

Abstract
Late Na+ current (INaL) is a major component of the action potential plateau in human and canine myocardium. Since INaL is increased in heart failure and ischemia, it represents a novel potential target for cardioprotection. However, the molecular identity of INaL remains unclear. We tested the hypothesis that the cardiac Na+ channel isoform (Nav1.5) is a major contributor to INaL in adult dog ventricular cardiomyocytes (VCs). Cultured VCs were exposed to an antisense morpholino-based oligonucleotide (Nav1.5 asOligo) targeting the region around the start codon of Nav1.5 mRNA or a control nonsense oligonucleotide (nsOligo). Densities of both transient Na+ current (INaT) and INaL (both in pA/pF) were monitored by whole cell patch clamp. In HEK293 cells expressing Nav1.5 or Nav1.2, Nav1.5 asOligo specifically silenced functional expression of Nav1.5 (up to 60% of the initial INaT) but not Nav1.2. In both nsOligo-treated controls and untreated VCs, INaT and INaL remained unchanged for up to 5 days. However, both INaT and INaL decreased exponentially with similar time courses (τ = 46 and 56 h, respectively) after VCs were treated with Nav1.5 asOligo without changes in 1) decay kinetics, 2) steady-state activation and inactivation, and 3) the ratio of INaL to INaT. Four days after exposure to Nav1.5 asOligo, INaT and INaL amounted to 68 ± 6% (mean ± SE; n = 20, P < 0.01) and 60 ± 7% (n = 11, P < 0.018) of those in VCs treated by nsOligo, respectively. We conclude that in adult dog heart Nav1.5 sodium channels have a “functional half-life” of ∼35 h (0.69τ) and make a major contribution to INaL.
Keywords: sodium channel functional half-life, antisense inhibition, patch clamp
slowly inactivating late sodium current (INaL) has been identified in ventricular cardiomyocytes (VCs) of a large variety of mammalian species, including humans (20, 22, 23), dogs (23, 41, 44, 51), guinea pigs (1, 15, 34), rabbits (48), and rats (3). In failing hearts INaL was found to be increased and was implicated in repolarization abnormalities as well as Na+-related Ca2+ overload (for review, see Ref. 24). Furthermore, augmented INaL is also reportedly involved in Na+ and Ca2+ overload in ischemic heart, and inhibition of Na+-related Ca2+ overload was suggested as a new cytoprotective principle (42, 46). Accordingly, INaL has recently resurged as a plausible target for cardioprotection (22, 24, 30). Despite its great fundamental and clinical importance, the molecular identity of INaL is still poorly understood.
Both tetrodotoxin (TTX)-sensitive (neuronal type) and TTX-resistant (cardiac type) Na+ channel (NaCh) isoforms have been suggested to contribute INaL. The idea that the neuronal-type NaCh may contribute to the action potential (AP) plateau was first brought up in 1979 (5). It has been shown that TTX shortened the AP in Purkinje fibers of dog heart in doses two orders of magnitude lower than needed to reduce the upstroke velocity (5). More recent studies claimed that at least some part of the late/persistent Na+ current is highly TTX and saxitoxin (STX) sensitive in rat cardiomyocytes (13, 33). Indeed, different transcripts of the main α-subunits of highly TTX-sensitive neuronal isoforms (Nav1.1, 1.3, 1.6) have recently been detected in mouse (18) and dog (11) heart (Nav1.1, 1.2, 1.3, 1.6). These neuronal NaCh isoforms were responsible for 10–20% of the peak transient Na+ current (INaT) responsible for the AP upstroke velocity in dog myocardial and Purkinje cells, respectively (10, 11). On the other hand, previous pharmacological studies of INaL suggested that this current is likely produced by the cardiac Nav1.5 NaCh isoform in human and dog cardiomyocytes, because it is blocked by cadmium ions and has relatively low sensitivities to TTX and STX (20, 24, 44). Furthermore, four splice variants of SCN5A gene encoding the cardiac-specific Nav1.5 with different biophysical properties have been reported in humans (40), indicating that the question about the exact molecular identity of INaL is even more complex than just separation of contributions from TTX-sensitive and -resistant NaCh isoforms.
The present study approaches this problem by direct molecular targeting of the mRNA encoding Nav1.5. More specifically, we studied both INaL molecular identity and the functional channel protein turnover, using antisense oligonucleotide (asOligo) inhibition of the mRNA encoding Nav1.5 in adult canine VCs. We found that Nav1.5 is a major INaL contributor in these cells.
MATERIALS AND METHODS
Cardiomyocyte isolation and culture.
This study conforms to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health and was approved by the Institutional Animal Care and Use Committee of the Henry Ford Health System. The five mongrel dogs were used for training purposes by the Department of Bioresources and served as heart donors for the present study. VCs were enzymatically isolated from slices of the apical third of the left ventricle from the midmyocardium as previously described (20). The yield of viable, Ca2+-tolerant, rod-shaped cells varied from 20% to 80%. The suspension of VCs was cultured in serum-free medium (M199, with Earle's salts, 25 mM HEPES, and bicarbonate, without l-glutamine) as previously described (21). The culturing M199 was supplemented with 2 mM carnitine, 5 mM creatine, 5 mM taurine, 2 g/l albumin (bovine, fraction V), 0.1 μM insulin, 100 U/ml penicillin, 100 μg/ml streptomycin, and 25 μg/ml gentamicin [CCT medium (47)]. The cells were incubated at 37°C in a humidified atmosphere of 95% O2-5% CO2. Under these conditions ∼80% of cells survived up to 5 days of incubation, similar to the survival reported previously for adult rabbit cardiomyocytes (28). During this time VCs undergo some changes in cell shape and morphology manifested in slightly rounded edges of the cultured cells. Nonsense oligonucleotide (nsOligo) or Nav1.5 asOligo did not further affect morphology of VCs (see Fig. 2A).
Fig. 2.

