ± 6.6 for serum-free medium, 171 ± 11.5 for 0.1 μg/μL HPSC-CM, 209 ± 12.7 for 0.25 μg/μL HPSC-CM, 261 ± 23.1 for 0.5 μg/μL HPSC-CM, and 371 ± 20.2 for 1.0 μg/μL HPSC-CM; P < 0.005, versus serum-free medium).
Conditioned medium from nonimmortalized HPSCs also stimulated tumor cell proliferation in a dose-dependent manner, from 0.25 to 1.0 μg/μL for both BxPC3 and Panc1 tumor cell lines (Fig. 2D). The stimulatory effects of HPSC-CM from nonimmortalized HPSCs on tumor cell migration, invasion, and soft-agar colony formation were comparable to HPSC-CM from immortalized HPSCs and are not shown.
Pancreatic stellate cells inhibit effects of chemotherapy and radiation on tumor cells
The addition of HPSC-CM to BxPC3 cells treated with gemcitabine (100 μmol/L) resulted in an inhibition of apoptosis (38.9 ± 3.1% apoptosis with serum-free medium versus 9.4 ± 1.1% apoptosis with HPSC-CM; P < 0.0001; Fig. 3). There was no significant difference in total cell number in the presence of gemcitabine with or without addition of HPSC-CM.
Figure 3.
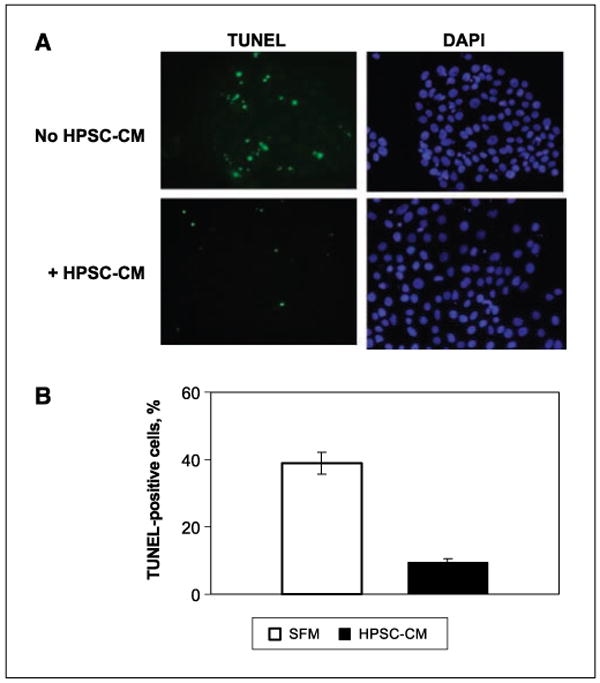
BxPC3 cells were treated with gemcitabine (100 μmol/L) with or without conditioned media from HPSCs (1 μg/μL) for 48 h. A, apoptosis was detected using TUNEL assay and cell nuclei were stained with DAPI. B, percentage of apoptotic cells was determined in 10 random fields for each condition. SFM, serum-free media.
Similarly, after exposure to 100-Gy radiation, BxPC3 cells that were treated with HPSC-CM (1 μg/μL) showed less apoptosis compared with cells grown in 10% DMEM (29.9% ± 6.9 versus 66.8 ± 7.8; P < 0.01; Fig. 4). Cells that were not exposed to radiation showed no difference in apoptosis in the presence or absence of HPSC-CM (not shown). The total number of viable nuclei was higher in cells that were exposed to radiation in the presence of HPSC-CM compared with cells without HPSC-CM (210.8 ± 13.6 versus 53.8 ± 5.6; P < 0.0001; Fig. 4).
Figure 4.
