

Abstract
Hemoglobin (Hb) potently inactivates the nitric oxide (NO) radical via a dioxygenation reaction forming nitrate (NO3−). This inactivation produces endothelial dysfunction during hemolytic conditions and may contribute to the vascular complications of Hb-based blood substitutes. Hb also functions as a nitrite (NO2−) reductase, converting nitrite into NO as it deoxygenates. We hypothesized that during intravascular hemolysis, nitrite infusions would limit the vasoconstrictive properties of plasma Hb. In a canine model of low- and high-intensity hypotonic intravascular hemolysis, we characterized hemodynamic responses to nitrite infusions. Hemolysis increased systemic and pulmonary arterial pressures and systemic vascular resistance. Hemolysis also inhibited NO-dependent pulmonary and systemic vasodilation by the NO donor sodium nitroprusside. Compared with nitroprusside, nitrite demonstrated unique effects by not only inhibiting hemolysis-associated vasoconstriction but also by potentiating vasodilation at plasma Hb concentrations of <25 μM. We also observed an interaction between plasma Hb levels and nitrite to augment nitroprusside-induced vasodilation of the pulmonary and systemic circulation. This nitrite reductase activity of Hb in vivo was recapitulated in vitro using a mitochondrial NO sensor system. Nitrite infusions may promote NO generation from Hb while maintaining oxygen delivery; this effect could be harnessed to treat hemolytic conditions and to detoxify Hb-based blood substitutes.
Keywords: blood flow, vascular endothelial function, blood substitute, vascular biology
endothelium-derived nitric oxide (NO) plays a central role in vascular physiology as an autocrine and paracrine vasodilator molecule that maintains basal vasomotor tone, mediates flow-mediated vasodilation, and inhibits platelet aggregation and endothelium adhesion molecule expression. NO's ability to diffuse as an endocrine molecule in blood is limited by an extremely rapid dioxygenation reaction with oxy-hemoglobin (oxy-Hb) to form the inert nitrate (NO3−) (20, 21, 35, 36, 42, 61, 62), as follows:
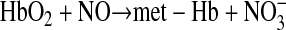 |
(1) |
Despite the near diffusion limited nature of this reaction in vitro, the rate of the NO dioxygenation reaction in vivo is limited by the compartmentalization of Hb within the erythrocyte (2, 6–8, 30, 42, 46–48, 72–74). This compartmentalization of Hb in the red blood cell reduces the rate of the inactivation reaction of NO by Hb by ∼1,000-fold, allowing NO to regulate vasodilation despite the presence of 10 mM intravascular red cell Hb.
During intravascular hemolysis, normal NO physiology is disrupted by the release of cell-free plasma Hb, leading to rapid scavenging of NO, vasoconstriction, and organ dysfunction (52, 79). Repeated episodes of intravascular hemolysis with NO scavenging may play a role in the long-term complications of sickle cell disease and other intravascular hemolytic disorders (1, 17, 19, 41, 59, 65). Similarly, the administration of cell-free Hb-based blood substitutes leads to excessive NO scavenging by Hb and may have contributed to the increased rates of pulmonary and systemic hypertension, decreased organ perfusion, gastrointestinal spasms, and increased mortality in clinical trials of cell-free Hb-based blood substitutes (14, 15, 28, 29, 77). In addition to NO scavenging, another potential mechanism that may contribute to the vasoconstrictive effects of cell-free Hb includes increased oxygen delivery to resistance vessels by cell-free Hb (9, 44).
In addition to endothelium-derived NO, accumulating evidence suggests that an intravascular NO storage molecule exists that can produce NO in hypoxic tissues independent of NO synthase pathways (10, 21, 25, 26, 31, 33, 39, 42, 56, 57, 71). Debate remains about the exact systemic storage form(s) of NO that can account for hypoxic vasodilation, but one potential candidate is the nitrite anion (NO2−) (10, 18, 21, 23, 27, 31, 33, 50, 60, 76). Multiple groups have recently reported vasodilatory effects of nitrite at both physiological and pharmacological doses (4, 10, 13, 34, 37, 43, 45, 49, 51, 53, 56). The proposed mechanisms of nitrite-induced vasodilation involve the reduction of nitrite to NO by acidic disproportionation, xanthine oxidoreductase activity, or the nitrite reductase activity of Hb (10, 11, 24, 31, 33, 49, 51, 54, 80). The first two mechanisms are likely to have limited physiological roles because they require a low pH and near anoxia (21). However, recent evidence suggests that the nitrite reductase activity of Hb likely plays an important role in hypoxic vasodilation and may have significant therapeutic implications for various human disease processes (10, 11, 31, 33).
The reduction of nitrite by Hb is enhanced during physiological hypoxia and as proton concentration increases (5, 16).
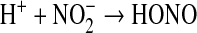 |
(2) |
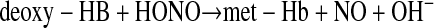 |
(3) |
Analogous to bacterial nitrite reductases (concerted electron and proton transfer to nitrite), the reduction of nitrite to NO requires a one-electron reduction of nitrous acid (33). Within the Hb tetramer, the proton dependence of nitrite reduction decreases from 1.0 to 0.88 secondary to a small stabilizing effect of the proton on the T state (tense; deoxy) of Hb (redox Bohr effect) (16). Hb functions as an allosteric nitrite reductase with a maximal NO generation rate around the intrinsic Hb P50 (50% Hb saturation with oxygen) due to the conformational change of Hb from the R (relaxed; oxy) state to the T state during deoxygenation from artery to vein (11, 31, 33). This dependence of the nitrite reductase activity of Hb on physiological hypoxia and acidosis allows for a metabolic “sensing” mechanism that leads to maximal NO generation in ischemic and hypoxic tissues (11, 21, 31, 33, 42). Supporting this theory, a number of laboratories have confirmed that deoxygenating Hb and myoglobin in the presence of nitrite generates NO in vivo and generates cGMP, causes vasodilation, and inhibits cytochrome c oxidase of the mitochondrial electron transport chain (an NO effect) in vitro (10, 11, 38, 56, 64, 66, 70). In a recent study, Rassaf and colleagues (64) infused nitrite in vivo in mice and measured the change in energy for ATP hydrolysis using MRI spectroscopy. They showed that nitrite generates NO in the heart and that this NO inhibits cellular respiration. Interestingly, this effect was only observed in myoglobin wild-type mice and not evident in myoglobin knockout mice, supporting a mechanism for nitrite reduction to NO that requires myoglobin (64, 70). Thus, Hb and myoglobin can be viewed as oxygen-regulated NO-processing molecules that act as NO scavengers under normoxia via the dioxygenase reaction (Eq. 1) and as NO generators during hypoxia via the nitrite reductase reaction (Eq. 3). We have recently found that this nitrite reductase reaction is coupled to a more complicated nitrite anydrase reaction that generates freely diffusible N2O3, which provides a mechanism for NO escape from the erythrocyte (3).
The allosteric nitrite reductase activity of deoxy-Hb may allow nitrite to have a specific therapeutic role during intravascular hemolysis and during the administration of cell-free Hb-based blood substitutes. According to this hypothesis, as Hb deoxygenates in the microcirculation, it may begin to generate NO as it releases oxygen. Such an effect could theoretically counter NO-scavenging effects of Hb and thus limit vasoconstriction and endothelial dysfunction during hemolysis or infusion of stroma-free Hb-based blood substitutes. To test these hypotheses, we used a previously developed canine model of acute intravascular hemolysis that is characterized by systemic NO scavenging, endothelial dysfunction, and vasculopathy (52). The sequella of intravascular hemolysis in this model manifests as systemic and pulmonary vasoconstriction, a process that reproduces the toxicities evident during the infusion of cell-free Hb-based blood substitutes and during episodes of acute intravascular hemolysis in patients with hemolytic disorders. We performed experiments to characterize the physiological effects of intravenous sodium nitrite and to determine if these effects of nitrite are influenced by the nitrite reductase activity of cell-free plasma Hb during intravascular hemolysis. Furthermore, we directly compared the effects of nitrite in this setting with a conventional NO donor medication, sodium nitroprusside.
MATERIALS AND METHODS
Experimental Design
All experiments were approved by the Animal Care and Use Committee of the Clinical Center of the National Institutes of Health. Thirty-two purpose-bred beagles (12–28 mo, 9–12 kg) were studied.
