

Abstract
Mechanical alternans in cardiac muscle is associated with intracellular Ca2+ alternans. Mechanisms underlying intracellular Ca2+ alternans are unclear. In previous experimental studies, we produced alternans of systolic Ca2+ under voltage clamp, either by partially inhibiting the Ca2+ release mechanism, or by applying small depolarizing pulses. In each case, alternans relied on propagating waves of Ca2+ release. The aim of this study is to investigate by computer modeling how alternans of systolic Ca2+ is produced. A mathematical model of a cardiac cell with 75 coupled elements is developed, with each element contains L-type Ca2+ current, a subspace into which Ca release takes place, a cytoplasmic space, sarcoplasmic reticulum (SR) release channels [ryanodine receptor (RyR)], and uptake sites (SERCA). Interelement coupling is via Ca2+ diffusion between neighboring subspaces via cytoplasmic spaces and network SR spaces. Small depolarizing pulses were simulated by step changes of cell membrane potential (20 mV) with random block of L-type channels. Partial inhibition of the release mechanism is mimicked by applying a reduction of RyR open probability in response to full stimulation by L-type channels. In both cases, systolic alternans follow, consistent with our experimental observations, being generated by propagating waves of Ca2+ release and sustained through alternation of SR Ca2+ content. This study provides novel and fundamental insights to understand mechanisms that may underlie intracellular Ca2+ alternans without the need for refractoriness of L-type Ca or RyR channels under rapid pacing.
Keywords: sarcoplasmic reticulum, computer modeling, diffusion, computer model
up to 50% of deaths seen in heart failure are sudden and likely, therefore, to be due to cardiac arrhythmias. In heart failure, up to 40% of patients exhibit a phenomenon known as mechanical alternans (19), when the force of contraction of the heart alternates between strong and weak. A related phenomenon is microvolt T-wave alternans, detectable in the ECG (1) and considered to be especially arrhythmogenic. Many cardiac arrhythmias are thought to be due to irregularity of Ca2+ handling (11, 30, 36) by the sarcoplasmic reticulum (SR), the main source of Ca2+ for cardiac contraction. Systolic Ca2+ release occurs via Ca2+-induced Ca2+ release (CICR), triggered by Ca2+ entry on L-type Ca2+ channels (3). In mechanical alternans, large and small contractions follow each other (10, 13, 27) due to alternation of systolic Ca2+ (21, 40). In isolated cardiac cells, alternans of systolic Ca2+ is associated with alternans of cell shortening (16). Systolic Ca alternans are also associated with alternans of action potential duration (6, 22, 31) and alternans of the T-wave of the ECG (31).
Changes in amplitude and timing of Ca release during systole can influence the action potential shape by acting on Ca-sensitive currents, e.g., Na/Ca exchange (NCX) causing ECG abnormalities and the arrhythmogenicity of systolic Ca alternans.
Previous work in our laboratory has focused on intracellular Ca2+ concentration ([Ca2+]i) alternans and suggests that SR Ca content fluctuation is key to cardiac alternans (7). We found that, in rat ventricular myocytes, small (20 mV) depolarizing pulses produced alternans of the amplitude of the Ca2+ transient. Confocal measurements of [Ca2+]i showed the larger transients involved waves of CICR. In addition, SR Ca2+ content alternated in phase with the alternans of Ca2+ transient amplitude. During alternans of systolic release, we found that the relationship between Ca2+ transient amplitude and SR Ca2+ content was extremely steep. We attributed this steeper relationship to the dependence on Ca wave propagation, as this requires a “threshold” SR Ca2+ content (8).
The work presented here attempts to build a theoretical framework to describe the observations of Ca2+ release profiles measured by confocal microscopy and to investigate detailed mechanisms underlying the emergence of Ca2+ alternans. The simulation results support conclusions from experiments that alternans can arise due to propagation of waves of Ca2+ release, and that this results from a large increase in the gain of the feedback controlling SR Ca2+ content. This study provides a novel and fundamental insight into a mechanism underlying the genesis of systolic Ca2+ alternans that does not require refractoriness of L-type Ca or ryanodine receptor (RyR) channel.
METHODS
Our model consists of a cluster of coupled RyRs for a cardiac cell (see Fig. 1A). The cell has a length of 150 μm, which is discretized into 75 units along the longitudinal direction. Each unit encompasses 2 μm in the longitudinal dimension, and the entire cell depth and height in other dimensions. The spatial resolution of 2 μm is similar to the longitudinal distance between adjacent Ca2+ release units of cardiac cells (1.94 ± 0.31 μm) (5) and is fine enough to produce stable numerical solutions for Ca2+ waves in the model. Each element has a cluster of unitary voltage-gated L-type Ca2+ channels (contributing to global L-type Ca2+ channel current in each element; hereafter referred to as L-type Ca2+ channel), a subspace under the sarcolemma, a cytoplasmic space, and a cluster of SR RyR channels (contributing global SR RyR channel release in each element; hereafter referred to as RyR channel) (see Fig. 1B). For each element, mathematical equations (see appendix) were developed to model Ca2+ cycling. Interelement coupling is via Ca2+ diffusion from subspaces to cytoplasmic spaces and via network SR spaces. In simulations, we have taken two approaches: 100-ms depolarizing pulses at 1 Hz from a holding potential of −40 mV were used to activate L-type channels to open in each element. In the first approach, the depolarizing pulse was from −40 to 0 mV, and we partially inhibited the Ca2+ release mechanism by increasing the threshold of RyR Ca2+ release to mimic the decreased sensitivity of RyR by tetracaine, as suggested by a previous study (15). In the second approach, the depolarizing pulse was small, from −40 to −20 mV. To simulate the decreased global L-type Ca2+ current in the cell in response to such a small stimulus pulse (7), the L-type Ca2+ current was blocked (setting to zero) in some elements randomly chosen from the cell, but remained the same as in the control for the nonblocked elements. Thus Ca2+ release was activated at only a few elements, with L-type Ca2+ current being fully activated. In simulations, the population of blocked elements varies (say 40 out of 75 elements) to produce different levels of L-type Ca2+ current amplitude reduction in the cell.
Fig. 1.

A: schematic model of a cluster of coupled ryanodine receptors (RyRs) in a cardiac cell. The cell has a length of 150 μm, with 75 elements, giving 2-μm spatial resolution. Coupling of these elements is via Ca2+ diffusion between neighboring cytoplasmic and network sarcoplasmic reticulum (SR) spaces, as described in the appendix. B: schematic model of Ca2+ cycling for each element of the cluster illustrated in A. Three main Ca2+ cycling processes were modeled, which included 1) Ca2+ entry by L-type Ca2+ channel and Ca2+-induced Ca2+ release (CICR); 2) Ca2+ removal by Na/Ca exchanger and ATP-dependent Ca2+ pump; and 3) Ca2+ diffusion and Ca2+ binding to its buffers.
Development of equations: table and choices of parameters.