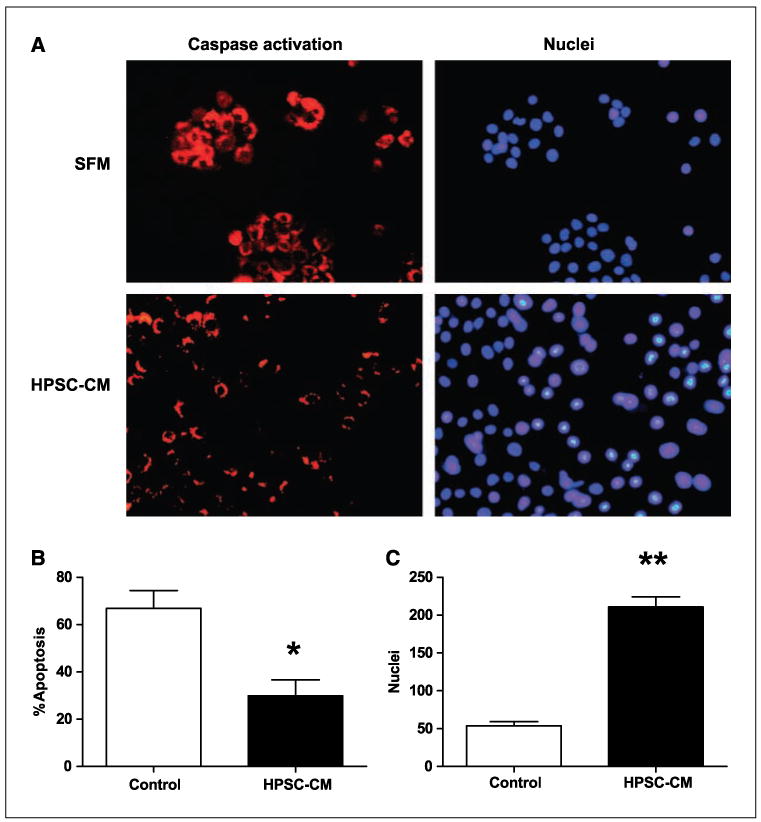
Conditioned media from HPSCs protects pancreatic tumor cells from apoptosis induced by radiation. BxPC3 cells were treated with 100 Gy of radiation in the presence of either media containing 10% FCS or conditioned media from HPSCs (1 μg/μL). After 24 h, cells were analyzed for caspase activation (A) and percentage of cells undergoing apoptosis (B) and total number of nuclei (C) were calculated. Control, DMEM with 10% FCS; XRT, radiation treatment. *, P < 0.01 versus control; **, P < 0.0001 versus control.
Secreted factors from pancreatic stellate cells stimulate activation of MAPK and Akt pathways in tumor cells
To determine which signaling pathways may be involved in the tumor-promoting effects of HPSCs, activation of MAPK and Akt in BxPC3 tumor cells treated with HPSC-CM was determined by Western blotting. Phosphorylated Erk1/2 was increased in cells treated with HPSC-CM compared with control cells beginning at 5 min and persisted until 120 min after treatment (Fig. 5). Similarly, addition of HPSC-CM activated Akt 5 min after stimulation and continued for 60 min. Compared with serum-free controls, stimulation with HPSC-CM resulted in 43% increase in Erk1/2 activation at 15 min and 125% increase in Akt activation at 5 min. Similar results were observed in Panc1 cells (data not shown).
Figure 5.
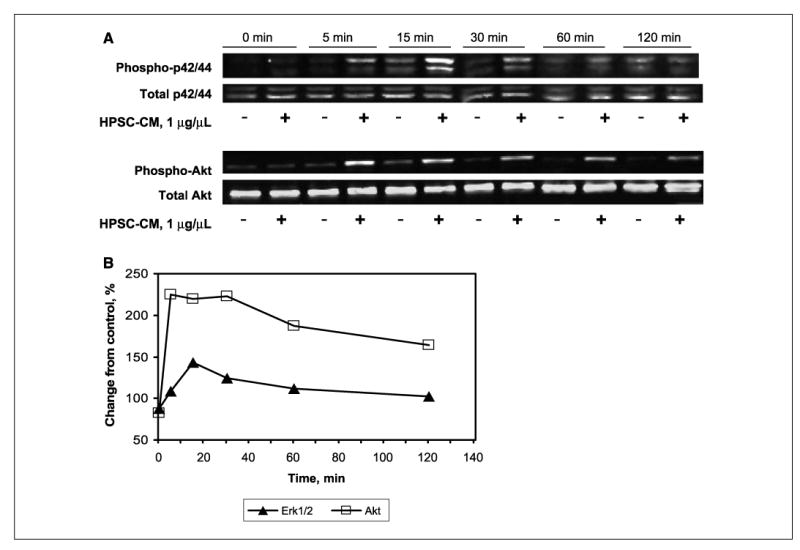
Protein lysates from BxPC3 cells treated with HPSC-CM (1 μg/μL) were analyzed for Erk1/2 and Akt activation by Western blotting at various time points (A) and quantifed by densitometry. Control cells were treated with serum-free media alone. B, change in levels of phosphorylated Erk1/2 and Akt was compared with serum-free controls.