All procedures were performed after the induction of anesthesia with halothane (1–4%) and the initiation of mechanical ventilation. Upon completion, the halogenated gas was terminated, and 100% oxygen was administered until the dog emerged from anesthesia and was extubated. Subsequently, the animal breathed room air spontaneously and was sedated throughout the duration of the experiments. Continuous infusions of medetomidine (sedation; 2–5 mcg·kg−1·h−1) and fentanyl (analgesia; 2.5–20 mcg·kg−1·h−1) were initiated postextubation and maintained for the study duration. Animals were monitored continuously, and signs of pain and distress were evaluated immediately and the infusions adjusted appropriately.
Nitrite infusion.
Using pilot experiments to characterize the beagle-specific pharmacokinetics of sodium nitrite, an infusion of 165 mg sodium nitrite over 6 h (27.5 mg/h) was chosen to be administered during the study to reach a targeted plasma nitrite concentration between 15 and 20 μM. Animals randomized to groups not receiving sodium nitrite received an equivalent rate and total volume infusion of 0.9% NaCl (normal saline) to serve as a placebo control.
Water-infusion intravascular hemolysis model.
A previously developed and validated canine model of water infusion-induced intravascular hemolysis was used in this study (52). Water-induced hemolysis produces direct intravascular hemolysis, thereby maintaining the same intravascular concentration of total Hb during hemolysis while altering the distribution of Hb between the red cell and plasma compartment. In this model, a 6-h infusion of water (rate: 16 ml·kg−1·h−1) produces clinically relevant levels of cell-free plasma Hb (20–300 μM heme), simulating an acute hemolytic episode. The extent of hemolysis increases over time, allowing for a graded physiological assessment of vasomotor dysfunction as plasma Hb levels rise. The final levels of plasma Hb would be analogous to those achieved following coronary bypass surgery or a hemolytic crisis induced by paroxysmal nocturnal hemoglobinuria or acute immune-mediated hemolysis. Control animals receive an equivalent rate and total volume infusion of 5% dextrose (D5W) to account for any potential hypotonic and volume effects of the water infusion on hemodynamics. We used a full-factorial study design with four groups of animals receiving either (1) D5W, (2) D5W plus intravenous sodium nitrite, (3) water, or (4) water plus intravenous sodium nitrite. This design allowed for the determination of the physiological effects of a sodium nitrite infusion, the physiological effects of intravascular hemolysis (water), and the assessment of an interaction between nitrite and hemolysis. Specifically, the interaction statistic tested if the effects of nitrite and Hb were influenced by the nitrite reductase activity of cell-free plasma Hb.
Paired experiments were performed in 20 animals (5 animals/group). In the first week, all animals underwent a baseline study and received a D5W infusion (16 ml·kg−1·h−1) through a central venous catheter to determine the physiological effects of the volume load in each animal. The D5W infusion did not cause hemolysis; it allowed each animal to serve as its own control for the effects of a hypotonic volume load in the model. One week later, animals underwent an intervention study and were randomized to receive a 6-h infusion through a central venous catheter of either (1) D5W (16 ml·kg−1·h−1), (2) D5W (16 ml·kg−1·h−1) plus nitrite (27.5 mg/h), (3) water (16 ml·kg−1·h−1), or (4) water (16 ml·kg−1·h−1) plus nitrite (27.5 mg/h). This paired experimental design allowed for the minimization of animal-to-animal variability by calculating the change for each measurement performed in each animal during the baseline and intervention experiments. Subsequent analyses calculate the differences across treatment groups by subtracting the previously calculated differences within animals (from baseline to intervention experiments) in one treatment group from another treatment group (i.e., comparison of the differences of the differences). This design allowed for analysis of the effects of hemolysis, the effects of sodium nitrite, and detection of any interaction between the two.
Preliminary analysis demonstrated a wider range of hemolysis than previously described secondary to the addition of salt-based therapies (sodium nitrite or sodium chloride) that affected the rate of hypotonic erythrocyte lysis. In these experiments, the 6-h water infusions produced low-rate hemolysis in 50% of the animals and rapid rate hemolysis in 50% of the animals. This created two equal-sized groups of animals with either low or high levels of cell-free plasma Hb, respectively. Both groups had peak cell-free plasma Hb levels that continued to be within a clinically relevant range (20–200 μM heme). Preliminary data analyses also suggested a possible interaction between sodium nitrite and hemolysis that was dependent on the amount of hemolysis (heme concentration <25 vs. >25 μM). Subsequently, we accounted for the variation in hemolytic rate and this potential interaction by calculating the number of animals needed to determine if there was an interaction between nitrite and hemolysis level (assuming a 1:1 ratio of low- to high-rate hemolysis in animals receiving a water infusion) and included the level of hemolysis in our final data analysis. The necessary additional paired experiments were then performed using the same treatment regimens with a weighted randomization scheme to the following groups: (1) D5W plus nitrite (n = 2), (2) water (n = 5), or (3) water plus nitrite (n = 5). Overall, these experiments used 32 animals.
Sodium nitroprusside challenge.
To determine the vascular responsiveness to exogenous NO in the presence and absence of hemolysis and sodium nitrite, all animals received a 20-min infusion of escalating doses of sodium nitroprusside, a direct NO donor (1, 3, 9, and 27 mcg·kg−1·min−1), at 5-min intervals before the conclusion of the study. These experiments allowed for comparison of a “traditional” NO donor with nitrite to determine if the observed nitrite effects (i.e., Hb-based nitrite reduction) were distinct from a pure NO vasodilatory effect.
Data Collection
Femoral arterial (20-gauge) and external jugular venous (8-French) catheters (Maxxim Medical, Athens, TX) were placed percutaneously under anesthesia using sterile techniques. Mean arterial pressure (MAP) and heart rate (HR) were obtained from the femoral artery catheter tracing. Additionally, a pulmonary artery thermodilution catheter (7-French, Abbott Critical Care, Chicago, IL) was introduced through the external jugular vein catheter to measure cardiac output (CO), mean pulmonary artery pressure (PAM), pulmonary artery occlusion pressure (PAOP), and central venous pressure (CVP). At the end of the first week's fluid control experiments, all catheters were removed and the animals recovered. At the end of the second week's intervention experiments, all animals were euthanized.
Hemodynamic measurements (MAP, CVP, PAM, CO, and PAOP) and laboratory measurements (hematocrit, Hb, serum chemistries, arterial blood gas analysis, spectrophotometric-based quantification of cell-free Hb concentration, and chemiluminescence-based assays of NO consumption and nitrite levels) were obtained at 0-, 1.5-, 3.0-, 4.5-, and 6.0-h time points. Hemodynamic measurements were also obtained at the end of each dose of sodium nitroprusside.
Plasma Nitrite and Hb Assays
Plasma nitrite levels were measured by an I -based chemiluminescent assay as previously described using the NO analyzer (model 280i NO analyzer, Seivers, Boulder, CO) (78). The total plasma Hb concentration (expressed in terms of heme groups, division by four gives the Hb concentration) was measured by visible absorbance spectrophotometry (HP8453 UV-Vis Diode Array Spectrophotometer, Hewlett Packard). The concentration of oxy-Hb and met-Hb were analyzed by deconvoluting the spectrum into components from basis spectra of canine Hb in PBS buffer using a least-square method as previously described, with subtraction of background plasma scattering (32).
-based chemiluminescent assay as previously described using the NO analyzer (model 280i NO analyzer, Seivers, Boulder, CO) (78). The total plasma Hb concentration (expressed in terms of heme groups, division by four gives the Hb concentration) was measured by visible absorbance spectrophotometry (HP8453 UV-Vis Diode Array Spectrophotometer, Hewlett Packard). The concentration of oxy-Hb and met-Hb were analyzed by deconvoluting the spectrum into components from basis spectra of canine Hb in PBS buffer using a least-square method as previously described, with subtraction of background plasma scattering (32).