We assume the global L-type Ca channel current (produced by a cluster of unitary L-type channels in each element) and intracellular Ca2+ cycling of RyRs have the same kinetics for all elements and are described by whole cell equations similar to the approach used in the study of Shiferaw and Karma (34). The equations of Ca2+ cycling were modified from the model developed by Kurata et al. (20) with parameters and equations derived from experimental data of ventricular myocytes and used in the Luo-Rudy (26) model of guinea pig ventricular myocytes. In the model, some parameters, such as the volumes of cell, subspace, cytoplasm, network SR, and junctional SR, were adjusted (3) to fit the rat ventricular cell in our experiments. Despite being simple, the model captures most general features of cardiac Ca cycling necessary for CICR wave propagation, which enables us to unravel the major determinants responsible for systolic Ca2+ alternans, i.e., propagation of CICR waves and steep relationship between RyR release and the SR Ca content. Detailed glossary and equations are given in the appendix.
The cardiac myocyte is a spatially extended object, within which Ca2+ diffuses. Our previous experimental results suggest Ca2+ diffusion is crucial during systolic Ca2+ alternans (7, 9). Spatially extended models have been developed to study Ca2+ sparks and waves (38). To simulate Ca2+ wave propagation and Ca2+ release alternans, we developed a more detailed model of Ca2+ spatial diffusion. It includes Ca2+ diffusion from the subspaces to myoplasmic spaces and between network SR spaces of neighboring elements. The diffusion parameters were chosen in such a way that the propagation of CICR waves matches our experimental data; these parameters are close to those used in previous studies (14, 15, 17, 20, 23, 26, 28, 32, 34, 35, 37, 38). Detailed comparisons of model parameter values with those of literature-based and other models are given in Table 1.
Table 1.
Comparison of parameters used in the model with experimental data and other models
| Parameters | Model Value | Literature-Based or Other Model Values | Ref. No. |
|---|---|---|---|
| tCa diff (time constant for Ca diffusion from subspace to cytoplasmic space) | 0.04 ms | 0.04 ms | 23 |
| 0.05–0.6 ms | 37 | ||
| 0.04 ms | 20 | ||
| 0.0007 ms | 32 | ||
| 0.0007 ms | 38 | ||
| 0.005 ms | 14 | ||
| 2 ms | 34 | ||
| 26.7 ms | 28 | ||
| τCa i diff (time constant for Ca diffusion between neighboring cytoplasmic spaces) | 4 ms | 5 ms | 34 |
| τCa up diff (time constant for Ca diffusion between neighboring network SR spaces) | 600 ms | 100 ms | 34 |
| Estimated Ca diffusion coefficient in the cytoplasmic space | 10.0 × 10−6 cm2/s | 1.0 × 10−6 cm2/s | 23 |
| 4.0 × 10−6 cm2/s | 2 | ||
| 7.0 × 10−7 to 7.9 × 10−6 cm2/s | 37 | ||
| 1.5 × 10−6 to 3.0 × 10−6 cm2/s | 34 | ||
| Measured Ca propagation velocity | 0.212 mm/s | 0.05–15 mm/s | 2 |
| τtr (time constant for Ca diffusion from network SR to junctional SR) | 60 ms | 1–100 ms | 35 |
| 50 ms | 34 | ||
| 60 ms | 20 | ||
| 180 ms | 26 | ||
| 34.48 ms | 17 | ||
| 10 ms | 38 | ||
| 3 ms | 14 | ||
| 0.5747 ms | 28 | ||
| 0.005 ms | 32 |
SR, sarcoplasmic reticulum.
RESULTS
Spatially uniform model.
Several models of Ca handling in cardiac muscle have been shown to produce systolic Ca alternans (18, 35, 37). Typically, these models lack a spatial dimension, and alternans is produced by raising the pacing frequency. We thought it important to show that our model can also produce similar behavior. Thus the traces in Fig. 2A show simulations run at 1 and 2 Hz in a single element of our model, i.e., there are now no other elements into which Ca can diffuse. At 1-Hz pacing, the Ca transient is uniform, with no variability from pulse to pulse. However, when the pacing frequency is raised to 2 Hz, clear, large alternans of systolic Ca are present. The full frequency dependence is shown in Fig. 2B, where the Ca transient amplitude is plotted as a function of the frequency of stimulation (the stimulus pulse is the same for all stimulus frequency, e.g., from −40 to 0 mV, with a duration of 100 ms). At a frequency of 2 Hz, alternans is maximal, but, as the stimulation frequency increases further, the ratio of alternans decreases until it reaches 1 again at 4 Hz. The plot (Fig. 2C) below shows how the SR Ca content varies with stimulation frequency. It decreases dramatically with the increase of stimulus rate, due to more frequent Ca release from the SR and less time interval for Ca being taken back to the SR between two successive stimuli.
Fig. 2.

Simulations from a single Ca2+ release unit showing the effects of pacing frequency. For all stimulus frequency, the stimulus pulse has the same amplitude (from −40 to 0 mV) and duration (100 ms). A: time traces of cytoplasmic Ca2+ transient at 1 Hz (left) and 2 Hz (right). Ca2+ release is uniform from pulse to pulse at 1 Hz, but shows clear alternans at 2 Hz. B: Ca transient amplitude as a function of frequency; bifurcation leads to cytoplasmic Ca2+ transient alternans. C: associated SR Ca2+ content. [Ca2+]i, intracellular Ca2+ concentration.
The other maneuver we have used in this study to elicit alternans is an increase of the half-maximal subspace [Ca2+] for Ca2+ release from junctional SR (Krel) (the subspace [Ca2+] required to stimulate RyR to open is raised by 20%) to mimic inhibition of CICR. In the uniform model (functionally equivalent to a single release site version of the model: Fig. 3A), there are two control stimuli, and raising Krel leads to alternans following two very small systolic transients. Junctional SR Ca2+ content (below) shows each control stimulus depletes the SR, followed by a refilling period. After raising Krel, the period with very small transients allows the SR to fill to the point where alternans can occur. Before small releases, SR content is less than before large releases. The plot in Fig. 3B shows how alternans is produced. There are two plots of SR Ca2+ release vs. SR Ca2+ content under control conditions (open symbols) and with Krel raised (solid symbols). These plots follow SR filling from the empty state (not shown). It is clear that, when Krel is raised, a higher SR Ca2+ content is reached (which requires more refilling time after a large release), and the steepness of the relationship is increased [the fitted lines have the formula systolic Ca = a + b(SR Ca)n; in control n = 3.5, rising to 15.0 when Krel is raised]. This extra steepness of this relationship allows large changes of released Ca2+ for relatively minor changes of diastolic SR content. The plot in Fig. 3C shows how the Ca transient amplitude (at 1 Hz) changes with increasing Krel. The 1:1 alternans that we illustrate throughout this study is, in fact, only seen in a narrow range of Krel values, around 1.2 times control: at lower values, two large releases accompany one small, and, at higher values, there are two small releases for each large. Examples of the Ca transients at each of the three values of Krel plotted (1.15, 1.2, and 1.3) are shown above.
Fig. 3.