Co-injection of HPSCs with tumor cells induces tumor progression and metastasis in an in vivo orthotopic model of pancreatic cancer
Pancreatic adenocarcinomas are notorious for having a significant fibrotic component to the tumor (16), which is not well recapitulated in orthotopic nude mouse models of pancreatic cancer that use injection of cultured human tumor cells. Thus, to evaluate the contribution of stromal fibroblasts to the growth of pancreatic cancer in vivo, we co-injected HPSCs with BxPC3 tumor cells. To follow tumor progression in real time, BxPC3 cells were labeled with firefly luciferase and bioluminescence was measured as previously described (15, 17). The proportion of HPSCs in the co-injection relative to tumor cells varied from 1:0 tumor-to-stroma ratio to 1:5, which reflects the greater abundance of stroma typically observed in human pancreatic adenocarcinomas.
The incidence of tumor formation increased when mice were injected with both HPSCs and tumor cells. In the group injected with equal numbers of tumor and stromal cells (0.5 × 106 cells/mouse), only 57% of mice formed tumors in the pancreas (Fig. 6A). However, in the groups injected with 5-fold higher number of stromal cells compared with tumor cells (tumor-to-stroma ratio 1:5), pancreatic tumor formation was 100%. No tumors developed when mice received HPSCs alone (0.5 or 1 × 106 per mouse).
Figure 6.
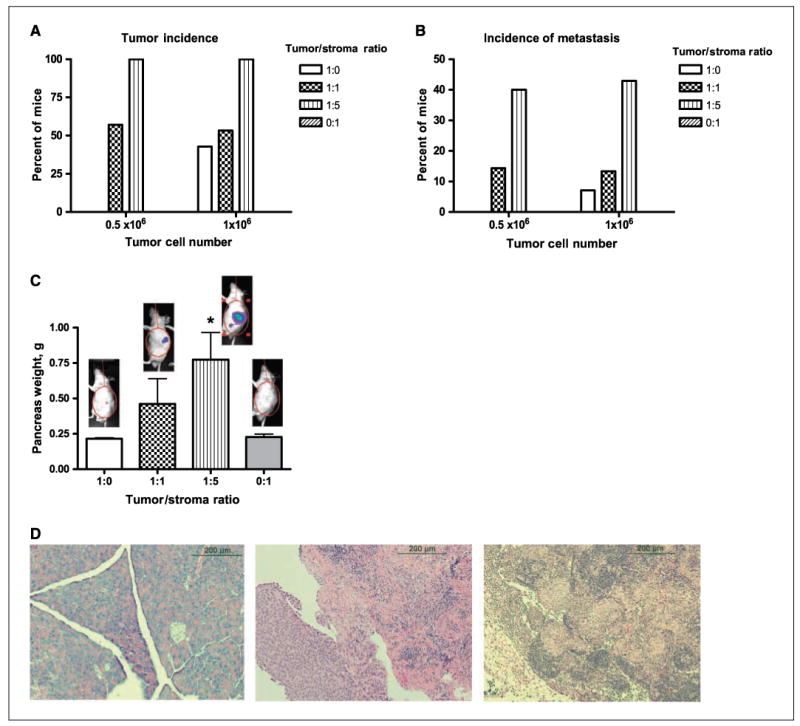
Co-injection of HPSCs with BxPC3 tumor cells in an orthotopic nude mouse model of pancreatic cancer results in increased tumor incidence, metastasis, and tumor size. HPSCs were injected with BxPC3 cells (either 0.5 × 106 or 1 × 106) in varying tumor/stroma ratios. At 8 wk, mice were sacrificed and primary tumor incidence (A) and metastasis (B) were quantified. C, pancreatic tumors were weighed and the luciferase signal from a representative mouse in each group is shown. D, H&E staining showed no tumors in the pancreas of mice injected with 0.5 × 106 BxPC3 cells alone (left) but confirmed tumor development in mice co-injected with BxPC3 and HPSCs (pancreas, center, and peritoneal metastases, right). Each experiment was repeated twice and a summary of all the results is shown. *, P < 0.01 versus tumor/stroma ratio 1:0 or 0:1.