In Vitro Mitochondrial Respiration Experiments
Male Sprague-Dawley rats (175–250 g) were used in accordance with the Animal Care and Use Committee of the National Heart Lung Blood Institute. Liver mitochondria were isolated by differential centrifugation in buffer consisting of sucrose (250 mM), Tris (10 mM), and EGTA (1 mM) as previously described (70). Mitochondrial respiration was measured by suspending isolated mitochondria (2 mg/ml) in respiration buffer (120 mM KCl, 25 mM sucrose, 10 mM HEPES, 1 mM EGTA, 1 mM KH2PO4, and 5 mM MgCl2) in a stirred sealed chamber fit with a Clark-type oxygen electrode (Instech) connected to a data recording device (DATAQ Systems). Mitochondria were supplemented with succinate (15 mM) and ADP (1 mM) to stimulate respiration. In experiments testing the effects of nitrite and Hb, sodium nitrite and human purified oxy-Hb (33) were incubated with mitochondria at the beginning of the experiment (70). In this experimental system, the rate of oxygen generation from the added Hb is less than the rate of oxygen consumption by the mitochondria so that the oxygen increase in the system after Hb addition (20 μM) is not detected by the oxygen electrode and not observed in the raw trace unless high concentrations of Hb are added, in which case a transient increase in the oxygen level may be detected. Note that in this system the chamber is opened to air and oxygen is diffusing into the system as well, but the rate of oxygen diffusion into the system is less than the rate of oxygen consumption by the mitochondria. Only after mitochondrial inhibition do the oxygen levels rise to detection by the electrode.
Statistical Analysis
Data were analyzed using ANOVA, with main effects for study (baseline and intervention), hemolysis (0 μM D5W, <25 μM heme, and >25 μM heme), nitrite, time, and animal (52). Two- and three-way interactions were included in the model. Analysis of responses to sodium nitroprusside were performed using ANOVA on percent changes in hemodynamic variables with increasing doses in the intervention experiments with main effects for hemolysis, nitrite, nitroprusside dose, and animal. Two-way interactions were included in the model. All values are depicted in the figures as means ± SE, and all Hb concentrations are expressed in terms of heme groups.
RESULTS
Physiological Effects of Intravenous Sodium Nitrite
While many groups have now confirmed that sodium nitrite is a potent vasodilator in vivo, to our knowledge no group has characterized more specifically its activity in vivo as a relative arterial versus venous vasodilator or its effects on inotropy and chronotropy. An intravenous infusion of sodium nitrite (27.5 mg/h) rapidly increased plasma nitrite levels to a steady-state concentration (range: 15–21 μM) that was maintained throughout the duration of the 6-h infusion (Fig. 1A). In animals receiving a D5W infusion, sodium nitrite increased the cardiac index (CI; P = 0.001) and decreased the systemic vascular resistance index (SVRI; P = 0.04), pulmonary vascular resistance index (PVRI; P = 0.001), MAP (P = 0.08), PAM (P = 0.09), CVP (P = 0.01), and PAOP (P = 0.65) compared with placebo (normal saline) (Fig. 1, B–H). These physiological effects suggest that low-dose sodium nitrite is a more potent arterial vasodilator than a venodilator and that nitrite increases cardiac performance by direct afterload reduction. Supporting evidence for the vasodilatory effects of nitrite can be derived by examining the components of CI in the log scale (Fig. 2) (68). In this format, the individual contributions of each component of CI are additive (normal scale: CI = SVI × HR, log scale: log CI = log SVI + log HR, where SVI is the stroke volume index). This transformation demonstrates that the nitrite-induced increase in CI is mediated predominantly through a sustained increase in the SVI and to a lesser extent by a chronotropic effect. This transformation also accounts for the rise in PAOP during the last 3 h of the experiment (Fig. 1H). The decrease in HR over time (an effect of anesthesia/analgesia seen in all groups in this model) (Fig. 2B) increases diastolic filling time in the ventricles, leading to higher end-diastolic volumes and pressures that translate into increases in PAOP and further increases in SVI. These data imply that nitrite enhances cardiac performance by afterload reduction through an arterial vasodilatory mechanism. These data also indicate that isolated measures of MAP in animal studies may fail to sensitively assess the magnitude of nitrite-dependent vasodilation because of the rise in the CI.
Fig. 1.

Cardiovascular effects of nitrite in nonhemolyzing animals. A: serial plasma nitrite levels (in μM) in nonhemolyzing animals. In animals receiving a 6-h infusion of 5% dextrose (D5W), a sodium nitrite infusion of 27.5 mg/h (○) led to a rapid rise and then a sustained plasma nitrite concentration (range: 15–21 μM) compared with a placebo infusion of 0.9% NaCl (•). B–H: in nonhemolyzing animals, sodium nitrite increased the cardiac index (CI; B) and decreased the systemic vascular resistance index (SVRI; C), pulmonary vascular resistance (PVRI; D), mean arterial pressure (MAP; E), mean pulmonary arterial pressure (PAM; F), central venous pressure (CVP; G), and pulmonary artery occlusion pressure (PAOP; H) compared with placebo. Intravenous nitrite enhanced cardiac performance (CI) by arterial vasodilation (SVRI, PVRI, MAP, and PAM) and caused venodilation (CVP).
Fig. 2.

Effects of nitrite on the components of the CI in nonhemolyzing animals. The CI (A) and its components (B) were transformed into the log scale to demonstrate the individual contribution of heart rate (HR) and stroke volume index (SVI) to CI in an additive fashion (normal scale: CI = SVI × HR, log scale: log CI = log SVI + log HR) (68). In animals receiving D5W and nitrite, the nitrite-induced increase in the CI is mediated predominantly through an increase in SVI and to a lesser extent by a chronotropic effect. Over time, the decrease in HR causes further increases in SVI by increasing diastolic filling time in the ventricles, leading to higher end-diastolic volumes. Furthermore, the higher end-diastolic volumes translate into higher end-diastolic pressures, which may explain the increase in PAOP over time (Fig. 1H).
Physiological Effects of Sodium Nitrite During Intravascular Hemolysis
In previous intravascular hemolysis experiments (52) and in the current experiments, cell-free plasma Hb increased systemic arterial pressure, PAM, SVRI, PVRI, and PAOP (Table 1; P = 0.04, 0.14, 0.06, 0.42, and 0.21, respectively, for the interaction of hemolysis levels and the mean change in each physiological variable during baseline and intervention experiments). If nitrite functioned purely as an NO donor medication, one would expect the vasoconstrictive effects of intravascular hemolysis to attenuate the vasodilatory effects of nitrite because any NO generated from nitrite would be readily scavenged by cell-free plasma Hb. However, in these experiments, the physiological effects of nitrite were not simply inhibited by increasing levels of hemolysis. In fact, the effect of nitrite was dependent on the level of intravascular hemolysis in an unusual way (Fig. 3). We detected a consistent U-shaped relationship between the physiological effects of nitrite and the levels of cell-free plasma Hb suggesting an interaction between the effects of nitrite and the amount of intravascular hemolysis (Fig. 3; P = 0.01 for a differing effect of nitrite at low-level hemolysis compared with 0 and high-level hemolysis across the 7 physiological variables combined). At low levels of hemolysis (Hb < 25 μM), the vasodilatory effects of nitrite were apparently potentiated, whereas with higher levels of hemolysis (Hb > 25 μM), the expected inhibition of the vasodilatory effects of nitrite were observed. These results suggest that there are two reactions that regulate the availability of NO at the smooth muscle: the reaction of Hb with NO and an opposing reaction of nitrite with deoxy-Hb that generates NO. Our results demonstrate that at low levels of Hb, the physiologic effects of the latter reaction are detected; however, with increasing Hb concentrations, the former reaction dominates. These effects are examined more closely and compared with the NO donor sodium nitroprusside in additional experiments described below.
Table 1.
Physiological effects of intravascular hemolysis
| Level of Hemolysis | Mean Systemic Arterial Pressure, mmHg | Mean Pulmonary Artery Pressure, mmHg | Systemic Vascular Resistance Index, dyn·s−1·cm5 | Pulmonary Vascular Resistance Index, dyn·s−1·cm5 | Pulmonary Artery Occlusion Pressure, mmHg |
|---|---|---|---|---|---|
| 0 μM | 6.7 | 3.0 | 15.1 | 1.3 | 2.9 |
| <25 μM | 7.9 | 4.2 | 28.1 | 3.0 | 2.8 |
| >25 μM | 16.4 | 4.8 | 33.9 | 2.3 | 3.8 |
n = 5 for the 0 μM hemolysis group, 4 for the <25 μM hemolysis group, and 4 for the >25 μM hemolysis group.
Fig. 3.