Model-generated Ca2+ alternans by increasing half-maximal Ca2+ subspace concentration for Ca2+ release from junctional SR (Krel) to mimic tetracaine. Ca transients were produced by a sequence of 1-Hz, 100-ms depolarizing pulses from a holding potential of −40 to 0 mV. Krel was increased uniformly by 20% after the first 2 pulses. Ca2+ alternans developed after a period of Ca release inhibition after Krel was increased. A: traces of cytoplasmic Ca2+ transients (top) and SR Ca2+ (bottom). B: relationship between SR Ca content and systolic Ca in the cytoplasmic space for control (○) and increased Krel (•) conditions as SR Ca recovers from empty state. Solid line was produced by curve fitting of systolic Ca = a + b(SR Ca)n, where n = 3.5 for control and rises to 15.0 for increased Krel. C: systolic Ca as a function of Krel. Bifurcation leads to cytoplasmic Ca2+ alternans. Increase in Krel results in various patterns of cytoplasmic Ca2+ alternans, such as 2:1 (i), 1:1 (ii), or 1:2 (iii) as shown in the inset.
The effect of adding a spatial dimension.
Introduction of a spatial dimension allows us to increase Krel in a random pattern across the 75 release unit array. On average, the increase of Krel is the same as in Fig. 3, but now it varies among the 75 release units randomly. This pattern of raised threshold also changes with each stimulus, to mimic in a simplified manner the random nature of CICR inhibition in RyR clusters. The line scan in Fig. 4A shows two control stimuli followed by two very small transients after Krel is raised. Raising Krel by an average of 20% is indicated by the line above the line scan. The line is initially dashed to indicate randomly changing Krel; thereafter, the solid line indicates a uniformly raised value. Raising Krel fragments the release profile, and local alternans follows. In agreement with our experimental data (9), large releases are associated with miniwaves of Ca2+ release (see inset A). Previously we have suggested that, associated with such behavior, there would be local variations in SR Ca2+ content caused by variations of local release and efflux. The model can give us this information; Fig. 4B shows local alternans of systolic Ca at the point marked by the arrow in Fig. 4A, the middle panel shows SR Ca2+ content from this area. During local alternans, SR Ca is relatively higher locally before large releases than before small releases. As the extent of alternans varies across the cell, the degree of alternans averaged over the whole cell is somewhat smaller that that shown in B (c.f., Fig. 4, B and C).
Fig. 4.

Model-generated Ca2+ alternans by increasing Krel nonuniformly across the 75 element array. Ca2+ transients were produced by a sequence of 1-Hz, 100-ms depolarizing pulses from a holding potential of −40 to 0 mV. Krel was increased in a random pattern by an average of 20% across the cell after the first 2 pulses (indicated above the line scan). The pattern in which Krel was raised also varied at random from stimulus to stimulus. The line above the line scan becoming solid indicates that Krel is uniformly 20% above the control level. A: line scan of cytoplasmic Ca2+ transient across the cell. After increase of Krel, localized and fragmented Ca2+ alternans developed after a short period of Ca2+ release inhibition. Inset A: propagation of local Ca2+ wave for the region marked by white box. B, top: traces of cytoplasmic Ca2+ recorded from one element marked by the arrow. Middle: associated trace of SR Ca2+ from the same element. Bottom: trace of Krel in the same element. C, top: trace of mean cytoplasmic Ca2+ averaged across the cell. Bottom: trace of SR Ca2+ averaged across the cell.
Cytoplasmic Ca2+ transients produced by small depolarizations.
Another experimental method that produces alternans is the use of small depolarizations (7). In the model, a series of small stimulus pulses lead to sustained alternans of cytoplasmic Ca2+ transients (e.g., Fig. 5A). However, the degree of alternans averaged over the whole cell is much greater than in Fig. 4. This is because either none of the cell releases Ca2+ (a in Fig. 5A), or all of the cell responds (b in Fig. 5A), i.e., there is no out-of-phase alternation taking place to reduce the overall degree of alternans. However, unlike Figs. 2 and 3, where similarly large alternans is present in the dimensionless model, the large release involves Ca2+ wave propagation (Figs. 5A and 7A), very similar to our experimental data (7). The steepness of the relationship between systolic Ca2+ and SR Ca2+ is also increased in this alternans. The fitted lines in Fig. 5B have n set to 3.5 in control (open symbols) and 18.4 during small depolarizations (solid symbols).
Fig. 5.

Model-generated Ca2+ alternans by small depolarizing pulses. The model is stimulated by a series of 1-Hz, 100-ms pulses from a holding potential of −40 to −20 mV. In addition, 40 out of 75 individual units of L-type Ca2+ channel current (ICa,L) were randomly blocked. A, top: cytoplasmic Ca2+ averaged across model of cell. Bottom: line scan images of cytoplasmic Ca2+ across cell that correspond to the transients marked a and b above. B: relationship between SR Ca2+ content and systolic Ca2+ in the cytoplasmic space for control (○) and small-pulse (•) conditions as the model recovers from the empty state. Solid line was produced by curve fitting of systolic Ca = a + b(SR Ca)n; n = 3.5 for control and 18.4 for small-pulse depolarization. C: systolic Ca as a function of ICa,L amplitude in the whole cell; bifurcation leads to cytoplasmic Ca2+ alternans. In simulations, different populations of elements are randomly chosen from the 75 units for block, producing different ICa,L amplitude in the cell (%ICa,L).
Fig. 7.

A: propagation of cytoplasmic Ca2+ wave produced by localized CICR in response to ICa,L trigger (with the same condition as in Fig. 6) and spatial diffusion of Ca2+. Solid bars: elements with fully active ICa,L channels. B: SR Ca2+ transients for some local elements 32 and 56. C: spatial distribution of SR Ca2+ content at end diastole.
In Fig. 5, A and B, 40 out of 75 elements in the cell are chosen randomly to have their L-type Ca current blocked. The remained current in the whole cell is 46% of control. Further studies have also been conducted to explore the general relationship between Ca2+ release and the reduction in the amplitude of L-type channel current in the whole cell via varying the number of blocked elements. The graph in Fig. 5C shows the full relationship between the remained averaged global L-type current (as percentage of control) in the whole cell and Ca release. A decrease of L-type channel current means the number of sites where CICR can take place decreases. At small current values (<60% of control), there is large alternans. At the current values between 30 and 50% of control, there is 1:1 alternans. With L-type currents <30% and between 50 and 60%, the alternans pattern produced is complicated and does not follow the 1:1 pattern shown in Fig. 5A (somewhat similar to the situation in Fig. 3C). Finally, >60% of L-type current amplitude alternans disappears.
Two phases of Ca2+ release.
The model also reproduces the biphasic release we see experimentally when using small pulses. The trace of systolic Ca2+ in Fig. 6A shows two successive stimuli each demonstrating a small, initial release (marked by small arrow). A second phase of release (produced by wave propagation; marked by big arrow) follows in the first stimulus. Again, this fits well with data from cells under similar experimental conditions. Both phases are due to CICR: the first represents release stimulated by L-type Ca channel activity, and the second, propagating CICR.
Fig. 6.

