

Abstract
We hypothesized that impaired nitric oxide (NO)-dependent dilation (endothelial dysfunction) in Type 2 diabetes results, in part, from elevated production of superoxide (O2•−) induced by the interaction of advanced glycation end products (AGE)/receptor for AGE (RAGE) and TNF-α signaling. We assessed the role of AGE/RAGE and TNF-α signaling in endothelial dysfunction in Type 2 diabetic (Leprdb) mice by evaluation of endothelial function in isolated coronary resistance vessels of normal control (nondiabetic, m Leprdb) and diabetic mice. Although dilation of vessels to the endothelium-independent vasodilator sodium nitroprusside (SNP) was not different between diabetic and control mice, dilation to the endothelium-dependent agonist acetylcholine (ACh) was reduced in diabetic vs. control mice. The activation of RAGE with RAGE agonist S100b eliminated SNP-potentiated dilation to ACh in Leprdb mice. Administration of a soluble form of RAGE (sRAGE) partially restored dilation in diabetic mice but did not affect dilation in control mice. The expression of RAGE in coronary arterioles was markedly increased in diabetic vs. control mice. We also observed in diabetic mice that augmented RAGE signaling augmented expression of TNF-α, because this increase was attenuated by sRAGE or NF-κB inhibitor MG132. Protein and mRNA expression of NAD(P)H oxidase subunits including NOX-2, p22phox, and p40phox increased in diabetic compared with control mice. sRAGE significantly inhibited the expression of NAD(P)H oxidase in diabetic mice. These results indicate that AGE/RAGE signaling plays a pivotal role in regulating the production/expression of TNF-α, oxidative stress, and endothelial dysfunction in Type 2 diabetes.
Keywords: coronary microcirculation, nitric oxide, reactive oxygen species
a hallmark of diabetic vasculopathy, which is postulated to lead to vascular disease, is abrogated endothelial dilation. We reported previously (10, 19, 29) that the inflammatory cytokine tumor necrosis factor-α (TNF-α) plays a pivotal role in endothelial dysfunction; the neutralizing antibody to TNF-α restored blunted dilation in diabetic rats (19), and diabetic mice null for TNF-α did not show endothelial dysfunction (10). TNF-α also affects intracellular insulin signaling in fat, skeletal muscle, and other insulin-sensitive tissues by inhibiting kinase activity in the proximal part of the insulin signaling pathway (20). We found that TNF-α contributes to oxidative stress in diabetes, which may explain, in part, elevations in oxidative stress in diabetes. However, there are other possible contributors to the increased oxidative stress in diabetes, which could serve to amplify the effects of TNF-α. Advanced glycation end products (AGE) and receptor for AGE (RAGE) signaling stimulates the production of superoxide (O2•−), which could further both the oxidative stress and the impaired bioavailability of nitric oxide (NO). In humans with diabetes, the increase in circulating AGE has been found to parallel the severity of diabetic kidney disease. AGE accumulate more quickly than normal in the blood and arteries of patients with diabetes (22). Diabetic rats treated with aminoguanidine (Pimagedine; prevents AGE formation) showed a reversal of inadequate blood flow to the nerves (13) and gradual improvement of the nerves' ability to transmit signals (27). This suggests that blockade of AGE formation may have potential for treating diabetic neuropathy. Theoretically, as AGE become self-perpetuating and well-established in certain tissues, and even if blood sugar is returned to normal, the AGE might continue to increase, thereby leading to diabetic complications. Recent evidence suggests that inflammation plays a role in the development of insulin resistance and is a predictor of the development of Type 2 diabetes mellitus (2).
Therefore, we proposed that AGE/RAGE contributes to endothelial dysfunction both directly and by regulating the production and expression of TNF-α in Type 2 diabetes. The latter proposition is based on observations showing that nuclear factor-κB (NF-κB), a transcription factor activated by inflammation and oxidative stress, plays a key role in TNF-α expression. Accordingly, we evaluated the expression of AGE/RAGE and TNF-α in coronary arterioles in Type 2 diabetic and normal control (nondiabetic) mice and determined whether AGE/RAGE signaling would compromise endothelial dilation and produce reactive oxygen species (ROS). We also tested whether AGE/RAGE signaling leads to TNF-α expression and production in diabetes.
MATERIALS AND METHODS
Animals.