Cardiovascular effects of nitrite during intravascular hemolysis. The cardiovascular effects of nitrite (27.5 mg/h) at different levels of cell-free plasma hemoglobin (Hb; 0, < 25 μM, and >25 μM) are shown in A–G. For each parameter, the isolated effect of nitrite is displayed after controlling for animal variability and the independent effects of hemolysis. The depicted value represents the mean change in the parameter from time 0 to 1.5, 3, 4.5, and 6 h for all animals within the specified hemolysis group (x-axis: 0 μM, •; <25 μM, ○; and >25 μM, ▾). According to previous experiments, if nitrite functioned purely as a nitric oxide (NO) donor, then there should be progressive attenuation of the vasodilatory effects of nitrite with increasing levels of hemolysis; the NO generated from nitrite should be progressively scavenged by the increasing levels of cell-free plasma Hb (52). In these experiments, the effect of nitrite was dependent on the level of intravascular hemolysis (P = 0.01 for a differing effect of nitrite at low-level hemolysis compared with zero and high-level hemolysis across the 7 physiologic variables combined). A consistent U-shaped relationship between the physiological effects of nitrite and the levels of cell-free plasma Hb was detected. At low levels of hemolysis (Hb concentration < 25 μM), the vasodilatory effects of nitrite were potentiated, whereas with higher levels of hemolysis (cell-free plasma Hb > 25 μM), the expected inhibition of the vasodilatory effects of nitrite were observed.
Nitrite Levels and Hb Species Formed During Intravascular Hemolysis
In animals receiving nitrite, plasma nitrite levels were similar and were maintained within a range of 16–20 μM throughout the 6-h experiment (Figs. 1A and 4). Intravascular hemolysis occurred at varying rates (Fig. 4). Animals receiving D5W and nitrite represent the zero-hemolysis control group with all measured cell-free plasma Hb levels < 5 μM (data not shown). In animals receiving water and nitrite infusions with low levels of hemolysis (Hb < 25 μM), the average peak cell-free plasma Hb level was 20 μM. In animals receiving water and nitrite infusions with high levels of hemolysis, the average peak cell-free plasma Hb level was 142 μM. In animals receiving D5W and nitrite (zero hemolysis), 81% of the measured cell-free plasma Hb was oxy-Hb (Fig. 4, values depicted as a dark gray reference line in B and E), consistent with observations in normal volunteers and sickle cell patients showing that plasma Hb is maintained largely in the reduced or ferrous-oxygen bound state (HbFe2+-O2). In hemolyzing animals, oxy-Hb accounted for 71% and 69% of the measured cell-free plasma Hb in animals with low and high levels of hemolysis, respectively (Fig. 4).
Fig. 4.

Nitrite levels and plasma Hb composition during intravascular hemolysis. A and D: intravascular hemolysis occurred at varying rates in animals receiving water and nitrite infusions. In the low-level hemolysis group (Hb < 25 μM; A), the average peak cell-free plasma Hb level was 20 μM; in the high-level hemolysis group (Hb > 25 μM; D), the average peak cell-free plasma Hb level was 142 μM. Animals receiving D5W and nitrite represent the zero-hemolysis control group with all measured cell-free plasma Hb levels <5 μM (data not shown). B and E: total plasma Hb composition in the low-level (B) and high-level (E) hemolysis groups (○, met-Hb; •, oxy-Hb). In animals receiving D5W and nitrite (zero hemolysis), 81% of the measured cell-free plasma Hb was oxy-Hb (depicted as dark gray reference lines) and 19% was met-Hb (depicted as light gray reference lines). With increasing Hb concentrations, the rate of met-Hb formation increased from 0 to 3 h (P = 0.0001), producing higher levels of met-Hb from 3 to 6 h (P = 0.0001) in animals with higher levels of hemolysis compared with animals with lower levels of hemolysis. These results can be explained by the fact that the overall reactions of nitrite with oxy- and deoxy-Hb are second order during their lag phases such that increasing Hb concentrations lead to increasing rates of reaction. C and F: in both the low-level (C) and high-level (F) hemolysis groups, plasma nitrite levels were similar and were maintained within a range of 16–21 μM throughout the 6-h experiment.
During nitrite infusions at low levels of hemolysis, nitrite reacted with Hb to form ∼30% met-Hb (Fig. 4B, values for the D5W + nitrite zero-hemolysis control are depicted as a light gray reference line in B and E). This reaction likely reflects two reactions of nitrite: the reaction of nitrite with oxy-Hb to form met-Hb and nitrate (NO2− + HbO2 → met-Hb + NO3−) and the reaction of nitrite with deoxy-Hb to form met-Hb and NO (as described in the Introduction, Eqs. 2 and 3). The former reaction decreases NO scavenging, and the latter reaction contributes to NO generation. Note that, paradoxically, there is sufficient oxy-Hb at the end of 6 h to almost completely scavenge and inhibit any NO that might form, yet the nitrite remains vasoactive and potentiated by low levels of Hb (Fig. 3). As shown in Fig. 4, B and E, with increasing Hb concentrations, the rate of met-Hb formation increases from 0 to 3 h (P = 0.0001), producing higher levels of met-Hb from 3 to 6 h (P = 0.0001) in animals with higher levels of hemolysis compared with animals with lower levels of hemolysis. This is because the overall reactions of nitrite and Hb are second order during their lag phases, meaning that as the Hb concentration increases, the rate of the reactions increases. Again, we see that the amount of oxy-Hb at the end of the reaction is sufficient to almost completely scavenge any NO that might be formed if nitrite acted as a pure NO donor.
Effects of Nitrite and Level of Hemolysis on Hemodynamic Responses to Sodium Nitroprusside, an Infused NO Donor
Sodium nitroprusside was administered to all animals to determine the physiological effects of a direct NO-donating agent in the setting of hemolysis with and without sodium nitrite. The physiological effects of sodium nitroprusside were dependent on the level (or dose) of hemolysis and the presence of nitrite. As expected, in animals that did not receive nitrite, sodium nitroprusside-induced increases in CI and decreases in SVRI and PVRI were progressively inhibited by increasing levels of hemolysis, suggesting progressive consumption of the donated NO by increasing levels of cell-free plasma Hb during intravascular hemolysis (Fig. 5). In contrast, the effects of sodium nitroprusside at the three levels of hemolysis were different in animals receiving nitrite compared with animals not receiving nitrite (Fig. 5). Compared with nonhemolyzing animals not receiving nitrite (zero hemolysis, no nitrite), nonhemolyzing animals receiving nitrite (zero hemolysis, nitrite) demonstrated blunted effects of sodium nitroprusside on CI, SVRI, and PVRI, suggesting a decreased vasodilator effect of donated NO in the presence of nitrite without hemolysis. If the effect of nitrite on the response to sodium nitroprusside during hemolysis was additive (i.e., the same effect at all levels of hemolysis), then the demonstrated relationship should be a similar linear relationship to the one demonstrated in animals not receiving nitrite, but starting at a smaller magnitude percent change due to the decreased vasodilator effect of donated NO from nitroprusside in the presence of nitrite (comparing zero hemolysis, no nitrite with zero hemolysis + nitrite). However, in animals receiving nitrite, the effects of sodium nitroprusside on CI, SVRI, and PVRI were accentuated with low levels of hemolysis (Hb < 25 μM, nitrite) and then attenuated with high levels of hemolysis (Hb > 25 μM, nitrite) compared with nonhemolyzing animals (zero hemolysis, nitrite) (Fig. 5; P = 0.09, 0.05, and 0.009 for the interaction, demonstrating a different relationship between level of hemolysis and nitrite on the effect of sodium nitroprusside for CI, SVRI, and PVRI respectively). Animals with low-level hemolysis demonstrated a similar or greater percent change on the physiological variables than zero hemolysis (instead of the expected smaller effect in an additive model) and animals with higher-level hemolysis demonstrated blunted physiological responses. This interaction is consistent with the U-shaped physiological effects of nitrite demonstrated during the 6-h hemolysis experiment; compared with the effect in animals with zero hemolysis, the physiological effect of nitrite was accentuated with low-level hemolysis and then attenuated at higher levels of hemolysis. This interaction may be explained by the additional nitrite reduction reaction with Hb contributing to vasodilation. Nitrite reacts with oxy- and deoxy-Hb to form met-Hb and met-Hb + NO, respectively (10). These nitrite reactions may lead to enhanced vasodilation by sodium nitroprusside in the setting of low levels of hemolysis by (1) minimizing the amount of oxy-Hb available in the plasma to consume the donated NO from sodium nitroprusside and (2) directly causing vasodilation secondary to the NO generated by the reaction of nitrite with deoxy-Hb. At higher levels of intravascular hemolysis, the nitrite reduction reaction with Hb may be overwhelmed by the large amounts of cell-free plasma Hb that consume any NO formed from the reaction. Consequently, the donated NO from sodium nitroprusside and the generated NO from the reaction of nitrite with deoxy-Hb are consumed by the excess oxy-Hb in the plasma.