Ca2+ content and efflux during Ca2+ alternans produced by small depolarization pulses with 40 out of 75 elements blocked. A: [Ca2+] in cytoplasmic space. Biphasic Ca release is obvious with a small initial release evoked by stimulus (small arrow) followed by a big release (large arrow). B: Ca2+ content in the SR. C: Na/Ca exchanger current (INCX). D: ICa,L.
SR Ca2+ content, NCX current, and Ca2+ transient.
Alternans following small depolarizations also relies on changes of SR Ca content; Fig. 6 shows SR Ca2+ content (averaged over all release units) and efflux via NCX current during large and small Ca2+ transients. A large Ca2+ transient was associated with a large SR Ca2+ release (i.e., junctional SR content falls more; Fig. 6B) and more Ca2+ removal by NCX (Fig. 6C), while a small Ca2+ transient was associated with less Ca2+ release and less Ca2+ removal by NCX. The dashed line in Fig. 6B illustrates that, before the small release, SR Ca2+ is lower than before the large release. This, too, is consistent with experimental observations (7). The L-type currents in Fig. 6D indicate that changes in SR Ca2+ are solely responsible for changes in systolic release as trigger Ca2+ is unchanged between large and small releases.
One question is: where is release triggered by L-type channel openings and where is it propagated following small depolarizations? Obviously, triggered release will only be possible where L-type Ca current is activated. The solid bars in Fig. 7A show the elements at which the L-type Ca channels open on this particular stimulus; where release is initiated (sites 28 and 29 and 31 and 32), there is full opening of L-type channels in the element. However, such L-type openings do not always trigger Ca release, e.g., sites 48, 49, and 50 and 56 and 58 have L-type openings but no CICR follows. These sites had to wait for the arrival of the wave initiated at sites 31 and 32 to release Ca.
One possible explanation for failure of L-type channels to trigger CICR is suggested by Fig. 7, B and C. Figure 7B shows changes in SR Ca at two different sites from the line scan, i.e., sites 32 and 56. At site 32, there is an early release of Ca due to activation of L-type Ca channels; at site 56, Ca release fails initially but is activated later by the arrival of the wave. Although sites 32 and 56 seem to have the same initial SR Ca content, Fig. 7C shows the initial values in more detail for each site, and it is clear that site 32 has a marginally higher content, making it more likely to release.
DISCUSSION
Our previous work has shown two maneuvers that produce local and global alternans of systolic Ca2+ release in ventricular cells (7, 9). Reducing RyR open probability in voltage-clamped cells with, e.g., tetracaine or acidic intracellular pH, produces alternans of systolic release that remains local, i.e., different parts of the cell show alternans out of phase with each other. Decreasing the amplitude of the L-type Ca current produces alternans that involve the whole cell. We have suggested that alternans is due to increased steepness of the relationship between SR Ca2+ content and release of Ca2+ (a measure of the gain of Ca release). Increased steepness in both local and global alternans is due to the appearance of waves, as these rely on the SR reaching a threshold Ca2+ content (8). This ought to lead to an essentially infinitely steep relationship: above threshold, waves occur and release is large; below threshold, there are no waves and release is small. In this paper, we present simulations of this behavior to determine the important factors underlying generation of alternans of systolic Ca release. Our major finding is that the fragmented Ca2+ wave propagation and steep relationship between SR Ca2+ content and release of Ca2+ play a major role in the genesis of Ca2+ alternans at low-stimulus frequency (1 Hz) without the need for refractoriness of L-type Ca or RyR channels. The alternans sustains through alternation of SR Ca2+ content. This is different from the mechanism underlying the genesis of Ca2+ alternans at rapid pacing, as reported by Shiferaw and Karma (34), where refractoriness of ion channels and electrical action potentials are involved to produce instable Ca diffusion, leading to symmetry breaking.
The simulations we have done here can reproduce all features of systolic Ca2+ alternans, local and global, that we have seen in cells. The two types of alternans we find share several features in common: on a large Ca transient, initial release is stimulated by L-type Ca current in only a few sites; the released Ca2+ initiates a propagating wave by inducing further SR Ca2+ release in regions that did not respond to the initial depolarization. Such a large Ca release locally ensures that the SR remains depleted at the next stimulus.
In the model, the reduced open probability of RyR can be produced by increasing the parameter Krel (the subspace Ca required to open the RyR, i.e., RyR Ca sensitivity). In Fig. 3, we increased Krel by 20%; this produces 1:1 alternans of systolic release. The plot in Fig. 3B shows why alternans has occurred; there is a greatly increased steepness of the relationship between Ca2+ release and diastolic SR Ca2+ content compared with the control. Thus for a small reduction of Ca2+ in the SR at end diastole, there is a very large reduction in systolic release. However, in this case, the increased steepness in this plot does not rely on wave propagation, as there is no spatial dimension in the model.
In our studies of raising Krel, we have concentrated on a value of 1.2 times control, as this produces an alternans ratio of 1:1, i.e., for each large transient, there is a small transient. However, it is clear from Fig. 3C that other patterns of alternans are possible. In all cases, the cell is maintaining Ca flux balance, i.e., the amount of Ca that enters the cell must also leave. However, it is clear that the number of stimulation cycles required to make this balance can vary. How altering Krel changes the pattern of alternans is not yet clear and warrants further studies.
Fragmentation of the release profile seen in cells when tetracaine or acid pH is applied can be reproduced in the full version of the model (75 release units in contact by diffusion between neighboring element's cytoplasmic spaces) by applying a random pattern of increase of Krel across the array of release units. Although L-type channel opening is uniform across the array, Ca2+ wave propagation was initiated at a limited number of release sites (Fig. 4A). If a region of the cell is not stimulated to release Ca by an L-type channel opening, it may then be able to support wave propagation. If Ca release takes place (triggered either way), on the next stimulus the SR in this region is depleted of Ca2+ (Fig. 4B) and so does not respond to either stimulus or arrival of a wave. This reliance on SR Ca does not, however, reflect a luminal effect (24), as no “sensor” of SR Ca content inside the SR has been built into the model, although it may reflect a change in the feedback of the CICR mechanism. Released Ca feeding back to activate more CICR will be more evident if SR Ca content is high.
Application of increased Krel in a random pattern across the cell requires explanation. Conditions throughout the cell are unlikely to be entirely uniform in experiments as they are in the model. Although other possibilities may exist, in real cells, fragmentation of the release profile might occur through small, local variations of SR Ca2+ due to, e.g., spontaneous sparks and/or local differences in Krel. In each case, a spatially inhomogeneous response of the SR to L-type current would be expected to result. The mechanism we propose for depletion and refilling of the SR would then maintain such inhomogeneity. This is confirmed in Fig. 4A, at the point where the dashed line (marking raised Krel) becomes solid, the random changes of Krel cease, i.e., Krel is raised by 20% uniformly across the array. Despite this, the fragmented release profile is maintained.
As in real cells, at the whole cell level, the degree of alternans in the model is rather small during local alternans (Fig. 4C). This results from the lack of synchrony of release, i.e., regions alternating to a lesser extent or out of phase partially balance each other. This is just as we saw in experiments on rat ventricular myocytes in the presence of tetracaine and acidic pH (9).