The procedures followed were approved by the Laboratory Animal Care Committee at University of Missouri, Columbia. Heterozygote controls (m Leprdb), wild-type (WT) controls, homozygote Type 2 diabetes (Leprdb, diabetic), and Leprdb null for TNF-α (dbTNF−/dbTNF−) mice were purchased from Jackson Laboratory and maintained on a normal rodent chow diet. Our studies utilized 12- to 16-wk-old 15- to 25-g m Leprdb and WT mice and 25- to 50-g Leprdb and dbTNF−/dbTNF− mice of either sex. We used the same strain (C57BL/6J) of m Leprdb and dbTNF−/dbTNF− mice to match the backgrounds of Leprdb mice. The dbTNF−/dbTNF− mice show the phenotype of hyperglycemia and obesity, the diabetic phenotype that is consistent with the penetrance of the leptin receptor mutation. The obese mice from the second round of breeding of Leprdb and TNF−/− were used in experimentation. We defined m Leprdb and WT mice as controls in this study because the results from m Leprdb and WT mice were identical. The Type 2 diabetic Leprdb mouse is designated as the diabetic mouse in this study.
Measurement of metabolic parameters.
The methods for measuring blood glucose, lipid level, and blood pressure (BP) were described in detail previously (10), and hemoglobin A1c (HbA1c) level, the index of AGE accumulation, was measured by a Cobas automatic analyzer (Roche Diagnostics, Indianapolis, IN). Briefly, blood was obtained from the vena cava after anesthesia with pentobarbital sodium (50 mg/kg ip) and exposure of the vein. Blood was collected, and plasma and serum were stored at −80°C until analysis. Mice were fasted for 4 h before blood was obtained via the retroorbital sinus. Plasma glucose levels were measured with a One Touch UltraSmart glucometer (Lifescan, Milpitas, CA). Plasma insulin levels were analyzed with a radioimmunoassay kit (Linco, St. Charles, MO). Plasma lipid levels were determined by the Louisiana State University Health Sciences Center Clinical Pathology Laboratory.
Treatment with TNF-α neutralization, soluble RAGE, and MG132.
The neutralizing antibody to TNF-α (anti-TNF) (10) was 2E2 monoclonal antibody (2E2 MAb 94021402; National Cancer Institute, Biological Resources Branch). At 12–16 wk of age, all mice received the neutralizing anti-TNF (2E2 MAb, 0.625 mg·ml−1·kg−1·day−1 ip, 3 days); dosage was based on our estimates of TNF-α expression (in the low nanogram or picogram range), and this dosage is able to neutralize 10- to 100-fold this amount of TNF-α.
Soluble RAGE (sRAGE), the extracellular two-thirds of the receptor, binds AGE and interferes with their ability to bind and activate cellular RAGE (26). To antagonize RAGE signaling, we administered sRAGE (a gift from A. M. Schmidt; 80 μg·mouse−1·day−1 ip) to control and diabetic mice for 10 days to determine the interaction between RAGE and TNF-α in Type 2 diabetes and to determine whether RAGE affects coronary arterial dilations mediated by NO.
MG132 is the proteasome inhibitor for NF-κB. The control and diabetic animals were injected intraperitoneally with 10 mg·kg−1·day−1 MG132 [Sigma; dissolved in 0.05 ml dimethyl sulfoxide (DMSO)] for 5 days (16).
Functional assessment of isolated coronary arterioles.
The heart was excised and immediately placed in cold (4°C) saline solution. Each coronary arteriole (40–100 μm in internal diameter) was carefully isolated and then used in the functional and molecular studies described below (14). The concentration-diameter relationships for the endothelium-dependent vasodilator ACh (1 nmol/l–10 μmol/l) and the endothelium-independent vasodilator and NO donor sodium nitroprusside (SNP, 1 nmol/l–1 μmol/l) were then established. ACh was used as an activator of endothelium-dependent NO-mediated vasodilation (9), and SNP was used to test the function of vascular smooth muscle cells.
To test the role of RAGE in ACh-induced NO-mediated vasodilation, we first studied responses to ACh before and after treatment with the RAGE agonist S100b (10 μg/ml, 60-min extraluminal incubation) in the presence and absence of SNP (0.1 μM) in both control and diabetic mice. We administered the dilator SNP because we were concerned that with such a small response to ACh in the diabetic mice, we might not be able to detect further inhibition of an already depressed response. Second, endothelium-dependent and -independent dilation was assessed in coronary arterioles from control, diabetic, sRAGE-treated control, and sRAGE-treated diabetic mice.
ELISA assay of serum AGE.