Fig. 5.

Effects of nitrite and intravascular hemolysis on cardiovascular responses to sodium nitroprusside. The physiological effects of sodium nitroprusside (a direct NO donor) were dependent on the level (or dose) of hemolysis and the presence of sodium nitrite (P = 0.09, 0.05, and 0.009 for the interaction between level of hemolysis and nitrite on the effect of sodium nitroprusside for CI, SVRI, and PVRI, respectively). Depicted values represent the mean percent changes in the parameter for all doses of nitroprusside for all animals within the specified hemolysis group (0 μM, •; <25 μM, ○, and >25 μM, ▾). As expected for a direct NO donor, in animals not receiving nitrite, sodium nitroprusside-induced increases in CI and decreases in SVRI and PVRI were progressively inhibited by increasing levels of hemolysis (A–C; no nitrite). Compared with nonhemolyzing animals not receiving nitrite (zero hemolysis, no nitrite), nonhemolyzing animals receiving nitrite (zero hemolysis, nitrite) demonstrated blunted effects of sodium nitroprusside on CI, SVRI, and PVRI, suggesting a decreased vasodilator effect of donated NO in the presence of nitrite (A–C). In animals receiving nitrite, the effects of sodium nitroprusside on CI, SVRI, and PVRI were accentuated with low levels of hemolysis (Hb < 25 μM, nitrite) and attenuated with high levels of hemolysis (Hb > 25 μM, nitrite) compared nonhemolyzing animals (zero hemolysis, nitrite) (A–C; nitrite). These findings may be explained by the nitrite reduction reaction with Hb (detailed in results).
Confirmatory In Vitro Mitochondrial Respiration Experiments
In vitro mitochondrial respiration experiments were performed with nitrite and cell-free Hb levels similar to those obtained in vivo to confirm that the effects of nitrite on vasoactivity during hemolysis are dependent on the reaction of nitrite and deoxy-Hb to generate NO. In these experiments, mitochondria serve as NO sensors because NO avidly binds to cytochrome c oxidase to inhibit respiration. In this experimental system, mitochondria suspended in a closed chamber respire until the chamber becomes anoxic (oxygen trace reads zero). Removal of the chamber lid allows oxygen diffusion into the chamber; however, the trace remains at zero due to rapid oxygen consumption by the respiring mitochondria. The oxygen trace deviates from zero only once the mitochondria stop respiring due to the exhaustion of substrate or inhibition (Fig. 6A). Time to inhibition (oxygen reaccumulation) should be dependent on the rate of NO production from reactions of nitrite with deoxy-Hb and the rate of NO consumption by excess oxy-Hb. With the addition of nitrite (18 μM) and low levels of Hb (10–20 μM), mitochondrial respiration was inhibited compared with mitochondria with nitrite or Hb alone. The shortest time to inhibition was observed with nitrite and 20 μM Hb, above which increasing concentrations of Hb resulted in longer times to inhibition (Fig. 6B). These mitochondrial inhibition experiments demonstrated a U-shaped relationship between nitrite and Hb levels consistent with the results of our in vivo experiments. Our animal experiments suggest an interaction between the effects of nitrite and the level of hemolysis such that low levels of hemolysis accentuate the vasodilatory effects of nitrite. These mitochondrial experiments demonstrate that NO generation and accumulation from nitrite reduction by Hb is maximal at low levels of hemolysis and decreases with higher levels of hemolysis. These results suggest that the in vivo accentuated vasodilatory effects of nitrite during low levels of hemolysis may be mediated by the generation of NO from the reduction of nitrite by Hb.
Fig. 6.

Nitrite- and Hb-dependent inhibition of mitochondrial respiration shows a U-shaped curve. A: mitochondria (2 mg/ml) were stimulated to respire in the presence of no treatment (solid trace), nitrite (18 μM, dark gray trace), oxygenated Hb (20 μM, mid-gray trace), or nitrite (18 μM) + Hb (20 μM) (light gray trace). Removal of the lid from the sealed chamber is denoted by the arrow. Time to inhibition was measured from removal of the lid to time the trace deviated from 0% oxygen. In these experiments, the oxygen trace deviates from zero once the mitochondria stop respiring due to the exhaustion of substrate or inhibition by NO produced by reactions of Hb with nitrite. B: quantification of several traces similar to those shown in A with different levels of Hb. Inhibition of mitochondrial respiration (secondary to NO production) occurred most rapidly with nitrite and low levels of Hb. The time to inhibition appears to be dependent on the rate of NO production from reactions of nitrite with deoxy-Hb and the rate of NO consumption by excess oxy-Hb. All data are means ± SE of at least 3 independent experiments. *P < 0.01 compared with nitrite alone.
DISCUSSION
Under physiological conditions, our experiments demonstrate that low-dose sodium nitrite is a potent arterial vasodilator that increases cardiac performance by direct afterload reduction with mild chronotropic effects. During hemolysis, we detected a consistent U-shaped relationship between the effects of nitrite and cell-free plasma Hb levels across three experimental settings, suggesting an interaction between nitrite and the level of intravascular Hb.
Nitrite reacts with oxy-Hb and deoxy-Hb to form met-Hb and met-Hb + NO, respectively (5, 11, 16, 31, 33). During low-level hemolysis, these reactions will minimize the amount of oxy-Hb available in the plasma that can consume NO (via the dioxygenation reaction) and generate NO by the reaction of nitrite with deoxy-Hb. The net result is accentuated vasodilation compared with no hemolysis. At higher levels of intravascular hemolysis, the large amounts of cell-free plasma Hb overwhelms the nitrite reductase reaction of Hb and consumes both the NO formed by nitrite reduction with Hb and endothelium-derived NO. The net result is vasoconstriction compared with low-level hemolysis and no hemolysis. During a sodium nitroprusside infusion with low-level hemolysis and nitrite, the nitrite-oxy-Hb reaction minimizes the oxy-Hb concentration and allows the donated NO from nitroprusside to cause vasodilation. This vasodilation is further accentuated by the production of additional NO from reactions of nitrite with deoxy-Hb. At higher levels of hemolysis, the vasodilatory effects of sodium nitroprusside are attenuated by the high levels of oxy-Hb, which consume both the NO donated from nitroprusside and the NO generated from the reaction of nitrite with deoxy-Hb. During the mitochondrial experiments, maximal NO production and accumulation occurred with nitrite and low levels of Hb because the excess heme groups at higher levels of Hb consumed the NO generated by the nitrite-deoxy-Hb reaction, leading to decreased NO accumulation.
Traditional NO donors, such as sodium nitroprusside, produce dose-dependent vasodilation that is inhibited by cell-free plasma Hb (52). In contrast, nitrite led to accentuated vasodilation during low-level hemolysis despite the presence of oxy-Hb levels sufficient to scavenge any NO that might be formed if nitrite acted as pure NO donor. The vasodilatory effect of nitrite clearly differs from traditional NO donors in the presence of Hb and can in part be explained by the nitrite reductase activity of Hb (11, 31, 33). Generation of NO from nitrite and Hb requires both hypoxia and an acidic environment, which are present in hypoxic tissues. This allows for maximal NO generation by the deoxyheme-nitrite allosteric reaction as Hb deoxygenates within the circulation.
This study provides in vivo evidence supporting the theory that Hb possesses a functional nitrite reductase activity. The notable interaction between nitrite and Hb in this study was markedly different from the behavior of a traditional NO donor, sodium nitroprusside. While the latter was inhibited in a dose-dependent manner, the former was potentiated by Hb at concentrations that produced NO in our in vitro mitochondrial NO sensor experiments. In another study with the Kim-Shapiro laboratory (3), we examined how NO could be generated from the nitrite reductase activity of Hb and then be able to escape heme autocapture (via Eq. 1 or an analogously fast reaction with deoxy-Hb). We found that nitrite can also bind to met-Hb to form a nitrite-met-Hb intermediate that possesses nitrogen-dioxide-ferrous Hb character. The NO that forms from nitrite reduction can react rapidly in a radical-radical reaction with nitrogen dioxide to form N2O3 (3). N2O3 is an uncharged, highly lipophilic, and diffusible molecule that is more stable than authentic NO. N2O3 can nitrosate thiols to form vasodilatory S-nitrosothiols, can homolyze back into NO, or can regenerate nitrite. The apparent inhibition of the nitrite effect at higher Hb concentrations in these experiments indicates that at least part of the mechanism must involve the regeneration of NO, which can be in part scavenged by excess Hb.