We can now ask why it is that, in real cells, alternans is not uniform, but is produced by propagation of miniwaves? It is clear from Fig. 3 that uniform alternans is possible (75 release units together with a uniform increase of Krel would also produce uniform alternans); however, if we compare the SR Ca values produced by the model required for uniform alternans with those when miniwaves produce alternans, we see that alternans in a single unit requires higher SR Ca levels (1.81 vs. 1.56 mM). Thus there are two probable factors that combine to prevent the appearance of uniform alternans in the cell: conditions are unlikely ever to be sufficiently uniform to allow them to develop, and, in any case, alternans due to miniwaves will develop at a lower SR Ca, thereby preventing SR Ca rising high enough.
Small depolarizations and alternans.
When alternans are produced by reducing the number of L-type Ca channels that respond to depolarization, rather than the number of RyR that respond to L-type current, the degree of alternans at the whole cell level is greater in both cells and the model (Fig. 5). In this case, a few elements with activated L-type channels trigger SR Ca release. This released Ca diffuses to neighboring sites, where it activates CICR. This two-stage process leads to the biphasic Ca2+ release in Fig. 6, averaged over the whole cell. The time taken for the second phase to rise is dictated by the propagation velocity of the wave of CICR.
In the model, the number of release sites responding to small depolarization pulse is reduced by the imposition of a block of L-type channel current locally in some of elements (see Limitations of the model), which are randomly chosen from the 75 release elements in the array, giving a different pattern of L-type channel activation with each stimulus. The different pattern of L-type channel activation on each stimulus results in different initiation sites for Ca release (not shown). However, the pattern of L-type channel opening is not the only influence on where RyR openings result, as many L-type channel openings do not produce SR release (Fig. 7). Where release triggered by L-type Ca channels is absent or small, SR Ca content is low at the time of stimulation, e.g., site 56. The model has no luminal site that could confer extra sensitivity of the release unit at high Ca content. Thus it appears that Ca released can feed back to stimulate further release, if SR content is high.
The importance of SR Ca content.
As we have reported before, during alternans, large releases deplete the SR due to greater activation of Ca2+ efflux pathways. This can be seen in Fig. 6, where the large NCX current depletes the SR of Ca. By the time of the next stimulus, the junctional SR has not completely refilled. This small depletion of the SR means no wave propagation is possible, and a small release results. As little efflux is activated by this small release, the SR is able to refill back to the level able to support wave propagation, and a large release results once again on the next cycle. A similar explanation of alternans can be used on a local scale to explain the simulations of the effects of tetracaine or acid pH. The large releases cause either global (Fig. 6B) or local (Fig. 4B) SR depletion, depending on how inhibition of RyR opening is applied. This depletion leads to reduced release of Ca and prevents wave propagation. Obviously, for local depletions of SR Ca to be able to influence the amount of Ca released, it requires that the depletion persist long enough. The intercoupling between neighboring SR release sites allows for local depletion to persist sufficiently long to maintain alternans.
The actual shape of the relationship between systolic [Ca] and frequency is interesting: when alternans first appears, the ratio is large, but, as frequency increases, the alternans ratio declines until systolic [Ca] is once again uniform from beat to beat. Associated with these changes is a fall of SR Ca content, which suggests that alternans is only possible over a narrow range of SR Ca content. Perhaps as SR Ca content falls, the steepness of the relationship between SR Ca release and diastolic SR Ca content falls too low to provide sufficient depletion of the SR in any one cycle. Obviously, real cells exhibit alternans when frequency is raised. In the past, our laboratory has produced models of alternans that rely on a very steep relationship between SR Ca2+ release and SR Ca2+ content (12). Such a model produces alternans (e.g., Figs. 2 and 3 in this study); however, we now consider any approach that does not include a spatial element incomplete, as it cannot produce behavior that involves propagation of waves. It would be interesting in future simulations to impose some heterogeneity on the model (perhaps by application of a resting frequency of sparks to produce small variations of SR Ca content) to determine whether this will cause the Ca release profile to fragment at higher frequencies and allow miniwaves of Ca release and alternans to appear.
A recent report has shown (29) that, although present in the majority of cells studied, systolic Ca alternans can result without apparent alternans of SR Ca content. The simulations presented here, however, are intended to address the situation where the release of Ca from the SR is compromised, either by inhibition of CICR or limitation of L-type Ca current. It is entirely possible that the mechanism put forward here of fragmentation of the Ca release profile, followed by propagation of waves, is not the only one capable of leading to systolic Ca alternans. It would be interesting in the future to model the conditions required for such alternans, independent of SR Ca content.
Limitations of the model.
The present model has several limitations. In the present study, the SR Ca release was modeled by quasi-steady-state approximation of RyR gating kinetics; however, this seems reasonable considering the rapid activation (0.07–0.27 ms) (42) and extremely slow inactivation of the RyR gate (43) compared with the L-type Ca channel and the low stimulus frequency (1 Hz). In simulations, the mechanism of reversible SR uptake was not incorporated in the model. However, simulations with an alternative reversible SR uptake equation (33) (see Eq. A18.1 in appendix) show similar results. We have made no attempt here to model electrophysiological consequences of systolic Ca2+ alternans, although it is clear from our previous work and here (Fig. 6) that currents generated as a result of the changes in Ca2+ release would be expected to alter action potential shape and/or duration. The model includes no influence of SR Ca content on the behavior of the Ca release channel (25); this is thought to play a role in control of Ca release, but should, if present, accentuate the effects of local differences of SR Ca content on Ca release. Importantly, the model uses whole cell behavior of L-type current in each of the elements rather than the behavior of single unitary L-type channels. As a result, the changes of voltage used in this paper reduce current flow through the L-type channel of each release unit rather than increase unitary channel current. Finally, the model is a one-dimensional simulation of a cell and, therefore, lacks the three-dimensional complexities of a whole cell, such as t-tubule networks (39) and spatial distribution of L-type current (4) and NCX current (41). Each of these limitations needs to be addressed in future studies.
Despite these limitations, the model demonstrates that propagation of waves of CICR following fragmentation of the systolic Ca release profile is able to produce systolic Ca alternans. In addition, the model highlights the importance of interactions between local SR Ca content, L-type Ca channel activity, and sensitivity to Ca of the Ca release channel of the SR in sustaining this behavior.
GRANTS
Work on this paper was funded in part by grants from the British Heart Foundation and Biotechnology and Biological Sciences Research Council.
Acknowledgments
We acknowledge the Aspen Centre for Physics for the use of its facilities.
APPENDIX
Glossary
- F
Faraday constant
- I
Ionic current
- R
Universal gas constant
- SR
Sarcoplasmic reticulum
- t
Time
- T
Absolute temperature
- V
Membrane potential (in mV)
Ionic channel currents.
- ICa,L
L-type Ca2+ channel current
- ICa,P
Sarcolemmal Ca2+ pump current
- INaCa
Na+/Ca2+ exchanger (NCX) current
Cell geometry.