Cell culture model and intracellular delivery of oligonucleotides. A: only slight changes in shape (rounded edges) of dog ventricular cardiomyocytes (VCs) were evident after 5 days of culture. 10 μm/div, distance between calibration dots. B: fluorescein-tagged oligo uptake by cardiomyocytes was monitored by confocal microscopy. Confocal images of live cardiomyocytes loaded with fluorescein-tagged oligo are shown. Optical slices were 0.5 μm (Zeiss Axiovert 100, Bio-Rad MRC 1024, excitation/emission wavelength 488/522 nm).
Heterologous expression of Nav1.5 and Nav1.2.
In this study we also used an HEK293 cell line (αHEK) stably expressing Nav1.5 or Nav1.2 as previously described (43). The wild-type human heart Nav1.5 clone (formerly hH1a) was kindly provided by Drs. H. A. Hartmann and A. M. Brown (Baylor College of Medicine, Houston, TX) (9).
Antisense oligonucleotide: design and delivery.
We chose morpholino-based 25-mer oligos over phosphorothioate (sugar based) oligos because of their superior properties for our experimental approach (38). Design of oligo sequences was based on high homology between mRNAs encoding Nav1.5 of human (8) and dog NaCh (GenBank AF017428 AY126477, AJ555547, AY126477). The Nav1.5 asOligo was designed to be around the start codon. The asOligo for Nav1.5 used in this study (5′-ccgaggtaataggaagtttgccatc; GenBank M77235) was aligned (BLAST 2 software) with gene transcripts encoding known human voltage-activated NaCh α-subunit isoforms (Navs): neuronal-type Nav1.7 (GenBank X82835), skeletal muscle Nav1.4 (GenBank M81758), brain-type 1 Nav1.1 (GenBank X65362), and type 2 Nav1.2 (GenBank M94055). Furthermore, we tested atypical Navs: Nav2.1 (GenBank M91556) as well as mouse Nav2.3 (GenBank L36179) and Norway rat SCL-11 (GenBank Y09164). No significant similarity was found. We used the nsOligo 5′-cctcttacctcagttacaatttaata as a control. We also did not find any significant similarity of the nsOligo with known α- and β-subunits of NaCh.
Given the well-known difficulty of using conventional nonviral methods to introduce exogenous DNA into terminally differentiated cells like cardiac myocytes (36), we chose a special delivery (SD) system based on ethoxylated polyethylenimine (EPEI) (29). The rationale of the SD system is that nonionic oligo is paired to the complementary DNA. The DNA is then bound electrostatically to weakly basic EPEI. This oligo-DNA-EPEI complex is efficiently endocytosed (29). The effect of asOligos (2.55 and 0.98 μM) on NaCh expression was tested both in stably transfected αHEK cells and VCs, respectively. VCs were cultured for 24 h before oligo delivery. Both αHEK cells and VCs were exposed to oligo-DNA-EPEI for 3 h and then washed out with CCT medium. About 98% of VCs cells were positive for the oligo, as monitored by fluorescence of the fluorescein-tagged oligo (see Fig. 2B). Other conventional gene transfection delivery systems were ineffective for the adult VCs, and limitations of our approach are discussed in Study limitations.
Patch-clamp technique and data analysis.
INa was recorded with a conventional whole cell patch-clamp technique (pCLAMP9, Axopatch 200A patch-clamp amplifier, Axon Instruments). The resistance of the borosilicate glass patch pipettes was 1.2–2.1 MΩ for HEK cells and 0.6–0.8 MΩ for VCs. The currents were low-pass filtered at 5 kHz and digitized at a sampling rate of 20 kHz. The quality of the voltage clamp was controlled in each cell as previously described (20, 44). Experiments were performed at 22–24°C.
INaL was elicited by a 2-s membrane depolarization to various potentials from a holding potential (Vh) of −130 mV applied with a pacing frequency of 0.1 Hz (see Table 1 for solutions). The “zero” current was determined after TTX (25 μM) application (20). The amplitude of INaL was determined from the averaged current measured at 200–220 ms after the onset depolarization to avoid contamination with INaT (43). INaT was measured at a symmetrical Na+ concentration of 5 mM (Table 1). All measurements were performed 10–25 min after the membrane rupture to allow for complete cell dialysis with intracellular recording solutions (26, 31, 32). Currents were normalized to the membrane electric capacitance (Cm) measured by a voltage ramp pulse with a slope (dV/dt) of −10 V/s from −80 mV to −100 mV (21). The fine structure of the INa time course was approximated by a double-exponential fit to INaT or INaL decay (19, 25):
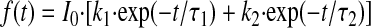 |
(1) |
where τ1 and τ2 are the time constants and I0 is the amplitude 0.2 ms after peak of INaT or the instant value of INaL at 40 ms after membrane depolarization; k1 and k2 are the contributions of each exponent (k1 + k2 = 1), respectively. Five to fifteen experimental traces were averaged to improve the quality of analysis for INaL. The decay of INaL was evaluated at membrane potential (Vm) = −30 mV (20), and τ1 and τ2 correspond to the burst (BM) and late scattered (LSM) modes of the late NaCh gating, respectively (19, 25).
Table 1.
Extracellular (bath) and intracellular (pipette) solutions used in study
| Pipette Solution INaT, INaL |
Bath Solutions |
||
|---|---|---|---|
| INaT for VCs | INaL for VCs, INaT for αHEK | ||
| NaCl | 5 | 5 | 140 |
| KCl | 5.4 | ||
| CsCl | 133 | 133 | 5.4 |
| CaCl2 | 1.8 | 1.8 | |
| MgCl2 | 2 | 2 | |
| MgATP | 2 | ||
| TEA | 20 | ||
| Nifedipine | 0.002 | 0.002 | |
| EGTA | 10 | ||
| HEPES | 5 | 5 | 5 |
| pH | 7.3 CsOH | 7.3 NaOH | 7.3 NaOH |
Concentrations are expressed in mM. INaT, peak transient Na+ current; INaL, late Na+ current; VCs, ventricular cardiomyocytes; αHEK, HEK293 cell line stably expressing Nav1.5 or Nav1.2; TEA, tetraethylammonium.