The levels of cell-free plasma Hb in the low hemolysis group of this study are consistent with the levels observed during sickle cell vasoocclusive crisis (40, 58, 65) and during other clinically relevant human hemolytic conditions such as cardiopulmonary bypass, malarial infection, hemolytic-uremic syndrome/thrombotic thrombocytopenic purpura, paroxysmal nocturnal hemoglobinuria, alloimmune hemolytic anemia, and rhabdomyolysis (myoglobin) (12, 40, 55, 58, 63, 65, 69). All of these conditions have now been associated with progressive vasculopathy and pulmonary hypertension and are associated with systemic NO scavenging by plasma Hb (22, 52, 67). Furthermore, hemolysis is associated with platelet activation and inhibition of NO-cGMP signaling in platelets (75). These represent processes in which the allosteric nitrite reductase activity of Hb may make nitrite an ideal therapeutic agent to attenuate the effects of accelerated NO scavenging by cell-free Hb released during intravascular hemolysis (1, 17, 19, 41, 52, 59, 65). In these clinical scenarios, the administration of low-dose nitrite will have minimal physiological effects in normal tissues. However, in tissues that have become hypoxic secondary to vasoconstriction from accelerated NO scavenging by cell-free Hb, low-dose nitrite may cause vasodilation by (1) reacting with oxy-Hb to form met-Hb, thereby preventing NO scavenging; and (2) reacting with deoxy-Hb to generate NO and met-Hb. The net effect would be hypoxic vasodilation in local tissues that have become ischemic from the vasoconstrictive effects of accumulating cell-free plasma Hb from ongoing low-level intravascular hemolysis.
In addition to low-level intravascular hemolysis, nitrite may also have a therapeutic role in minimizing the vascular toxicities of more severe episodes of intravascular hemolysis (cell-free plasma Hb levels > 50 μM) and the administration of several types of cell-free Hb-based blood substitutes (cell-free plasma Hb levels > 600 μM) (14, 15, 28, 29, 77). In these clinical scenarios, the ability of nitrite to attenuate the physiological effects of cell-free plasma Hb will be overwhelmed by the accelerated NO consumption caused by the large amounts of plasma Hb. However, in these scenarios, the affected tissues and organs will subsequently develop areas of hypoxia and acidosis. Within these areas, there will be accelerated reduction of nitrite by deoxygenated cell-free plasma Hb leading to local NO generation and vasodilation. Therapeutic strategies to deliver Hb-based blood substitutes will either require increasing the molar ratio of nitrite to Hb or will require modulating the reaction kinetics by increasing the concentration of nitrite bound to met-Hb (to facilitate the formation of N2O3) (3), decreasing the Hb oxygen affinity (so that there is more deoxyheme to reduce nitrite), or decreasing the redox potential of the heme-based blood substitute (to increase the reactivity with nitrite). Therefore, despite not reversing global physiological changes during high-level canine hemolysis, nitrite may be able to limit organ damage and dysfunction during severe hemolytic episodes and during the administration of Hb-based blood substitutes. In these situations, the optimal dose of nitrite with Hb for resuscitation that maximizes NO generation and minimizes Hb oxidation (which would limit oxygen transport of Hb) has yet to be determined.
Conclusions
Nitrite is a unique vasodilator. Under normal physiological conditions, low-dose nitrite is potent arterial vasodilator that increases cardiac performance by direct afterload reduction. During hemolysis, the allosteric nitrite reductase activity of Hb produces an accentuated vasodilatory response to nitrite during low levels of hemolysis that is overwhelmed during high levels of hemolysis. These unique properties of nitrite and Hb may allow nitrite to be used as therapy during episodes of acute intravascular hemolysis or potentially to detoxify cell-free Hb-based blood substitutes.
GRANTS
This work was funded by the Intramural Research Division of the National Heart, Lung, and Blood Institute (NHLBI) and the Critical Care Medicine Department, Clinical Center, of the National Institutes of Health (NIH) and by extramural NHLBI Grant K22-HL-089041-01. Further support was provided from the Office of Rare Diseases and the Clinical Center of the NIH via a bench to bedside grant.
DISCLOSURES
M. T. Gladwin is named as a coinventor on an NIH government provisional patent application for the use of nitrite salts in the treatment of cardiovascular diseases.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Aessopos A, Stamatelos G, Skoumas V, Vassilopoulos G, Mantzourani M, Loukopoulos D. Pulmonary hypertension and right heart failure in patients with beta- thalassemia intermedia. Chest 107: 50–53, 1995. [DOI] [PubMed] [Google Scholar]
- 2.Azarov I, Huang KT, Basu S, Gladwin MT, Hogg N, Kim-Shapiro DB. Nitric oxide scavenging by red blood cells as a function of hematocrit and oxygenation. J Biol Chem 280: 39024–39032, 2005. [DOI] [PubMed] [Google Scholar]
- 3.Basu S, Grubina R, Huang J, Conradie J, Huang Z, Jeffers A, Jiang A, He X, Azarov I, Seibert R, Mehta A, Patel R, King SB, Hogg N, Ghosh A, Gladwin MT, Kim-Shapiro DB. Catalytic generation of N2O3 by the concerted nitrite reductase and anhydrase activity of hemoglobin. Nat Chem Biol 3: 785–794, 2007. [DOI] [PubMed] [Google Scholar]
- 4.Bjorne HH, Petersson J, Phillipson M, Weitzberg E, Holm L, Lundberg JO. Nitrite in saliva increases gastric mucosal blood flow and mucus thickness. J Clin Invest 113: 106–114, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brooks J The action of nitrite on haemoglobin in the absence of oxygen. Proc R Soc Med 123: 368–382, 1937. [Google Scholar]
- 6.Butler AR, Megson IL, Wright PG. Diffusion of nitric oxide and scavenging by blood in the vasculature. Biochim Biophys Acta 1425: 168–176, 1998. [DOI] [PubMed] [Google Scholar]
- 7.Carlsen E, Comroe JH Jr. The rate of uptake of carbon monoxide and of nitric oxide by normal human erythrocytes and experimentally produced spherocytes. J Gen Physiol 42: 83–107, 1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coin JT, Olson JS. The rate of oxygen uptake by human red blood cells. J Biol Chem 254: 1178–1190, 1979. [PubMed] [Google Scholar]
- 9.Cole RH, Vandegriff KD, Szeri AJ, Savas O, Baker DA, Winslow RM. A quantitative framework for the design of acellular hemoglobins as blood substitutes: implications of dynamic flow conditions. Biophys Chem 128: 63–74, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, Yang BK, Waclawiw MA, Zalos G, Xu X, Huang KT, Shields H, Kim-Shapiro DB, Schechter AN, Cannon RO 3rd, Gladwin MT. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med 9: 1498–1505, 2003. [DOI] [PubMed] [Google Scholar]
- 11.Crawford JH, Isbell TS, Huang Z, Shiva S, Chacko BK, Schechter AN, Darley-Usmar VM, Kerby JD, Lang JD Jr, Kraus D, Ho C, Gladwin MT, Patel RP. Hypoxia, red blood cells, and nitrite regulate NO-dependent hypoxic vasodilation. Blood 107: 566–574, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis CL, Kausz AT, Zager RA, Kharasch ED, Cochran RP. Acute renal failure after cardiopulmonary bypass in related to decreased serum ferritin levels. J Am Soc Nephrol 10: 2396–2402, 1999. [DOI] [PubMed] [Google Scholar]