None of the mice that received BxPC3 cells only at 0.5 × 106 per mouse developed pancreatic tumors, but when the number of BxPC3 cells was increased to 1 × 106, 43% of mice developed tumors. Thus, the presence of HPSC stromal fibroblasts contributed to the initiation of tumor formation, particularly with a low number of tumor cells. When HPSCs were present in equal or greater numbers to tumor cells, the incidence of tumor formation was increased compared with tumor cells alone, and the incidence was comparable to the higher tumor cell number (1 × 106) for every given tumor-to-stroma ratio. These results suggest that HPSCs compensate for a lower tumor cell number and can promote tumor initiation to result in tumor development equivalent to the higher tumor cell number.
Interestingly, the proportion of mice that developed distant metastases also increased with the number of HPSCs injected. With a low tumor cell number (0.5 × 106 per mouse), none of the mice developed metastases (Fig. 6B). When the same number of tumor cells was mixed with HPSCs, the incidence of metastasis increased in a dose-dependent manner with the proportion of stromal fibroblasts (14.3% versus 40%; tumor-to-stroma ratio, 1:1 versus 1:5, respectively). Metastases were predominantly intraperitoneal, lymphatic, and hepatic; none were visible grossly or with IVIS imaging in other organs. At a higher tumor cell number (1 × 106 per mouse), the incidence of metastasis was higher than with a lower starting number but was further increased in a concentration-dependent manner by the presence of stellate cells. As expected, no metastatic lesions were identified in mice that received HPSCs only.
The presence of stromal fibroblasts affected not only tumor incidence but also tumor size. As the proportion of HPSCs increased relative to tumor cells, the total weight of the pancreas including tumor increased (Fig. 6C). In the mice that received tumor cells alone (0.5 × 106 per mouse), the weight of the pancreas was 0.22 ± 0.01 g and not significantly different than the pancreas weight of mice that received HPSCs only. As the proportion of HPSCs increased in the mixture, the weight of the pancreas increased [0.46 ± 0.18 versus 0.82 ± 0.22 g; tumor-to-stroma ratio, 1:1 versus 1:5, respectively (means ± SE)]. The mean pancreatic weight in the group with tumor-to-stroma ratio 1:5 was significantly different from the group with tumor cells only (P < 0.05). The increasing tumor burden was reflected in the IVIS signal (representative images of each group at 8 weeks shown in Fig. 6C). The histology of primary and metastatic tumors was confirmed with H&E staining (Fig. 6D).
Discussion
The importance of the local tumor microenvironment to tumor progression has been recognized for many years and highlighted in several reviews (2, 3, 18). Insight into the role of tumor-associated stroma in breast and prostate cancer has recently been described (4, 19–27); however, in contrast, there is little known about the possible contribution of the stroma to pancreatic cancer growth and metastasis. This is particularly unfortunate as pancreatic cancer has an especially abundant stroma. Using a new model of human cancer-associated stellate cells, we found that conditioned medium from HPSCs stimulated pancreatic tumor cell proliferation, migration, invasion, and anchorage-independent growth in a dose-dependent manner. Moreover, conditioned medium from HPSCs inhibited the response of tumor cells to chemotherapy and radiation. These observations indicate that soluble factors are produced by stellate cells that stimulate proliferation and survival of pancreatic cancer cells. We also observed that co-injection of stellate cells increased tumor incidence, growth, and metastasis in an orthotopic model of pancreatic cancer. Taken together, these studies indicate that the abundant stroma of pancreatic cancer plays an important role in the aggressiveness of this disease.