The steady-state availability (or inactivation, SSI) parameters [the midpoint (V½A) and the slope (kA) of the relationship] were measured by a standard double-pulse protocol with the 2-s-duration prepulse (Vp) ranging from −130 mV to −40 mV. The data points of INa normalized to INa measured at −30 and −130 mV prepulse were fitted to a Boltzmann function A(Vp):
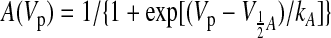 |
(2) |
The data points of the normalized current in the current-voltage relationships were fitted (by Origin 7.0 software, Microcal Software) to the following function (21):
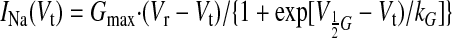 |
(3) |
where Gmax is a normalized maximum Na+ conductance, Vr is a reversal potential, Vt is testing voltage, and V½G and kG are the midpoint and the slope of the respective Boltzmann function underlying the NaCh steady-state activation (SSA). The time course of both INaT and INaL reduction by the asOligo was evaluated by the best single-exponential fit to the data points:
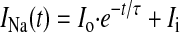 |
(4) |
where τ is the time constant and Io and Ii are the amplitudes and asymptotic levels, respectively. The functional half-life of NaCh protein was assessed as t1/2 = 0.69τ.
Statistical analysis.
Data are reported as means ± SE, with n representing the number of cells. Multiple comparisons between treatment groups were made with one-way analysis of variance (ANOVA) followed by Bonferroni's post hoc test. The significance of SSA and SSI changes was evaluated with an F-test (StatMost, Data-Most, Salt Lake City, UT) for tabulated values predicted by the model (Eqs. 2 and 3) at a confidence level of 0.95. Differences for both experimental data and model predictions were considered statistically significant at P < 0.05.
Chemicals.
Enzymes protease type XXIV and hyaluronidase type IV-S were purchased from Sigma (St. Louis, MO) and collagenase type II (291 U/mg) from Worthington (Freehold, NJ). asOligo and nsOligo as well as the EPEI-based SD system were purchased from Gene Tools (Philomath, OR; www.gene-tools.com). All other chemicals were purchased from Sigma.
RESULTS
Probing efficacy of Nav1.5 antisense oligonucleotide in a heterologous expression system.
In αHEK cells we tested the efficacy of our Nav1.5 asOligo to reduce functional expression of Nav1.5 but not Nav1.2. Treatment with Nav1.5 asOligo caused a significant decrease of INaT density without changes in midpoint potential of activation and inactivation (Fig. 1, A–C; Table 2) or the decay kinetics (Fig. 1D). Analysis of the time course of INaT reduction by Nav1.5 asOligo revealed an exponential decrease (Eq. 4) of the INaT density with a time constant τ of 52 h (Fig. 1E) and Ii ∼27% of the initial INaT. The t½ for NaCh evaluated as 0.69τ was found to be 36 h. Control cells loaded with nsOligo did not show any significant changes in INaT over this time (Fig. 1E), indicating stable expression of NaCh. We performed additional tests with the HEK cells stably transfected with the brain NaCh isoform Nav1.2 to demonstrate specificity of our Nav1.5 asOligo. Indeed, Nav1.5 asOligo did not affect the maximum Na+ current density [14.2 ± 3.1 (n = 16) vs. 15.1 ± 2.5 (n = 13) pA/pF, nsOligo vs. asOligo, respectively, at Vm = −10 mV], SSI and SSA (Table 2). Thus these control experiments in αHEK cells proved the effectiveness of the Nav1.5 asOligo to selectively silence (by ∼70%) functional expression of Nav1.5.
Fig. 1.

Potency of antisense oligonucleotide (asOligo) to silence Nav1.5 expression in HEK293 cell line stably expressing Nav1.5. A: representative raw traces of transient Na+ current (INaT) were recorded at different membrane potentials (Vm) in control conditions [nonsense oligonucleotide (nsOligo)] and in the presence of Nav1.5 asOligo. B: average data for peak INaT-voltage (V) relationship 5 days after nsOligo or Nav1.5 asOligo delivery. Solid lines show theoretical I-V curves fitted in accordance with Eq. 3. asOligo reduced the maximum INaT conductance (Gmax) from 12.5 × 10−3 to 5.1 × 10−3 nS/pF (P < 0.05, F-test) without changes in steady-state activation (SSA) parameters (see Table 2). *Statistically significant (P < 0.05) differences in data points. C: average experimental data of steady-state inactivation (SSI) along with their theoretical Boltzmann fits (Eq. 2, solid and dashed lines). D: asOligo did not affect INaT decay time course evaluated by the double-exponential fit (Eq. 1). Shown are data points of 2 time constants (τfast, τslow) at different Vm, pooled from 9 or 10 cells. E: INaT decreases in exponential manner in response to asOligo delivery. Shown are data points along with the linear regression (solid line for nsOligo) and a single-exponential fit (asOligo, Eq. 4). Exponential time constant (τ) value is indicated. Data points in B–E are means ± SE. A and C, insets: voltage-clamp protocols. Vh, holding potential; Vp, prepulse. Statistically significant difference (P < 0.05) in B and E was evaluated by ANOVA followed by Bonferroni's post hoc test. Detailed statistics for all SSA and SSI parameters of the theoretical fits shown in B and C are presented in Table 2. Equations 1–4 are given in materials and methods.
Table 2.