- 13.Dejam A, Hunter CJ, Pelletier MM, Hsu LL, Machado RF, Shiva S, Power GG, Kelm M, Gladwin MT, Schechter AN. Erythrocytes are the major intravascular storage sites of nitrite in human blood. Blood 106: 734–739, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doherty DH, Doyle MP, Curry SR, Vali RJ, Fattor TJ, Olson JS, Lemon DD. Rate of reaction with nitric oxide determines the hypertensive effect of cell-free hemoglobin. Nat Biotechnol 16: 672–676, 1998. [DOI] [PubMed] [Google Scholar]
- 15.Dou Y, Maillett DH, Eich RF, Olson JS. Myoglobin as a model system for designing heme protein based blood substitutes. Biophys Chem 98: 127–148, 2002. [DOI] [PubMed] [Google Scholar]
- 16.Doyle MP, Hoekstra JW. Oxidation of nitrogen oxides by bound dioxygen in hemoproteins. J Inorg Biochem 14: 351–358, 1981. [DOI] [PubMed] [Google Scholar]
- 17.Du ZD, Roguin N, Milgram E, Saab K, Koren A. Pulmonary hypertension in patients with thalassemia major. Am Heart J 134: 532–537, 1997. [DOI] [PubMed] [Google Scholar]
- 18.Duranski MR, Greer JJ, Dejam A, Jaganmohan S, Hogg N, Langston W, Patel RP, Yet SF, Wang X, Kevil CG, Gladwin MT, Lefer DJ. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J Clin Invest 115: 1232–1240, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eberhardt RT, McMahon L, Duffy SJ, Steinberg MH, Perrine SP, Loscalzo J, Coffman JD, Vita JA. Sickle cell anemia is associated with reduced nitric oxide bioactivity in peripheral conduit and resistance vessels. Am J Hematol 74: 104–111, 2003. [DOI] [PubMed] [Google Scholar]
- 20.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288: 373–376, 1980. [DOI] [PubMed] [Google Scholar]
- 21.Gladwin MT, Raat NJ, Shiva S, Dezfulian C, Hogg N, Kim-Shapiro DB, Patel RP. Nitrite as a vascular endocrine nitric oxide reservoir that contributes to hypoxic signaling, cytoprotection, and vasodilation. Am J Physiol Heart Circ Physiol 291: H2026–H2035, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Gladwin MT, Sachdev V, Jison ML, Shizukuda Y, Plehn JF, Minter K, Brown B, Coles WA, Nichols JS, Ernst I, Hunter LA, Blackwelder WC, Schechter AN, Rodgers GP, Castro O, Ognibene FP. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med 350: 886–895, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Gladwin MT, Schechter AN, Kim-Shapiro DB, Patel RP, Hogg N, Shiva S, Cannon RO 3rd, Kelm M, Wink DA, Espey MG, Oldfield EH, Pluta RM, Freeman BA, Lancaster JR Jr, Feelisch M, Lundberg JO. The emerging biology of the nitrite anion. Nat Chem Biol 1: 308–314, 2005. [DOI] [PubMed] [Google Scholar]
- 24.Godber BL, Doel JJ, Sapkota GP, Blake DR, Stevens CR, Eisenthal R, Harrison R. Reduction of nitrite to nitric oxide catalyzed by xanthine oxidoreductase. J Biol Chem 275: 7757–7763, 2000. [DOI] [PubMed] [Google Scholar]
- 25.Gow AJ, Luchsinger BP, Pawloski JR, Singel DJ, Stamler JS. The oxyhemoglobin reaction of nitric oxide. Proc Natl Acad Sci USA 96: 9027–9032, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gow AJ, Stamler JS. Reactions between nitric oxide and haemoglobin under physiological conditions. Nature 391: 169–173, 1998. [DOI] [PubMed] [Google Scholar]
- 27.Hataishi R, Rodrigues AC, Neilan TG, Morgan JG, Buys E, Shiva S, Tambouret R, Jassal DS, Raher MJ, Furutani E, Ichinose F, Gladwin MT, Rosenzweig A, Zapol WM, Picard MH, Bloch KD, Scherrer-Crosbie M. Inhaled nitric oxide decreases infarction size and improves left ventricular function in a murine model of myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 291: H379–H384, 2006. [DOI] [PubMed] [Google Scholar]
- 28.Hess JR, MacDonald VW, Brinkley WW. Systemic and pulmonary hypertension after resuscitation with cell-free hemoglobin. J Appl Physiol 74: 1769–1778, 1993. [DOI] [PubMed] [Google Scholar]
- 29.Hess JR, Macdonald VW, Gomez CS, Coppes V. Increased vascular resistance with hemoglobin-based oxygen carriers. Artif Cells Blood Substit Immobil Biotechnol 22: 361–372, 1994. [DOI] [PubMed] [Google Scholar]
- 30.Huang KT, Han TH, Hyduke DR, Vaughn MW, Van Herle H, Hein TW, Zhang C, Kuo L, Liao JC. Modulation of nitric oxide bioavailability by erythrocytes. Proc Natl Acad Sci USA 98: 11771–11776, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang KT, Keszler A, Patel N, Patel RP, Gladwin MT, Kim-Shapiro DB, Hogg N. The reaction between nitrite and deoxyhemoglobin. Reassessment of reaction kinetics and stoichiometry. J Biol Chem 280: 31126–31131, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Huang Z, Louderback JG, Goyal M, Azizi F, King SB, Kim-Shapiro DB. Nitric oxide binding to oxygenated hemoglobin under physiological conditions. Biochim Biophys Acta 1568: 252–260, 2001. [DOI] [PubMed] [Google Scholar]
- 33.Huang Z, Shiva S, Kim-Shapiro DB, Patel RP, Ringwood LA, Irby CE, Huang KT, Ho C, Hogg N, Schechter AN, Gladwin MT. Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. J Clin Invest 115: 2099–2107, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hunter CJ, Dejam A, Blood AB, Shields H, Kim-Shapiro DB, Machado RF, Tarekegn S, Mulla N, Hopper AO, Schechter AN, Power GG, Gladwin MT. Inhaled nebulized nitrite is a hypoxia-sensitive NO-dependent selective pulmonary vasodilator. Nat Med 10: 1122–1127, 2004. [DOI] [PubMed] [Google Scholar]
- 35.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci USA 84: 9265–9269, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ignarro LJ, Byrns RE, Buga GM, Wood KS. Endothelium-derived relaxing factor from pulmonary artery and vein possesses pharmacologic and chemical properties identical to those of nitric oxide radical. Circ Res 61: 866–879, 1987. [DOI] [PubMed] [Google Scholar]
- 37.Ignarro LJ, Gruetter CA. Requirement of thiols for activation of coronary arterial guanylate cyclase by glyceryl trinitrate and sodium nitrite: possible involvement of S-nitrosothiols. Biochim Biophys Acta 631: 221–231, 1980. [DOI] [PubMed] [Google Scholar]
- 38.Isbell TS, Gladwin MT, Patel RP. Hemoglobin oxygen fractional saturation regulates nitrite-dependent vasodilation of aortic ring bioassays. Am J Physiol Heart Circ Physiol 293: H2565–H2572, 2007. [DOI] [PubMed] [Google Scholar]
- 39.Jia L, Bonaventura C, Bonaventura J, Stamler JS. S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature 380: 221–226, 1996. [DOI] [PubMed] [Google Scholar]
- 40.Kaul DK, Hebbel RP. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J Clin Invest 106: 411–420, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaul DK, Liu XD, Chang HY, Nagel RL, Fabry ME. Effect of fetal hemoglobin on microvascular regulation in sickle transgenic-knockout mice. J Clin Invest 114: 1136–1145, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim-Shapiro DB, Schechter AN, Gladwin MT. Unraveling the reactions of nitric oxide, nitrite, and hemoglobin in physiology and therapeutics. Arterioscler Thromb Vasc Biol 26: 697–705, 2006. [DOI] [PubMed] [Google Scholar]
- 43.Kozlov AV, Staniek K, Nohl H. Nitrite reductase activity is a novel function of mammalian mitochondria. FEBS Lett 454: 127–130, 1999. [DOI] [PubMed] [Google Scholar]
- 44.Kunert MP, Liard JF, Abraham DJ, Lombard JH. Low-affinity hemoglobin increases tissue Po2 and decreases arteriolar diameter and flow in the rat cremaster muscle. Microvasc Res 52: 58–68, 1996. [DOI] [PubMed] [Google Scholar]
- 45.Li H, Samouilov A, Liu X, Zweier JL. Characterization of the magnitude and kinetics of xanthine oxidase-catalyzed nitrite reduction. Evaluation of its role in nitric oxide generation in anoxic tissues. J Biol Chem 276: 24482–24489, 2001. [DOI] [PubMed] [Google Scholar]