Previously, a relevant cell model to study pancreatic cancer–associated stromal fibroblasts did not exist. Although the pancreatic stellate cell has been identified as the cell responsible for stroma production in both chronic pancreatitis and pancreatic cancer (5, 6, 28), few studies have used pancreatic stellate cells from human pancreatic adenocarcinoma samples due to the relative paucity of available fresh tumor specimens and the limited life span of primary cells. Immortalization of rat (10) and human (29) stellate cells has previously been found to be useful because primary cells grow slowly and eventually senesce after 10 to 15 passages. Our study is the first report of the use of immortalized primary HPSCs derived from pancreatic adenocarcinoma. Previous studies in other solid tumors have suggested that phenotypic differences exist between stromal cells derived from tumors and normal tissues (23). Therefore, these tumor-derived cells are the most relevant cell model to study the role of pancreatic cancer–associated stromal fibroblasts.
We found that the effects of HPSCs on pancreatic cancer were dose dependent. In vitro, this was manifest as a direct concentration dependence of the effects of conditioned media. In vivo, a tumor-to-stroma ratio of at least 1:1 or higher resulted in the most dramatic increase in tumor growth and metastasis. The higher proportion of stromal cells compared with tumor cells is consistent with the histologic appearance of human pancreatic adenocarcinoma, in which the desmoplastic stroma is overwhelmingly abundant relative to neoplastic epithelial cells (16). This is the first study to co-inject HPSCs with cancer cells in an orthotopic model of pancreatic cancer; however, other investigators have also found that the presence of pancreatic stellate cells increases growth of pancreatic cancer cells in s.c. xenograft models (12, 30, 31).
The extensive stromal component of solid tumors has been postulated by other groups to influence tumor response to chemotherapy (32, 33). In the current study, we have confirmed this hypothesis. We observed HPSC-mediated inhibition of pancreatic cancer cell responses to gemcitabine and radiation. Interestingly, Ohuchida et al. (34) also found that radiation treatment of stromal fibroblasts increases the invasiveness of pancreatic cancer cells even more than nonirradiated fibroblasts. Thus, by including the stromal contribution to pancreatic cancer, our xenograft model with co-injection of HPSCs and pancreatic tumor cells may be a more clinically relevant model of pancreatic adenocarcinoma than previous xenograft models of tumor cells alone.
The mechanism responsible for the increased survival of pancreatic cancer cells treated with conditioned medium from stellate cells is not certain. We observed that such treatments resulted in the activation of both proliferation related (Erks) and survival related (Akt) pathways; these processes may be mediated by interleukin-1β and transforming growth factor-β, both of which are secreted by pancreatic stellate cells (35). Additional candidate molecules that may mediate tumor-stromal interactions have been identified by gene expression profiling of stromal fibroblast cocultured with CFPAC1 pancreatic cancer cells (36). Of the genes identified, COX-2 was markedly augmented in both cancer cells and fibroblasts when cultured together and was shown to mediate invasion of CFPAC1 cells. In contrast, other stromal-derived factors may suppress tumor growth. Sato et al. (37) found that SPARC was overexpressed in stromal fibroblasts in pancreatic cancer and exogenous SPARC decreased proliferation of pancreatic cancer cells in vitro. Thus, the interactions between tumor and stromal fibroblasts are quite complex and further studies will be necessary to positively identify the factors and signaling mechanisms involved in these interactions.
Another important observation in the current study was that the presence of HPSCs increased the incidence of tumor formation when limiting numbers of cancer cells were injected. Inclusion of stellate cells allowed the injection of low numbers of cancer cells that otherwise were unable to form tumors. This supports the hypothesis that stellate cells provide a microenvironment that is particularly advantageous to pancreatic cancer cells. It is possible that stromal fibroblasts may interact with pancreatic cancer stem cells, which have recently been described by Li et al. (38). The tumor vasculature, another important component of the tumor microenvironment, was recently shown to influence the number of brain cancer stem cells (nestin+ and CD133+; ref. 39). Thus, targeting the “niche” or unique microenvironment of the cancer stem cells may be an effective therapeutic strategy. The mechanisms responsible for this effect are not completely understood, but the observation suggests an important role for stellate cells in tumor initiation.