Steady-state activation and inactivation for INa in heterologously expressed NaCh isoforms and in dog ventricular cardiomyocytes
| Conditions |
SSI Parameters |
SSA Parameters
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| V1/2A, mV | kA, mV | n | V1/2G, mV | kG, mV | n | |||||||
| Heterologously expressed Nav1.2, INaT | ||||||||||||
| nsOligo, 5 days | −70.0±2.6 | −7.7±0.9 | 7 | −21.4±1.5 | 5.1±0.8 | 5 | ||||||
| asOligo, 5 days | −68.9±2.1 | −8.0±0.9 | 7 | −24.1±1.1 | 4.5±0.3 | 5 | ||||||
| Heterologously expressed Nav1.5, INaT | ||||||||||||
| nsOligo, 5 days | −81.4±1.4 | −5.8±0.4 | 9 | −39.7±1.2 | 5.1±0.5 | 7 | ||||||
| asOligo, 5 days | −82.9±0.8 | −5.9±0.3 | 8 | −41.7±1.2 | 5.1±0.3 | 9 | ||||||
| Dog ventricular cardiomyocytes, INaT | ||||||||||||
| Freshly isolated | −80.6±0.7 | −5.7±0.3 | 8 | −38.8±1.5 | 5.1±0.3 | 9 | ||||||
| 5 Days in culture | −81.7±0.9 | −6.2±0.4 | 7 | −36.3±1.5 | 6.3±0.2 | 9 | ||||||
| nsOligo, 5 days | −82.9±0.9 | −6.2±0.2 | 11 | −38.3±1.2 | 5.9±0.2 | 13 | ||||||
| asOligo, 5 days | −82.8±1.2 | −6.9±0.6 | 7 | −37.9±1.1 | 6.3±0.3 | 11 | ||||||
| Dog ventricular cardiomyocytes, INaL | ||||||||||||
| Freshly isolated | −80.0363±0.9 | −6.3±0.5 | 19 | −39.4±0.6 | 7.1±0.1 | 6 | ||||||
| 5 Days in culture | −81.2822± | −5.9±0.6 | 8 | −39.5±0.7 | 7.3±0.5 | 6 | ||||||
| nsOligo, 5 days | −82.0061±1.2 | −5.0±0.8 | 8 | −36.8±1.2 | 6.3±0.3 | 11 | ||||||
| asOligo, 5 days | −80.3534± | −5.1±0.7 | 8 | −37.5±0.7 | 6.4±0.9 | 9 | ||||||
Data are means ± SE; n is cell number. Steady-state inactivation (SSI) and activation (SSA) parameters were obtained from INa data fit to Eqs. 2 and 3 (materials and methods), respectively. We did not find statistically significant differences between parameters within the experimental groups as evaluated by ANOVA followed by Bonferroni's post hoc test. nsOligo, nonsense oligonucleotide; asOligo, antisense oligonucleotide; V1/2A and kA, midpoint and slope of Boltzmann function A curve; V1/2G and kG, midpoint and slope of conductance curve.
INaL and INaT in cardiomyocytes in culture.
We first addressed how culturing conditions may affect cell morphology and levels of NaCh expression. VCs underwent well-known changes in cell shape (28), mainly observed as slightly rounded cell edges during the first 5 days of culture (Fig. 2A). Delivery of nsOligo or Nav1.5 asOligo did not further affect the appearance of VCs (Fig. 2A). Moreover, these culturing conditions did not affect NaCh expression as judged from INaT and INaL over the time in culture (Figs. 3 and 4, Table 2). Thus these control experiments of the time course of Na+ current density confirmed the usefulness of cultured adult dog cardiomyocytes for experiments to further address the molecular identity of INaL.
Fig. 3.

INaT remains unchanged in cultured adult dog cardiomyocytes. A: representative raw current trace recorded in freshly isolated VCs (left) and after 5 days in culture (right) at different Vm. B: peak INaT-V relationship obtained in freshly isolated cells and cells cultured for 5 days. Theoretical curves fit to I-V (Eq. 3, solid and dashed lines) were not statistically different. C: SSI data points along with the fit to a Boltzmann function (Eq. 2, solid and dashed lines, respectively). No statistically significant difference was found when SSI curves were compared. D: decay time constants of INaT (double-exponential fit, Eq. 1) were evaluated at the different Vm. E: maximum density of INaT remained unchanged in these conditions. Data in B–D are means ± SE and were pooled from 5–8 cells. Detailed statistics for all SSA and SSI parameters of the theoretical fits shown in B and C are presented in Table 2. B and C, insets: voltage-clamp protocols.
Fig. 4.

Cultured adult dog cardiomyocytes represent a useful model to study INaL. A: data points of INaL-V relationships for freshly isolated and cultured cells along with their theoretical fits (Eq. 3, solid and dashed lines). Inset: typical examples of raw INaL traces recorded in fresh and cultured cells are shown superimposed. There was no statistical difference between the data points or theoretical fit of the I-V curves. B and C: density and SSI of INaL remain unchanged in cultured cells. C: data points of SSI are shown along with their theoretical fits (Eq. 2, solid and dashed lines). Inset: our voltage-clamp protocol. D: decay time course of INaL was evaluated by the 2-exponential fit (Eq. 1) and remained unchanged in the cultured cells. All data are means ± SE. Detailed statistics for all SSA and SSI parameters of the theoretical fits shown in A and C are presented in Table 2.
Effects of antisense oligonucleotide on INaT and INaL in cultured cardiomyocytes.
We tested the effects of asOligo in cultured VCs, assuming high homology of mRNAs encoding cardiac human and canine NaCh. Indeed, alignment of Nav1.5 with partial sequence of the dog heart NaCh (GenBank AF017428; Ref. 49) revealed 92% identity with cardiac human Nav1.5 NaCh. Visualization of Nav1.5 asOligo delivery was based on fluorescence of fluorescein-tagged oligo. Typical examples of confocal images of cardiomyocytes loaded with the nsOligo or Nav1.5 asOligo are shown in Fig. 2B. After 3 h of incubation, we found diffused distribution of the OLIGO in VC cytoplasm. Using fluorescence microscopy (not shown), we estimated that 98% of VCs take up oligos. However, it is important to note that treatment with oligos was toxic to the cells, and <50% of treated cells survived.
Voltage-clamp experiments showed that Nav1.5 asOligo substantially reduced density of INaT in contrast to that of the control nsOligo-treated VCs (Fig. 5, A and B). Importantly, the decay kinetics (Fig. 5D) and SSA or SSI (Fig. 5, B and C) remained unchanged. Data on the SSA and SSI parameters are summarized in Table 2. Similar to αHEK cells, in VCs treated with asOligo INaT decreased exponentially, with τ = 46 h and Ii = 62.8% of the initial INaT (Fig. 5E). The evaluated functional half-life was t½ = 32 h. There was moderate (∼1%/day) and negligible change of INaT in the control pool of cells treated by nsOligo (Fig. 5E). The data confirmed efficacy of the asOligo to reduce Nav1.5 expression in adult dog cardiomyocytes.