- Vcell
Cell volume
- Vi
Myoplasmic volume available for Ca2+ diffusion
- Vrel
Volume of junctional SR (Ca2+ release store)
- Vsub
Subspace volume
- Vup
Volume of network SR (Ca2+ uptake store)
Ionic concentrations.
- [Ca2+]i
Myoplasmic Ca2+ concentration
- [Ca2+]o
Extracellular Ca2+ concentration
- [Ca2+]rel
Ca2+ concentration in junctional SR
- [Ca2+]sub
Subspace Ca2+ concentration
- [Ca2+]up
Ca2+ concentration in network SR
- [Mg2+]i
Intracellular Mg2+ concentration
- [Na+]i
Intracellular Na+ concentration
- [Na+]o
Extracellular Na+ concentration
Equilibrium (reversal) potentials.
- ECa,L
Apparent reversal potential of ICa,L
Sarcolemmal ionic currents.
- dL
Activation gating variable for ICa,L
- dL,∞
Steady-state dL
- dNaCa
Denominator constant for INaCa
- fL
Inactivation gating variable for ICa,L
- fL,∞
Steady-state fL
- gCa,L
Maximum ICa,L conductance
- kNaCa
Scaling factor for INaCa
- yNaCa
Position of Erying rate theory energy barrier controlling voltage dependence of INaCa
- α
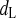
Opening rate constant of dL
- α
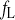
Opening rate constant of fL
- β
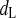
Closing rate constant of dL
- β
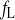
Closing rate constant of fL
- τ
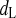
Time constant of dL
- τ
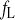
Time constant of fL
Ca2+ diffusion flux.
- JCa diff
Ca2+ diffusion flux from subspace to myoplasm
- JCa i diff
Ca2+ transfer flux between neighboring myoplasm
- JCa up diff
Ca2+ transfer flux between neighboring network SR
- τCa diff
Time constant for Ca2+ diffusion from subspace to myoplasm
- τCa i diff
Time constant for Ca2+ diffusion between neighboring myoplasmic units
- τCa up diff
Time constant for Ca2+ diffusion between neighboring network SR units
SR function.
- Jrel
Ca2+ release flux from junctional SR to subspace
- Jtr
Ca2+ transfer flux from network SR to junctional SR
- Jup
Ca2+ uptake flux from myoplasm to network SR
- Krel
Half-maximal [Ca2+]sub for Ca2+ release from junctional SR
- Kup
Half-maximal [Ca2+]i for Ca2+ uptake by Ca2+ pump in network SR
- Prel
Rate constant for Ca2+ release from junctional SR
- Pup
Rate constant for Ca2+ uptake by Ca2+ pump in network SR
- τtr
Time constant for Ca2+ transfer from network SR to junctional SR
Ca2+ buffering.
- [CQ]tot
Total calsequestrin concentration
- [CM]tot
Total calmodulin concentration
- kb CM
Ca2+ dissociation constant for calmodulin
- kb CQ
Ca2+ dissociation constant for calsequestrin
- kb TC
Ca2+ dissociation constant for troponin-Ca complex
- kb TMC
Ca2+ dissociation constant for troponin-Mg complex
- kb TMM
Mg2+ dissociation constant for troponin-Mg complex
- kf CM
Ca2+ association constant for calmodulin
- kf CQ
Ca2+ association constant for calsequestrin
- kf TC
Ca2+ association constant for troponin
- kf TMC
Ca2+ association constant for troponin-Mg complex
- kf TMM
Mg2+ association constant for troponin-Mg complex
- [TC]tot
Total concentration of troponin-Ca complex
- [TMC]tot
Total concentration of troponin-Mg complex
- fCMi,∞
Fractional occupancy of calmodulin by Ca2+ in myoplasm
- fCMi rate
Derivative of fCMi with respect to (WRT) time: dfCMi/dt
- fCMs,∞
Fractional occupancy of calmodulin by Ca2+ in subspace
- fCMs rate
Derivative of fCMs WRT time: dfCMs/dt
- fCQ,∞
Fractional occupancy of calsequestrin by Ca2+
- fCQ rate
Derivative of fCQ WRT time: dfCQ/dt
- fTC,∞
Fractional occupancy of the troponin-Ca site by Ca2+
- fTC rate
Derivative of fTC WRT time: dfTC/dt
- fTMC,∞
Fractional occupancy of the troponin-Mg site by Ca2+
- fTMC rate
Derivative of fTMC WRT time: dfTMC/dt
- fTMM,∞
Fractional occupancy of the troponin-Mg site by Mg2+
- fTMM rate
Derivative of fTMM WRT time: dfTMM/dt
Equations
Sarcolemmal ionic current.
L-TYPE Ca2+ CHANNEL CURRENT.
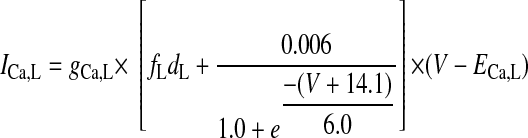 |
(A1) |
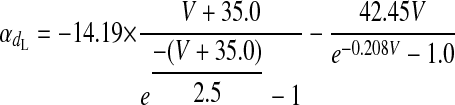 |
(A2) |
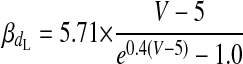 |
(A3) |
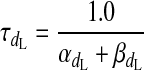 |
(A4) |
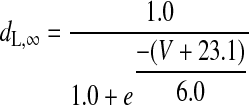 |
(A5) |
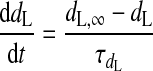 |
(A6) |
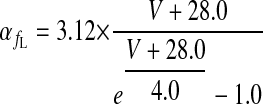 |
(A7) |
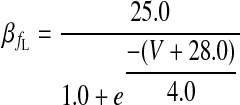 |
(A8) |
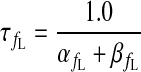 |
(A9) |
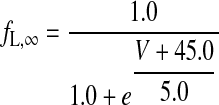 |
(A10) |
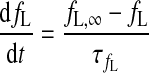 |
(A11) |
BACKGROUND, PUMP, AND EXCHANGER CURRENTS.
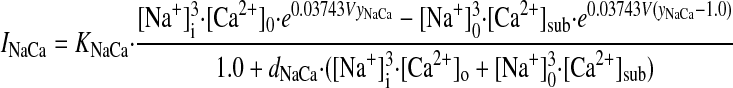 |
(A12) |
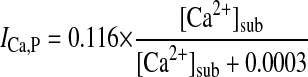 |
(A13) |
Intracellular Ca2+ dynamics.
Ca2+ DIFFUSION FLUX.
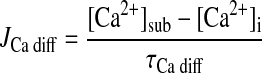 |
(A14) |
Ca2+ DIFFUSION FLUX BETWEEN NEIGHBORED ELEMENTS.
For each coupled element array, indexed by k (k varies between 1 and 75):
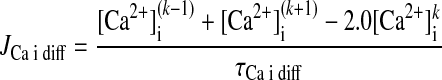 |
(A15) |
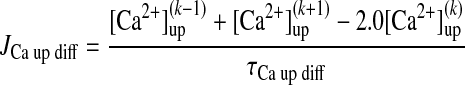 |
(A16) |
Ca2+ HANDLING BY SR.