The current study is limited to a single HPSC line isolated from a single patient with pancreatic adenocarcinoma. The effects of stromal fibroblasts from different individuals may vary, as has been shown in prostate cancer (40). Our lab has isolated HPSCs from several other patients with pancreatic adenocarcinoma but these have not yet been immortalized. Another potential limitation of this study is the two-dimensional nature of the in vitro experiments. It is increasingly recognized that two-dimensional models of cancer can differ significantly from three-dimensional models in cell morphology and behavior of tumor and stromal cells (20, 41–43). It is likely that in three-dimensional cultures, HPSCs may express genes that were not identified in our study, either alone or in coculture with tumor cells. Finally, our study focuses on cancer-associated fibroblasts as the sole stromal element interacting with tumor cells. However, many other cell types in the stroma are known to influence tumor cells, of which the best studied are the endothelial cells and pericytes that compose the vascular compartment. Other cell types in the stroma, such as immune cells (44) and adipocytes (45), also interact with tumor cells to mediate malignant behavior. Thus, a more physiologically relevant model for human pancreatic adenocarcinoma would be one that includes all stromal cell types along with tumor cells in a three-dimensional environment, a model which is the subject of active investigation in our laboratory.
In conclusion, our data provide evidence that stromal fibroblasts and tumor cells interact to promote tumor progression in pancreatic cancer. The stroma also impairs tumor cell response to chemotherapy and radiation. Moreover, in an orthotopic model of pancreatic cancer, stromal fibroblasts enhanced tumor growth, metastasis, and initiation. Our lab is currently investigating several stroma-specific factors associated with pancreatic cancer and we hope to elucidate the specific mechanisms of action of these molecules. With this information, targeting the stroma in pancreatic cancer may not only be effective in treating the primary tumor and metastases but may also play a role in prevention of tumor development as well. The identity of these stellate cell derived factors and their mechanisms of action are the subject of ongoing investigations and may lead to novel therapies directed toward the microenvironment of pancreatic cancer.
Acknowledgments
Grant support: National Pancreas Foundation, Society for Surgery of the Alimentary Tract Career Development Award, and the Topfer Fund (R.F. Hwang) and NIH grant R01DK052067-08 (C.D. Logsdon).
References
- 1.Evans DB, Abbruzzese J, Willett C. Cancer of the pancreas. In: Devita V Jr, Hellman S, Rosenberg S, editors. Cancer: principles and practice of oncology. Philadelphia: Lippincott; 2001. pp. 1126–61. [Google Scholar]
- 2.Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411:375–9. doi: 10.1038/35077241. [DOI] [PubMed] [Google Scholar]
- 3.Fidler IJ. The organ microenvironment and cancer metastasis. Differentiation. 2002;70:498–505. doi: 10.1046/j.1432-0436.2002.700904.x. [DOI] [PubMed] [Google Scholar]
- 4.De Wever O, Mareel M. Role of tissue stroma in cancer cell invasion. J Pathol. 2003;200:429–47. doi: 10.1002/path.1398. [DOI] [PubMed] [Google Scholar]
- 5.Apte MV, Haber PS, Applegate TL, et al. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut. 1998;43:128–33. doi: 10.1136/gut.43.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Apte MV, Park S, Phillips PA, et al. Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas. 2004;29:179–87. doi: 10.1097/00006676-200410000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Blomhoff R, Wake K. Perisinusoidal stellate cells of the liver: important roles in retinol metabolism and fibrosis. FASEB J. 1991;5:271–7. doi: 10.1096/fasebj.5.3.2001786. [DOI] [PubMed] [Google Scholar]