Fig. 5.

asOligo effectively knocks down Nav1.5 expression in adult dog cardiomyocytes. A: asOligo caused decrease in peak INaT. Representative raw traces of INaT were recorded at different membrane potentials in control conditions (nsOligo, right) and with asOligo (left). B: peak INaT-V relationship obtained in cells treated with nsOligo (control) and asOligo. *Statistically significant differences in data points (P < 0.05) compared at different Vm. Solid (nsOligo) and dashed (asOligo) lines show theoretical curves of I-V (Eq. 3) fitted to data points. asOligo significantly reduced Gmax from 1.39 to 1.01 pS/pF (P < 0.05, F-test). C: asOligo did not affect the SSI of INaT. Shown are data points of SSI along with their theoretical fits (Eq. 2; solid lines nsOligo and dashed lines asOligo; see Table 2 for SSI parameters). D: asOligo did not affect the decay time course of INaT. The two time constants, τfast and τslow (double-exponential fit, Eq. 1), vs. membrane and the relative contribution of τslow (kslow) are given at top and bottom, respectively. E: time course of INaT decrease in response to Nav1.5 knockdown by asOligo. Data points are shown along with the linear regression (solid line for nsOligo) and exponential fit (asOligo). Time constant is indicated; n is the number of cardiomyocytes. The initial level of INaT (100%) was 47.3 ± 9 pA/pF (n = 25). Statistically significant difference (P) in A and E was evaluated by ANOVA followed by Bonferroni's post hoc test. Detailed statistics for all SSA and SSI parameters of the theoretical fits shown in A and C are presented in Table 2. Bars in B and D and points in A and C represent means ± SE.
asOligo affected INaL in a similar way as observed for INaT. Indeed, Nav1.5 asOligo resulted in a substantial and statistically significant decrease of INaL density (Fig. 6A) without changes in the ratio of INaL to INaT (Fig. 6B), decay time course (Fig. 6C), and SSA or SSI (Table 2). The data on the time course of the Nav1.5 asOligo effect on INaL are summarized in Fig. 6D. In VCs treated with Nav1.5 asOligo but not with nsOligo, the amplitude of INaL decreased over time in culture and are well described by a single-exponential decay (Eq. 4) with τ = 56 h and an Ii ∼58.3% of the initial INaL. The estimated functional half-life t1/2 for the pool of NaCh underlying INaL was 39 h. Thus the time course and degree of Nav1.5 asOligo-induced density decrease in VCs were similar for both INaT and INaL (compare Figs. 5E and 6D).
Fig. 6.

Molecular identity of INaL in dog ventricular cardiomyocytes assessed by asOligo specific to mRNA encoding Nav1.5. A: INaL density is statistically significantly reduced by the asOligo (P < 0.001). Inset: superimposed representative raw INaL traces recorded in the presence of nsOligo and asOligo, respectively. B: knockdown of SCN5A gene by asOligo leads to the parallel decrease of both INaT and INaL, as INaL-to-INaT ratio remains unchanged. C: asOligo does not affect the fine structure of INaL decay evaluated by the 2-exponential fit (Eq. 1). D: statistical analysis of the time course of INaL density changes in response to asOligo. Data points are shown along with the linear regression (solid line for nsOligo) and the exponential fit (asOligo). The time constant is indicated; the initial level (100%) of INaL current density was 0.223 ± 0.025 pA/pF (n = 18). Statistically significant difference (P) in A and E was evaluated by ANOVA followed by Bonferroni's post hoc test. Detailed statistics for all SSA and SSI parameters of the theoretical fits shown in A and C are presented in Table 2. Bars in A–C and points in D represent means ± SE.
Theoretical evaluation of INaL that is contributed by Nav1.5.
Approximation of the components of the entire INaL time course by a double-exponential function (Eq. 1) allows interpretation of the whole cell data in terms of gating modes of late NaCh openings (19). According to a numerical model (25) of Na+ channel gating based on the single-channel data of late openings, the entire INaL time course is well described as a sum of two exponentials: BM generates the fast, exponentially decaying INaL component (IBM), whereas LSM generates a relatively slow INaL decaying component (ILSM):
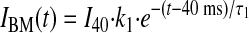 |
(5) |
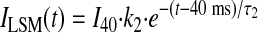 |
(6) |
where I40, k1, k2, τ1, and τ2 are from Eq. 1 and correspond to the whole cell currents (IBM and ILSM) produced by BM and LSM, respectively. The results of the theoretical approximation of the idealized, average late currents that are generated by these modes are shown in Fig. 7. The analysis of INaL in cells treated with Nav1.5 asOligo and nsOligo thus revealed that both BM (Fig. 7A) and LSM (Fig. 7B) of the late NaCh openings are produced by the Nav1.5 channel. The total average INaL in adult dog cardiomyocytes determined by Nav1.5 asOligo is shown in Fig. 7C.
Fig. 7.

Theoretical evaluation of the fine structure of INaL determined by Nav1.5 in adult dog cardiomyocytes. Parameters (time constants and densities) for the idealized, 2-exponential decay time course of INaL were assigned from the averaged experimental data measured in cultured cardiomyocytes treated with nsOligo (density 0.276 pA/pF at 200 ms, τ1 = 48.6, τ2 = 502 ms, k2 = 0.268) or asOligo (0.132 pA/pF, τ1 = 38.8, τ2 = 497 ms, k2 = 0.305). Currents generated by the burst (IBM in A) and late scattered openings (ILSM in B) were obtained from Eqs. 5 and 6 (results), respectively, and the total INaL (C) was obtained as the sum of these two. The difference currents between nsOligo and asOligo-treated cells are shown as shaded areas.
DISCUSSION
Result summary, conclusions, and possible importance.