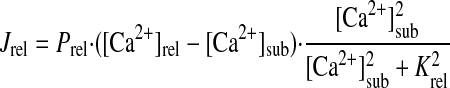 |
(A17) |
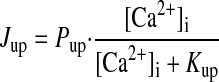 |
(A18) |
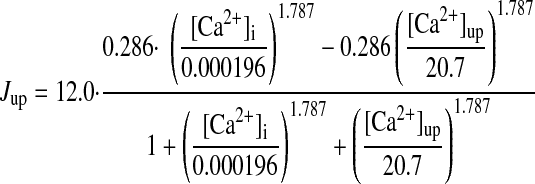 |
(A18.1) |
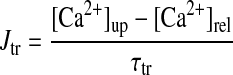 |
(A19) |
INTRACELLULAR ION CONCENTRATIONS.
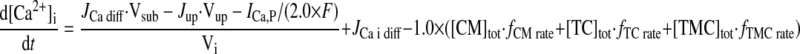 |
(A20) |
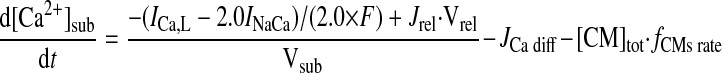 |
(A21) |
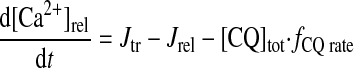 |
(A22) |
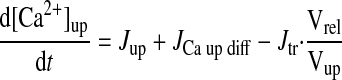 |
(A23) |
Ca2+ BUFFERING.
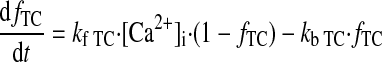 |
(A24) |
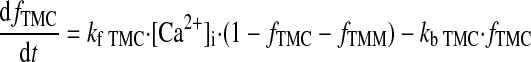 |
(A25) |
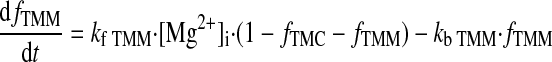 |
(A26) |
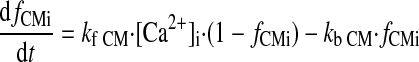 |
(A27) |
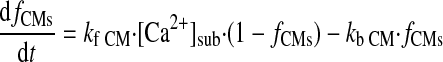 |
(A28) |
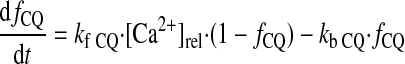 |
(A29) |
Model constants.
See Table 2 for model parameters and values.
Table 2.
Model parameters and values
| Model Parameters | Values |
|---|---|
| [Ca2+]o, mM | 2 |
| [CM]tot, mM | 0.045 |
| [CQ]tot, mM | 10 |
| dNaCa | 0.0001 |
| ECa,L, mV | 46.4 |
| F, C/M | 96,484.6 |
| gCa,L | 0.06588648 |
| kb CM, ms−1 | 0.542 |
| kb CQ, ms−1 | 0.445 |
| kb TC, ms−1 | 0.446 |
| kb TMC, ms−1 | 0.00751 |
| kb TMM, ms−1 | 0.751 |
| kf CM, mM−1·ms−1 | 227.7 |
| kf CQ, mM−1·ms−1 | 0.534 |
| kf TC, mM−1·ms−1 | 88.8 |
| kf TMC, mM−1·ms−1 | 227.7 |
| kf TMM, mM−1·ms−1 | 2.277 |
| kNaCa | 0.883584 × 10−5 |
| Krel, mM | 0.0012 |
| Kup, mM | 0.00045 |
| [Mg2+]i, mM | 2.5 |
| [Na+]i, mM | 8 |
| [Na+]o, mM | 140 |
| Prel, ms−1 | 1.8 |
| Pup, mM/ms | 0.005 |
| R, J·mol−1·K−1 | 8.314472 |
| T, Kelvin | 310 |
| [TC]tot, mM | 0.031 |
| [TMC]tot, mM | 0.062 |
| Vcell, pl | 36.8 |
| Vi, pl | 13 |
| Vrel, pl | 0.2352 |
| Vsub, pl | 2.4 |
| Vup, pl | 1.624 |
| τtr, ms | 60 |
| τCa diff, ms | 0.04 |
| τCaidiff, ms | 4 |
| τCa up diff, ms | 600 |
| yNaCa | 0.5 |
See Glossary for definition of terms.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Armoundas AA, Tomaselli GF, Esperer HD. Pathophysiological basis and clinical application of T-wave alternans. J Am Coll Cardiol 40: 207–217, 2002. [DOI] [PubMed] [Google Scholar]
- 2.Backx PH, de Tombe PP, van Deen JHK, Mulder BJM, ter Keurs HEDJ. A model of propagating calcium-induced calcium release mediated by calcium diffusion. J Gen Phyisol 93: 963–977, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bers DM Excitation-Contraction Coupling and Cardiac Contractile Force (2nd Ed.). Dordrecht, the Netherlands: Kluwer Academic, 2001.