- 8.de Leeuw AM, McCarthy SP, Geerts A, Knook DL. Purified rat liver fat-storing cells in culture divide and contain collagen. Hepatology. 1984;4:392–403. doi: 10.1002/hep.1840040307. [DOI] [PubMed] [Google Scholar]
- 9.Yokoi Y, Namihisa T, Kuroda H, et al. Immunocytochemical detection of desmin in fat-storing cells (Ito cells) Hepatology. 1984;4:709–14. doi: 10.1002/hep.1840040425. [DOI] [PubMed] [Google Scholar]
- 10.Sparmann G, Hohenadl C, Tornoe J, et al. Generation and characterization of immortalized rat pancreatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2004;287:G211–9. doi: 10.1152/ajpgi.00347.2003. [DOI] [PubMed] [Google Scholar]
- 11.Phillips PA, McCarroll JA, Park S, et al. Rat pancreatic stellate cells secrete matrix metalloproteinases: implications for extracellular matrix turnover. Gut. 2003;52:275–82. doi: 10.1136/gut.52.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bachem MG, Schunemann M, Ramadani M, et al. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology. 2005;128:907–21. doi: 10.1053/j.gastro.2004.12.036. [DOI] [PubMed] [Google Scholar]
- 13.Erkan M, Kleeff J, Reiser C, et al. Preoperative acute pancreatitis in periampullary tumors: implications for surgical management. Digestion. 2007;75:165–71. doi: 10.1159/000106799. [DOI] [PubMed] [Google Scholar]
- 14.Kowolik CM, Liang S, Yu Y, Yee JK. Cre-mediated reversible immortalization of human renal proximal tubular epithelial cells. Oncogene. 2004;23:5950–7. doi: 10.1038/sj.onc.1207801. [DOI] [PubMed] [Google Scholar]
- 15.Amos K, Moore T, Arumugam T, Logsdon C, Hwang RF. Pancreatic stellate cells promote malignant potential in pancreatic cancer. 97th Annual Meeting, American Association for Cancer Research; Washington, D.C.. 2006. [Google Scholar]
- 16.Seymour AB, Hruban RH, Redston M, et al. Allelotype of pancreatic adenocarcinoma. Cancer Res. 1994;54:2761–4. [PubMed] [Google Scholar]
- 17.Arumugam T, Simeone DM, Van Golen K, Logsdon CD. S100P promotes pancreatic cancer growth, survival, and invasion. Clin Cancer Res. 2005;11:5356–64. doi: 10.1158/1078-0432.CCR-05-0092. [DOI] [PubMed] [Google Scholar]
- 18.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 19.Park CC, Bissell MJ, Barcellos-Hoff MH. The influence of the microenvironment on the malignant phenotype. Mol Med Today. 2000;6:324–9. doi: 10.1016/s1357-4310(00)01756-1. [DOI] [PubMed] [Google Scholar]
- 20.Weaver VM, Petersen OW, Wang F, et al. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137:231–45. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shekhar MP, Werdell J, Santner SJ, Pauley RJ, Tait L. Breast stroma plays a dominant regulatory role in breast epithelial growth and differentiation: implications for tumor development and progression. Cancer Res. 2001;61:1320–6. [PubMed] [Google Scholar]
- 22.Tuxhorn JA, Ayala GE, Smith MJ, Smith VC, Dang TD, Rowley DR. Reactive stroma in human prostate cancer: induction of myofibroblast phenotype and extracellular matrix remodeling. Clin Cancer Res. 2002;8:2912–23. [PubMed] [Google Scholar]
- 23.Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59:5002–11. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cunha GR, Hayward SW, Wang YZ. Role of stroma in carcinogenesis of the prostate. Differentiation. 2002;70:473–85. doi: 10.1046/j.1432-0436.2002.700902.x. [DOI] [PubMed] [Google Scholar]
- 25.Bhowmick NA, Chytil A, Plieth D, et al. TGF-β signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–51. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- 26.Tlsty TD. Stromal cells can contribute oncogenic signals. Semin Cancer Biol. 2001;11:97–104. doi: 10.1006/scbi.2000.0361. [DOI] [PubMed] [Google Scholar]
- 27.Bissell MJ, Radisky DC, Rizki A, Weaver VM, Petersen OW. The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation. 2002;70:537–46. doi: 10.1046/j.1432-0436.2002.700907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bachem MG, Schneider E, Gross H, et al. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology. 1998;115:421–32. doi: 10.1016/s0016-5085(98)70209-4. [DOI] [PubMed] [Google Scholar]