We used Nav1.5 asOligos to determine molecular identity of the INaL in adult dog VCs. We demonstrated that culturing of these cells (up to 5 days) does not significantly alter either INaT or INaL. This fact allowed the use of the cell culture as a model for the experiments with gene expression knockdown by the Nav1.5 asOligo. The asOligo that was designed with the sequence of human mRNA encoding cardiac Nav1.5 was also effective in dog cardiomyocytes. Knockdown of cardiac Nav1.5 in dog VCs resulted in exponential simultaneous decrease of INaT and INaL (to ∼60% of their initial levels after 100 h; Figs. 5E and 6D) without changes in INaL-to-INaT ratio, decay kinetics (Figs. 5D and 6D), and the parameters of SSA and SSI (Table 2). The t½ for INaL was similar to that for INaT (32–36 h). Our data suggest that the cardiac Nav1.5 isoform is the major contributor to INaL in cardiomyocytes of the adult dog, and we believe that this finding is important in the context of recent interest in INaL as a therapeutic target in heart failure and ischemia (4, 24, 30). It is also important to note that our previous studies of INaL (see Ref. 24 for review) and the fact that our asOligo was highly effective for human and dog Nav1.5 (this study) indicate that functional and molecular properties of cardiac NaCh (both early and late currents) are very close in humans and dogs so that canine models of heart failure or ischemia are helpful in studying the roles of INaT and INaL in human heart disease.
A new paradigm in heart failure is that INaL is increased while INaT is reportedly decreased (45, 50). With this in mind, further blockade of INaT is proarrhythmic as it worsens existing conduction problems of the failing myocardium. The results of the present study showing that the same NaCh likely produces INaT and INaL indicate that specific pharmacological targeting of INaL will be challenging because many drugs will block both INaT and INaL. One approach to solving the problem is based on properties of specific drugs to change specific gating properties of NaCh (see Ref. 23 for review). For example, amiodarone and the new antianginal drug ranolazine interact with specific gating states of NaCh and thereby predominantly block INaL vs. INaT (22, 41). Another approach is based on indirect targeting of INaL via its multiple known modulator factors of NaCh environment [affecting late NaCh gating modes (25, 43)] such as cytoskeleton, phospholipids, auxiliary subunits, and Ca2+ (see Refs. 19, 24 for review). In any case, an established molecular identity of INaL is critical to approach the unresolved problem.
Functional half-life: comparison with other plasmalemmal proteins.
asOligos have been used previously to study turnover of plasmalemmal proteins, such as Na+/Ca2+ exchanger (NCX). For example, a relatively short t1/2 (<12 h) for the NCX has been reported in some studies that utilized relatively high asOligo concentrations (3–10 μM), resulting in inhibition of NCX function by from 30% (2) up to 80% (16). The apparent fast turnover of NCX at higher doses of oligo could be due to either side effects of the oligo or the existence of a fraction of NCX1 protein with a relatively fast turnover (6). Alternatively, with lower doses of asOligo (0.5–2.0 μM), slower NCX protein turnover rates (t½ > 30 h) were reported in neonatal and adult cardiomyocytes (7, 37), which is in line with t½ of 20–40 h for several other plasmalemmal transport proteins (14, 39) and NaCh (this study and Ref. 35).
Cultured adult cardiomyocytes as a model to study NaCh function.
Primary cultures of cardiomyocytes are a useful model because they produce a homogeneous population of terminally differentiated cells free of extracellular matrix and neurohumoral factors. This is an attractive model for studies that utilize electrophysiological (including patch clamp) and molecular biology approaches. On the other hand, numerous studies have shown that over the culturing period adult cardiomyocytes undergo changes in their morphology and protein expression levels including those of ion channels (see Ref. 28 for review). Accordingly, results gathered over prolonged periods (≥6 days) in these cells should be treated with caution. These morphological and functional changes can be minimized by the design of an appropriate culturing medium (so-called CCT medium used in the present study) that does not contain serum and glutamate and is supplemented with creatine, l-carnitine, and taurine. In CCT medium the survival rate increased up to 14 days for rod-shaped myocytes. The loss of some contractile and cytoskeleton proteins may be linked to the absence of an appropriate humoral factor (47). Reduction of contractile function over the period of culture was linked to the atrophy of contractile proteins (in rat and rabbit but not in feline cardiomyocytes) and a reduced number of T tubules (27). Fairly rapid internalization of the intercalated disks (within 24 h) was reported in rat VCs (12). The present study clearly demonstrates that dog VCs in culture represent a relatively stable and useful model to study cardiac NaCh (using electrophysiological and/or molecular biology techniques), because of the stable intrinsic functional expression of these channels during a relatively long period of time (up to 100 h of cell culture).
Molecular identity of INaL.
Culture-specific changes of cells can affect ion channel expression, particularly for proteins that are localized within T tubules and intercalated disks. Indeed, it has been shown that brain-type isoforms Nav1.1, Nav1.3, and Nav1.6 are located in T tubules and Nav1.5 at the intercalated disks of rat VCs (17). In dog VCs Nav1.1 was localized at the intercalated disks and Nav1.2 was present at the Z lines (11). Unlike other ion channels (see Ref. 28 for review), sodium ion channels underlying both INaT and INaL did not change during the 5 days of culture (Figs. 3 and 4). Simultaneous inhibition of both INaT and INaL in response to the Nav1.5 asOligo knockdown (Figs. 5 and 6) supports the idea that Nav1.5 generates both INaT and INaL. This idea is also supported by previous pharmacological studies using NaCh-specific toxins, cadmium ions, and other experimental conditions to distinguish between NaCh isoforms (20, 23). Furthermore, the present study demonstrates that Nav1.5 is responsible for both burst and late scattered gating mode components determining the fine structure of the total INaL time course (Fig. 7).
Study limitations.
The delivery of oligos to VCs has limitations that are common to all antisense methods [discussed in detail elsewhere (36)]. A major common limitation is a significant reduction in the number of viable VCs, although surviving cells demonstrate inclusion of the oligo (in ∼98% of cells in our experiments). The low number of surviving cells was suitable for our patch-clamp experiments but makes biochemical analysis (i.e., Western blotting) much more challenging. While we conclude that Nav1.5 is the major contributor to INaL, a possible role for neuronal isoforms especially in disease states could not be completely excluded based on the data presented in this study.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Grants HL-53819 and HL-074238 (A. Undrovinas) and RO1-HL-65661 (J. W. Kyle) and by American Heart Association Grant-in-Aid 0350472Z (A. Undrovinas).