- 4.Brette F, Salle L, Orchard CH. Quantification of calcium entry at the T-tubules and surface membrane in rat ventricular myocytes. Biophys J 90: 381–389, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen-Izu Y, McCulle SL, Ward CW, Soeller C, Allen BM, Rabang C, Cannell MB, Balke CW, Izu LT. Three-dimensional distribution of ryanodine receptor clusters in cardiac myocytes. Biophys J 91: 1–13, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi BR, Salama G. Simultaneous maps of optical action potentials and calcium transients in guinea-pig hearts: mechanisms underlying concordant alternans. J Physiol 529: 171–188, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Díaz ME, O'Neill SC, Eisner DA. Sarcoplasmic reticulum calcium content fluctuation is the key to cardiac alternans. Circ Res 94: 650–656, 2004. [DOI] [PubMed] [Google Scholar]
- 8.Díaz ME, Trafford AW, O'Neill SC, Eisner DA. Measurement of sarcoplasmic reticulum Ca2+ content and sarcolemmal Ca2+ fluxes in isolated rat ventricular myocytes during spontaneous Ca2+ release. J Physiol 501: 3–16, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Díaz ME, Eisner DA, O'Neill SC. Depressed ryanodine receptor activity increases variability and duration of the systolic Ca2+ transient in rat ventricular myocytes. Circ Res 91: 585–593, 2002. [DOI] [PubMed] [Google Scholar]
- 10.Dumitrescu C, Narayan P, Efimov IR, Cheng Y, Radin MJ, McCune SA, Altschuld RA. Mechanical alternans and restitution in failing SHHF rat left ventricles. Am J Physiol Heart Circ Physiol 282: H1320–H1326, 2002. [DOI] [PubMed] [Google Scholar]
- 11.Eisner DA, Isenberg G, Sipido KR. Normal and pathological excitation-contraction coupling in the heart–an overview. J Physiol 546: 3–4, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eisner DA, Choi HS, Díaz ME, O'Neill SC, Trafford AW. Integrative analysis of calcium cycling in cardiac muscle. Circ Res 87: 1087–1094, 2000. [DOI] [PubMed] [Google Scholar]
- 13.Euler DE Cardiac alternans: mechanisms and pathophysiological significance. Cardiovasc Res 42: 583–590, 1999. [DOI] [PubMed] [Google Scholar]
- 14.Greenstein JL, Winslow RL. An integrative model of the cardiac ventricular myocytes incorporating local control of Ca2+ release. Biophys J 83: 2918–2945, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gyorke S, Lukyanenko V, Gyorke I. Dual effects of tetracaine on spontaneous calcium release in rat ventricular myocytes. J Physiol 500: 297–309, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hüser J, Wang YG, Sheehan KA, Cifuentes F, Lipsius SL, Blatter LA. Functional coupling between glycolysis and excitation-contraction coupling underlies alternans in cat heart cells. J Physiol 524: 795–806, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jafri MS, Rice JJ, Winslow RL. Cardiac Ca2+ dynamics: the roles of ryanodine receptor adaptation and sarcoplasmic reticulum load. Biophys J 74: 1149–1166, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jordan PN, Christini DJ. Action potential morphology influences intracellular calcium handling stability and the occurrence of alternans. Biophys J 90: 672–680, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kodama M, Kato K, Hirono S, Okura Y, Hanawa H, Yoshida T, Hayashi M, Tachikawa H, Kashimura T, Watanbe K, Aizawa Y. Linkage between mechanical and electrical alternans in patients with chronic heart failure. J Cardiovasc Electrophysiol 15: 295–299, 2004. [DOI] [PubMed] [Google Scholar]
- 20.Kurata Y, Hisatome I, Imanishi S, Shibamoto T. Dynamical description of sinoatrial node pacemaking: improved mathematical model for primary pacemaker cell. Am J Physiol Heart Circ Physiol 283: H2074–H2101, 2002. [DOI] [PubMed] [Google Scholar]
- 21.Lab MJ, Lee JA. Changes in intracellular calcium during mechanical alternans in isolated ferret ventricular muscle. Circ Res 66: 585–595, 1990. [DOI] [PubMed] [Google Scholar]
- 22.Lakireddy V, Baweja P, Syed A, Bub G, Boutjdir M, El Sherif N. Contrasting effects of ischemia on the kinetics of membrane voltage and intracellular calcium transient underlie electrical alternans. Am J Physiol Heart Circ Physiol 288: H400–H407, 2005. [DOI] [PubMed] [Google Scholar]
- 23.Langer GA, Peskoff A. Calcium concentration and movement in the diadic cleft space of the cardiac ventricular cells. Biophys J 70: 1169–1182, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laver DR Coupled calcium release channels and their regulation by luminal and cytosolic ions. Eur Biophys J 34: 359–368, 2005. [DOI] [PubMed] [Google Scholar]
- 25.Lukyanenko V, Györke I, Györke S. Regulation of calcium release by calcium inside the sarcoplasmic reticulum in ventricular myocytes. Pflügers Arch 432: 1047–54, 1996. [DOI] [PubMed] [Google Scholar]
- 26.Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ Res 74: 1071–1096, 1994. [DOI] [PubMed] [Google Scholar]
- 27.Orchard CH, McCall E, Kirby MS, Boyett MR. Mechanical alternans during acidosis in ferret heart muscle. Circ Res 68: 69–76, 1991. [DOI] [PubMed] [Google Scholar]
- 28.Pandit SV, Clark RB, Giles WR, DEmir SS. A mathematical model of action potential heterogeneity in adult rat left ventricular myocytes. Biophys J 81: 3029–3051, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Picht E, Desantiago J, Blatter LA, Bers DM. Cardiac alternans do not rely on diastolic sarcoplasmic reticulum calcium content fluctuations. Circ Res 99: 740–748, 2006. [DOI] [PubMed] [Google Scholar]
- 30.Pogwizd SM, Hoyt RH, Saffitz JE, Corr PB, Cox JL, Cain ME. Reentrant and focal mechanisms underlying ventricular tachycardia in the human heart. Circulation 86: 1872–1887, 1992. [DOI] [PubMed] [Google Scholar]
- 31.Pruvot EJ, Katra RP, Rosenbaum DS, Laurita KR. Role of calcium cycling versus restitution in the mechanism of repolarization alternans. Circ Res 94: 1083–1090, 2004. [DOI] [PubMed] [Google Scholar]
- 32.Rice JJ, Jafri MS, Winslow RL. Modeling gain and gradedness of Ca2+ release in the functional unit of the cardiac diadic space. Biophys J 77: 1871–1884, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shannon TR, Wang F, Bers DM. Regulation of cardiac sarcoplasmic reticulum Ca release by luminal [Ca] and altered gating assessed with a mathematical model. Biophys J 89: 4096–4110, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shiferaw Y, Karma A. Turing instability mediated by voltage and calcium diffusion in paced cardiac cells. Proc Natl Acad Sci USA 103: 5670–5675, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shiferaw Y, Watanabe MA, Garfinkel A, Weiss JN, Karma A. Model of intracellular calcium cycling in ventricular myocytes. Biophys J 85: 3666–3686, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sipido KR, Volders PGA, de Groot SHM, Verdonck F, Van de Werf F, Wellens HJJ, Vos MA. Enhanced Ca2+ release and Na/Ca exchange activity in hypertrophied canine ventricular myocytes: a potential link between contractile adaptation and arrhythmogenesis. Circulation 102: 2137–2144, 2000. [DOI] [PubMed] [Google Scholar]
- 37.Snyder SM, Palmer BM, Moore RL. A mathematical model of cardiocyte Ca2+ dynamics with a novel representation of sarcoplasmic reticulum Ca2+ control. Biophys J 79: 94–115, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sobie EA, Dilly KW, Dos Santos CJ, Lederer WJ, Jafri MS. Termination of cardiac Ca2+ sparks: an investigative mathematical model of calcium-induced calcium release. Biophys J 83: 59–78, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soeller C, Cannell MB. Examination of the transverse tubular system in living cardiac rat myocytes by 2-photon microscopy and digital image-processing techniques. Circ Res 84: 266–275, 1999. [DOI] [PubMed] [Google Scholar]
- 40.Wu Y, Clusin WT. Calcium transient alternans in blood-perfused ischemic hearts: observations with fluorescent indicator fura red. Am J Physiol Heart Circ Physiol 273: H2161–H2169, 1997. [DOI] [PubMed] [Google Scholar]
- 41.Yang Z, Pascarel C, Steele DS, Komukai K, Brette F, Orchard CH. Na+-Ca2+ exchange activity is localized in the T-tubules of rat ventricular myocytes. Circ Res 91: 315–322, 2002. [DOI] [PubMed] [Google Scholar]
- 42.Zahradnikova A, Zahradnik I, Gyorke I, Gyorke S. Rapid activation of the cardiac ryanodine receptor by submillisecond calcium stimuli. J Gen Physiol 114: 787–798, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zahradnikova A, Zahradnik I. A minimal gating model for the cardiac calcium release channel. Biophys J 71: 2996–3012, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]