- 29.Jesnowski R, Furst D, Ringel J, et al. Immortalization of pancreatic stellate cells as an in vitro model of pancreatic fibrosis: deactivation is induced by Matrigel and N-acetylcysteine. Lab Invest. 2005;85:1276–91. doi: 10.1038/labinvest.3700329. [DOI] [PubMed] [Google Scholar]
- 30.Neesse A, Wagner M, Ellenrieder V, Bachem M, Gress TM, Buchholz M. Pancreatic stellate cells potentiate proinvasive effects of SERPINE2 expression in pancreatic cancer xenograft tumors. Pancreatology. 2007;7:380–5. doi: 10.1159/000107400. [DOI] [PubMed] [Google Scholar]
- 31.Schneiderhan W, Diaz F, Fundel M, et al. Pancreatic stellate cells are an important source of MMP-2 in human pancreatic cancer and accelerate tumor progression in a murine xenograft model and CAM assay. J Cell Sci. 2007;120:512–9. doi: 10.1242/jcs.03347. [DOI] [PubMed] [Google Scholar]
- 32.Miyamoto H, Murakami T, Tsuchida K, Sugino H, Miyake H, Tashiro S. Tumor-stroma interaction of human pancreatic cancer: acquired resistance to anticancer drugs and proliferation regulation is dependent on extracellular matrix proteins. Pancreas. 2004;28:38–44. doi: 10.1097/00006676-200401000-00006. [DOI] [PubMed] [Google Scholar]
- 33.Muerkoster S, Wegehenkel K, Arlt A, et al. Tumor stroma interactions induce chemoresistance in pancreatic ductal carcinoma cells involving increased secretion and paracrine effects of nitric oxide and interleukin-1β. Cancer Res. 2004;64:1331–7. doi: 10.1158/0008-5472.can-03-1860. [DOI] [PubMed] [Google Scholar]
- 34.Ohuchida K, Mizumoto K, Murakami M, et al. Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor-stromal interactions. Cancer Res. 2004;64:3215–22. doi: 10.1158/0008-5472.can-03-2464. [DOI] [PubMed] [Google Scholar]
- 35.Aoki H, Ohnishi H, Hama K, et al. Autocrine loop between TGF-β1 and IL-1β through Smad3- and ERK-dependent pathways in rat pancreatic stellate cells. Am J Physiol Cell Physiol. 2006;290:C1100–8. doi: 10.1152/ajpcell.00465.2005. [DOI] [PubMed] [Google Scholar]
- 36.Sato N, Maehara N, Goggins M. Gene expression profiling of tumor-stromal interactions between pancreatic cancer cells and stromal fibroblasts. Cancer Res. 2004;64:6950–6. doi: 10.1158/0008-5472.CAN-04-0677. [DOI] [PubMed] [Google Scholar]
- 37.Sato N, Fukushima N, Maehara N, et al. SPARC/osteonectin is a frequent target for aberrant methylation in pancreatic adenocarcinoma and a mediator of tumor-stromal interactions. Oncogene. 2003;22:5021–30. doi: 10.1038/sj.onc.1206807. [DOI] [PubMed] [Google Scholar]
- 38.Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–7. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 39.Calabrese C, Poppleton H, Kocak M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 40.Tuxhorn JA, McAlhany SJ, Dang TD, Ayala GE, Rowley DR. Stromal cells promote angiogenesis and growth of human prostate tumors in a differential reactive stroma (DRS) xenograft model. Cancer Res. 2002;62:3298–307. [PubMed] [Google Scholar]
- 41.Yamada KM, Cukierman E. Modeling tissue morphogenesis and cancer in 3D. Cell. 2007;130:601–10. doi: 10.1016/j.cell.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 42.Kenny HA, Krausz T, Yamada SD, Lengyel E. Use of a novel 3D culture model to elucidate the role of mesothelial cells, fibroblasts and extra-cellular matrices on adhesion and invasion of ovarian cancer cells to the omentum. Int J Cancer. 2007;121:1463–72. doi: 10.1002/ijc.22874. [DOI] [PubMed] [Google Scholar]
- 43.Nelson CM, Bissell MJ. Modeling dynamic reciprocity: engineering three-dimensional culture models of breast architecture, function, and neoplastic transformation. Semin Cancer Biol. 2005;15:342–52. doi: 10.1016/j.semcancer.2005.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 45.Iyengar P, Espina V, Williams TW, et al. Adipocyte-derived collagen VI affects early mammary tumor progression in vivo, demonstrating a critical interaction in the tumor/stroma microenvironment. J Clin Invest. 2005;115:1163–76. doi: 10.1172/JCI23424. [DOI] [PMC free article] [PubMed] [Google Scholar]