Acknowledgments
Present address of V. A. Maltsev: Gerontology Research Center, National Institute on Aging, NIH, 560 Nathan Shock Dr., Baltimore, MD 21224.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Belardinelli L, Shryock JC, Fraser H. Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart 92: iv6–iv14, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bridge JH, Smolley J, Spitzer KW, Chin TK. Voltage dependence of sodium-calcium exchange and the control of calcium extrusion in the heart. Ann NY Acad Sci 639: 34–47, 1991. [DOI] [PubMed] [Google Scholar]
- 3.Chattou S, Coulombe A, Diacono J, Le Grand B, John G, Feuvray D. Slowly inactivating component of sodium current in ventricular myocytes is decreased by diabetes and partially inhibited by known Na+-H+ exchange blockers. J Mol Cell Cardiol 32: 1181–1192, 2000. [DOI] [PubMed] [Google Scholar]
- 4.Conti CR Inhibition of sodium-dependent calcium overload to treat myocardial ischemia. Clin Cardiol 29: 141–143, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coraboeuf E, Deroubaix E, Coulombe A. Effect of tetrodotoxin on action potentials of the conducting system in the dog heart. Am J Physiol Heart Circ Physiol 236: H561–H567, 1979. [DOI] [PubMed] [Google Scholar]
- 6.Egger M, Porzig H, Niggli E, Schwaller B. Rapid turnover of the “functional” Na+-Ca2+ exchanger in cardiac myocytes revealed by an antisense oligodeoxynucleotide approach. Cell Calcium 37: 233–243, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Eigel BN, Hadley RW. Antisense inhibition of Na+/Ca2+ exchange during anoxia/reoxygenation in ventricular myocytes. Am J Physiol Heart Circ Physiol 281: H2184–H2190, 2001. [DOI] [PubMed] [Google Scholar]
- 8.Gellens ME, George AL Jr, Chen LQ, Chahine M, Horn R, Barchi RL, Kallen RG. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc Natl Acad Sci USA 89: 554–558, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hartmann HA, Tiedeman AA, Chen SF, Brown AM, Kirsch GE. Effects of III–IV linker mutations on human heart Na+ channel inactivation gating. Circ Res 75: 114–122, 1994. [DOI] [PubMed] [Google Scholar]
- 10.Haufe V, Camacho JA, Dumaine R, Gunther B, Bollensdorff C, von Banchet GS, Benndorf K, Zimmer T. Expression pattern of neuronal and skeletal muscle voltage-gated Na+ channels in the developing mouse heart. J Physiol 564: 683–696, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haufe V, Cordeiro JM, Zimmer T, Wu YS, Schiccitano S, Benndorf K, Dumaine R. Contribution of neuronal sodium channels to the cardiac fast sodium current INa is greater in dog heart Purkinje fibers than in ventricles. Cardiovasc Res 65: 117–127, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Jacobson SL, Piper HM. Cell cultures of adult cardiomyocytes as models of the myocardium. J Mol Cell Cardiol 18: 661–678, 1986. [DOI] [PubMed] [Google Scholar]
- 13.Ju YK, Saint DA, Gage PW. Inactivation-resistant channels underlying the persistent sodium current in rat ventricular myocytes. Proc R Soc Lond B Biol Sci 256: 163–168, 1994. [DOI] [PubMed] [Google Scholar]
- 14.Karin NJ, Cook JS. Turnover of the catalytic subunit of Na,K-ATPase in HTC cells. J Biol Chem 261: 10422–10428, 1986. [PubMed] [Google Scholar]
- 15.La C, You Y, Zhabyeyev P, Pelzer DJ, McDonald TF. Ultraviolet photoalteration of late Na+ current in guinea-pig ventricular myocytes. J Membr Biol 210: 43–50, 2006. [DOI] [PubMed] [Google Scholar]
- 16.Lipp P, Schwaller B, Niggli E. Specific inhibition of Na+-Ca2+ exchange function by antisense oligodeoxynucleotides. FEBS Lett 364: 198–202, 1995. [DOI] [PubMed] [Google Scholar]
- 17.Maier S, Westenbroek R, Yu FH, Vien T, Scheuer T, Catterall WA. Functional expression and localization of brain Nav1.1 sodium channel α-subunits in single cardiac myocytes (Abstract). Circulation 104: II-309, 2001. [Google Scholar]
- 18.Maier SK, Westenbroek RE, Schenkman KA, Feigl EO, Scheuer T, Catterall WA. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci USA 99: 4073–4078, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maltsev VA, Reznikov V, Undrovinas NA, Sabbah HN, Undrovinas A. Modulation of late sodium current by Ca2+, calmodulin, and CaMKII in normal and failing dog cardiomyocytes: similarities and differences. Am J Physiol Heart Circ Physiol 294: H1597–H1608, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maltsev VA, Sabbah HN, Higgins RSD, Silverman N, Lesch M, Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation 98: 2545–2552, 1998. [DOI] [PubMed] [Google Scholar]
- 21.Maltsev VA, Sabbah HN, Undrovinas AI. Down-regulation of sodium current in chronic heart failure: effects of long-term therapy with carvedilol. Cell Mol Life Sci 59: 1561–1568, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maltsev VA, Sabbah HN, Undrovinas AI. Late sodium current is a novel target for amiodarone: studies in failing human myocardium. J Mol Cell Cardiol 33: 923–932, 2001. [DOI] [PubMed] [Google Scholar]
- 23.Maltsev VA, Silverman N, Sabbah HN, Undrovinas AI. Chronic heart failure slows late sodium current in human and canine ventricular myocytes: implications for repolarization variability. Eur J Heart Fail 9: 219–227, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maltsev VA, Undrovinas A. Late sodium current in failing heart: friend or foe? Prog Biophys Mol Biol 96: 421–451, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maltsev VA, Undrovinas AI. A multi-modal composition of the late Na+ current in human ventricular cardiomyocytes. Cardiovasc Res 69: 116–127, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mathias RT, Cohen IS, Oliva C. Limitations of the whole cell patch clamp technique in the control of intracellular concentrations. Biophys J 58: 759–770, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitcheson JS, Hancox JC, Levi AJ. Action potentials, ion channel currents and transverse tubule density in adult rabbit ventricular myocytes maintained for 6 days in cell culture. Pflügers Arch 431: 814–827, 1996. [DOI] [PubMed] [Google Scholar]