- 46.Liao JC, Hein TW, Vaughn MW, Huang KT, Kuo L. Intravascular flow decreases erythrocyte consumption of nitric oxide. Proc Natl Acad Sci USA 96: 8757–8761, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu X, Miller MJ, Joshi MS, Sadowska-Krowicka H, Clark DA, Lancaster JR Jr. Diffusion-limited reaction of free nitric oxide with erythrocytes. J Biol Chem 273: 18709–18713, 1998. [DOI] [PubMed] [Google Scholar]
- 48.Liu X, Samouilov A, Lancaster JR Jr, Zweier JL. Nitric oxide uptake by erythrocytes is primarily limited by extracellular diffusion not membrane resistance. J Biol Chem 277: 26194–26199, 2002. [DOI] [PubMed] [Google Scholar]
- 49.Lundberg JON, Weitzberg E, Lundberg JM, Alving K. Intragastric nitric oxide production in humans: measurements in expelled air. Gut 35: 1543–1546, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McMahon TJ Hemoglobin and nitric oxide. N Engl J Med 349: 402–405, 2003. [PubMed] [Google Scholar]
- 51.Millar TM, Stevens CR, Benjamin N, Eisenthal R, Harrison R, Blake DR. Xanthine oxidoreductase catalyses the reduction of nitrates and nitrite to nitric oxide under hypoxic conditions. FEBS Lett 427: 225–228, 1998. [DOI] [PubMed] [Google Scholar]
- 52.Minneci PC, Deans KJ, Zhi H, Yuen PS, Star RA, Banks SM, Schechter AN, Natanson C, Gladwin MT, Solomon SB. Hemolysis-associated endothelial dysfunction mediated by accelerated NO inactivation by decompartmentalized oxyhemoglobin. J Clin Invest 115: 3409–3417, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mittal CK, Arnold WP, Murad F. Characterization of protein inhibitors of guanylate cyclase activation from rat heart and bovine lung. J Biol Chem 253: 1266–1271, 1978. [PubMed] [Google Scholar]
- 54.Modin A, Bjorne H, Herulf M, Alving K, Weitzberg E, Lundberg JO. Nitrite-derived nitric oxide: a possible mediator of “acidic-metabolic” vasodilation. Acta Physiol Scand 171: 9–16, 2001. [DOI] [PubMed] [Google Scholar]
- 55.Murakami F, Usui A, Hiroura M, Kawamura M, Koyama T, Murase M. Clinical study of totally roller pumpless cardiopulmonary bypass system. Artif Organs 21: 803–807, 1997. [DOI] [PubMed] [Google Scholar]
- 56.Nagababu E, Ramasamy S, Abernethy DR, Rifkind JM. Active nitric oxide produced in the red cell under hypoxic conditions by deoxyhemoglobin mediated nitrite reduction. J Biol Chem 278: 46349–46356, 2003. [DOI] [PubMed] [Google Scholar]
- 57.Nagababu E, Ramasamy S, Rifkind JM. S-nitrosohemoglobin: a mechanism for its formation in conjunction with nitrite reduction by deoxyhemoglobin. Nitric Oxide 15: 20–29, 2006. [DOI] [PubMed] [Google Scholar]
- 58.Naumann HN, Diggs LW, Barreras L, Williams BJ. Plasma hemoglobin and hemoglobin fractions in sickle cell crisis. Am J Clin Pathol 56: 137–147, 1971. [DOI] [PubMed] [Google Scholar]
- 59.Nolan VG, Wyszynski DF, Farrer LA, Steinberg MH. Hemolysis-associated priapism in sickle cell disease. Blood 106: 3264–3267, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Okamoto M, Tsuchiya K, Kanematsu Y, Izawa Y, Yoshizumi M, Kagawa S, Tamaki T. Nitrite-derived nitric oxide formation following ischemia-reperfusion injury in kidney. Am J Physiol Renal Physiol 288: F182–F187, 2005. [DOI] [PubMed] [Google Scholar]
- 61.Palmer RM, Ashton DS, Moncada S. Vascular endothelial cells synthesize nitric oxide from l-arginine. Nature 333: 664–666, 1988. [DOI] [PubMed] [Google Scholar]
- 62.Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 327: 524–526, 1987. [DOI] [PubMed] [Google Scholar]
- 63.Pepper JR, Mumby S, Gutteridge JM. Transient iron-overload with bleomycin-detectable iron present during cardiopulmonary bypass surgery. Free Radic Res 21: 53–58, 1994. [DOI] [PubMed] [Google Scholar]
- 64.Rassaf T, Flogel U, Drexhage C, Hendgen-Cotta U, Kelm M, Schrader J. Nitrite reductase function of deoxymyoglobin: oxygen sensor and regulator of cardiac energetics and function. Circ Res 100: 1749–1754, 2007. [DOI] [PubMed] [Google Scholar]
- 65.Reiter CD, Wang X, Tanus-Santos JE, Hogg N, Cannon RO, Schechter AN, Gladwin MT. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med 8: 1383–1389, 2002. [DOI] [PubMed] [Google Scholar]
- 66.Rifkind JM, Nagababu E, Barbiro-Michaely E, Ramasamy S, Pluta RM, Mayevsky A. Nitrite infusion increases cerebral blood flow and decreases mean arterial blood pressure in rats: a role for red cell NO. Nitric Oxide 16: 448–456, 2007. [DOI] [PubMed] [Google Scholar]
- 67.Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA 293: 1653–1662, 2005. [DOI] [PubMed] [Google Scholar]
- 68.Rowland T, Goff D, Martel L, Ferrone L, Kline G. Normalization of maximal cardiovascular variables for body size in premenarcheal girls. Pediatr Cardiol 21: 429–432, 2000. [DOI] [PubMed] [Google Scholar]
- 69.Shimono T, Shomura Y, Tani K, Shimamoto A, Hioki I, Tokui T, Onoda K, Takao M, Shimpo H, Yada I. Clinical evaluation of a silicone coated hollow fiber oxygenator. ASAIO J 43: M735–M739, 1997. [PubMed] [Google Scholar]
- 70.Shiva S, Huang Z, Grubina R, Sun J, Ringwood LA, MacArthur PH, Xu X, Murphy E, Darley-Usmar VM, Gladwin MT. Deoxymyoglobin is a nitrite reductase that generates nitric oxide and regulates mitochondrial respiration. Circ Res 100: 654–661, 2007. [DOI] [PubMed] [Google Scholar]
- 71.Stamler JS, Jia L, Eu JP, McMahon TJ, Demchenko IT, Bonaventura J, Gernert K, Piantadosi CA. Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science 276: 2034–2037, 1997. [DOI] [PubMed] [Google Scholar]
- 72.Vaughn MW, Huang KT, Kuo L, Liao JC. Erythrocyte consumption of nitric oxide: competition experiment and model analysis. Nitric Oxide 5: 18–31, 2001. [DOI] [PubMed] [Google Scholar]
- 73.Vaughn MW, Huang KT, Kuo L, Liao JC. Erythrocytes possess an intrinsic barrier to nitric oxide consumption. J Biol Chem 275: 2342–2348, 2000. [DOI] [PubMed] [Google Scholar]
- 74.Vaughn MW, Kuo L, Liao JC. Effective diffusion distance of nitric oxide in the microcirculation. Am J Physiol Heart Circ Physiol 274: H1705–H1714, 1998. [DOI] [PubMed] [Google Scholar]
- 75.Villagra J, Shiva S, Hunter LA, Machado RF, Gladwin MT, Kato GJ. Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension, and nitric oxide scavenging by cell-free hemoglobin. Blood 110: 2166–2172, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Webb A, Bond R, McLean P, Uppal R, Benjamin N, Ahluwalia A. Reduction of nitrite to nitric oxide during ischemia protects against myocardial ischemia-reperfusion damage. Proc Natl Acad Sci USA 101: 13683–13688, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Winslow RM αα-Crosslinked hemoglobin: was failure predicted by preclinical testing? Vox Sang 79: 1–20, 2000. [DOI] [PubMed] [Google Scholar]
- 78.Yang BK, Vivas EX, Reiter CD, Gladwin MT. Methodologies for the sensitive and specific measurement of S-nitrosothiols, iron-nitrosyls, and nitrite in biological samples. Free Radic Res 37: 1–10, 2003. [DOI] [PubMed] [Google Scholar]
- 79.Yu B, Raher MJ, Volpato GP, Bloch KD, Ichinose F, Zapol WM. Inhaled nitric oxide enables artificial blood transfusion without hypertension. Circulation 117: 1982–1990, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zweier JL, Wang P, Samouilov A, Kuppusamy P. Enzyme-independent formation of nitric oxide in biological tissues. Nat Med 1: 804–809, 1995. [DOI] [PubMed] [Google Scholar]