- 28.Mitcheson JS, Hancox JC, Levi AJ. Cultured adult cardiac myocytes: future applications, culture methods, morphological and electrophysiological properties. Cardiovasc Res 39: 280–300, 1998. [DOI] [PubMed] [Google Scholar]
- 29.Morcos PA Achieving efficient delivery of morpholino oligos in cultured cells. Genesis 30: 94–102, 2001. [DOI] [PubMed] [Google Scholar]
- 30.Noble D, Noble PJ. Late sodium current in the pathophysiology of cardiovascular disease: consequences of sodium-calcium overload. Heart 92: iv1–iv5, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oliva C, Cohen IS, Mathias RT. Calculation of time constants for intracellular diffusion in whole cell patch clamp configuration. Biophys J 54: 791–799, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pusch M, Neher E. Rates of diffusional exchange between small cells and a measuring patch pipette. Pflügers Arch 411: 204–211, 1988. [DOI] [PubMed] [Google Scholar]
- 33.Saint DA, Ju YK, Gage PW. A persistent sodium current in rat ventricular myocytes. J Physiol 453: 219–231, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakmann BF, Spindler AJ, Bryant SM, Linz KW, Noble D. Distribution of a persistent sodium current across the ventricular wall in guinea pigs. Circ Res 87: 910–914, 2000. [DOI] [PubMed] [Google Scholar]
- 35.Schmidt JW, Catterall WA. Palmitylation, sulfation, and glycosylation of the alpha subunit of the sodium channel. Role of post-translational modifications in channel assembly. J Biol Chem 262: 13713–13723, 1987. [PubMed] [Google Scholar]
- 36.Shubeita HE, Martinson EA, Van Bilsen M, Chien KR, Brown JH. Transcriptional activation of the cardiac myosin light chain 2 and atrial natriuretic factor genes by protein kinase C in neonatal rat ventricular myocytes. Proc Natl Acad Sci USA 89: 1305–1309, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Slodzinski MK, Blaustein MP. Na+/Ca2+ exchange in neonatal rat heart cells: antisense inhibition and protein half-life. Am J Physiol Cell Physiol 275: C459–C467, 1998. [DOI] [PubMed] [Google Scholar]
- 38.Summerton J Morpholino antisense oligomers: the case for an RNase H-independent structural type. Biochim Biophys Acta 1489: 141–158, 1999. [DOI] [PubMed] [Google Scholar]
- 39.Tamkun MM, Fambrough DM. The (Na++K+)-ATPase of chick sensory neurons. Studies on biosynthesis and intracellular transport. J Biol Chem 261: 1009–1019, 1986. [PubMed] [Google Scholar]
- 40.Tan BH, Valdivia CR, Rok BA, Ruwaldt KM, Tester DJ, Ackerman MJ, Makielski JC. Common human SCN5A polymorphisms have altered electrophysiology when expressed in Q1077 splice variants. Heart Rhythm 2: 741–747, 2005. [DOI] [PubMed] [Google Scholar]
- 41.Undrovinas AI, Belardinelli L, Undrovinas NA, Sabbah HN. Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J Cardiovasc Electrophysiol 17: S169–S177, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Undrovinas AI, Fleidervish IA, Makielski JC. Inward sodium current at resting potentials in single cardiac myocytes induced by the ischemic metabolite lysophosphatidylcholine. Circ Res 71: 1231–1241, 1992. [DOI] [PubMed] [Google Scholar]
- 43.Undrovinas AI, Maltsev VA, Kyle JW, Silverman NA, Sabbah HN. Gating of the late Na+ channel in normal and failing human myocardium. J Mol Cell Cardiol 34: 1477–1489, 2002. [DOI] [PubMed] [Google Scholar]
- 44.Undrovinas AI, Maltsev VA, Sabbah HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cell Mol Life Sci 55: 494–505, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, Kamp TJ, Makielski JC. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol 38: 475–483, 2005. [DOI] [PubMed] [Google Scholar]
- 46.Ver Donck L, Borgers M, Verdonck F. Inhibition of sodium and calcium overload pathology in the myocardium: a new cytoprotective principle. Cardiovasc Res 27: 349–357, 1993. [DOI] [PubMed] [Google Scholar]
- 47.Volz A, Piper HM, Siegmund B, Schwartz P. Longevity of adult ventricular rat heart muscle cells in serum-free primary culture. J Mol Cell Cardiol 23: 161–173, 1991. [DOI] [PubMed] [Google Scholar]
- 48.Wu L, Shryock JC, Song Y, Belardinelli L. An increase in late sodium current potentiates the proarrhythmic activities of low-risk QT-prolonging drugs in female rabbit hearts. J Pharmacol Exp Ther 316: 718–726, 2006. [DOI] [PubMed] [Google Scholar]
- 49.Yue L, Melnyk P, Gaspo R, Wang Z, Nattel S. Molecular mechanisms underlying ionic remodeling in a dog model of atrial fibrillation. Circ Res 84: 776–784, 1999. [DOI] [PubMed] [Google Scholar]
- 50.Zicha S, Maltsev VA, Nattel S, Sabbah HN, Undrovinas AI. Post-transcriptional alterations in the expression of cardiac Na+ channel subunits in chronic heart failure. J Mol Cell Cardiol 37: 91–100, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zygmunt AC, Eddlestone GT, Thomas GP, Nesterenko VV, Antzelevitch C. Larger late sodium conductance in M cells contributes to electrical heterogeneity in canine ventricle. Am J Physiol Heart Circ Physiol 281: H689–H697, 2001. [DOI] [PubMed] [Google Scholar]