To measure AGE product, we used the competitive AGE-ELISA assay method. The competitive AGE-ELISA procedure was performed as described previously (6). In brief, competing antigen in dilution buffer was added, followed by anti-AGE MAb (1:1,000, a gift from A. M. Schmidt) in dilution buffer containing 2% normal rabbit serum (Pierce). Secondary antibody [rabbit anti-chicken horseradish peroxidase (HRP) IgY, Biomeda, Foster City, CA] in dilution buffer with 1% normal rabbit serum was then added to each well, and the plates were incubated at 37°C. Optical density (OD) at 405 nm was determined by a SPECTRA Fluor Plus TECAN reader. Results were calculated as experimental OD − background OD (i.e., no antibody)/total OD (i.e., no competitor) − background OD.
mRNA expression of RAGE and NAD(P)H oxidase subunits by real-time PCR.
Total RNA was extracted from coronary arteries with TRIzol reagent (Life Technologies) (17, 19, 29) and was processed directly to cDNA synthesis with SuperScript III reverse transcriptase (Life Technologies). The primers of RAGE and NAD(P)H oxidase subunits, p22phox, p40phox, p47phox, p67phox, NOX-1, NOX-2 (gp91phox), and NOX-4, were designed (primer 3 software) and synthesized (Qiagen). Data were calculated by the 2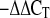 method (where CT is threshold cycle) (17) and are presented as fold change of transcripts for RAGE gene in diabetic mice normalized to β-actin, compared with control mice (defined as 1.0-fold) (19).
method (where CT is threshold cycle) (17) and are presented as fold change of transcripts for RAGE gene in diabetic mice normalized to β-actin, compared with control mice (defined as 1.0-fold) (19).
Protein expression of RAGE, TNF-α, and NF-κB by Western blot analyses.
For Western blot analysis, coronary arterioles (6–8 vessels/group) (19) were separately homogenized and sonicated in lysis buffer (Cellytic MT Mammalian Tissue Lysis/Extraction Reagent, Sigma). RAGE, TNF-α, and NF-κB protein expression were detected by Western blot analysis with the use of TNF-α primary antibodies (Santa Cruz) or RAGE or NF-κB primary antibody (Abcam) in control mice, diabetic mice, diabetic mice treated with anti-TNF (0.625 mg·ml−1·kg−1·day−1 ip, 3 days), diabetic mice treated with the free radical scavenger TEMPOL (100 mg·kg−1·day−1 ip, 7 days), diabetic mice treated with sRAGE (80 μg·mouse−1·day−1 ip, 10 days), and diabetic mice treated with NF-κB inhibitor MG132 (10 mg·kg−1·day−1 ip, 5 days). HRP-conjugated secondary antibodies were accordingly used. Signals were visualized by enhanced chemiluminescence (ECL, Santa Cruz) and quantified by Quantity One (Bio-Rad Versadoc imaging system).
Chemicals.
All drugs were obtained from Sigma, except as specifically stated. ACh, SNP, TEMPOL, aminoguanidine, and S100b were dissolved in physiological salt solution (PSS) for molecular and functional studies. These drugs were then diluted in PSS to obtain the desired final concentration. Vehicle control studies indicated that the final concentrations of solvent had no effect on the arteriolar function.
Statistical analyses.
At the end of each experiment, the vessel was relaxed with 100 μmol/l SNP to obtain its maximal diameter at intraluminal pressure of 60 cmH2O (10). All diameter changes in response to agonists were normalized to the vasodilation in response to 100 μmol/l SNP and expressed as a percentage of maximal dilation. All data are presented as means ± SE, except as specifically stated (e.g., as means ± SD for molecular study). Statistical comparisons of vasomotor responses under various treatment conditions were performed with one-way or two-way ANOVA, and intergroup differences were tested with Bonferroni inequality. Significance was accepted at P < 0.05.
RESULTS
Fasting glucose, blood pressure, body weight, abdominal girth, HbA1c, and lipid level.
Metabolic parameters were measured at 12–16 wk in the different strains of mice (Table 1). Fasting glucose, lipid level, body weight, and abdominal girth were higher in diabetic, dbTNF−/dbTNF−, and diabetic mice treated with anti-TNF than those in control mice, but there were no differences in BP among the four groups on the day of surgery. HbA1c was higher in diabetic vs. control mice, but there were no differences in dbTNF−/dbTNF− mice and diabetic mice treated with anti-TNF compared with control mice.
Table 1.
Baseline metabolic parameters
| Control | Diabetic | Diabetic Anti-TNF | dbTNF−/dbTNF− | |
|---|---|---|---|---|
| Body weight, g | 25±7 | 49±5* | 50±8* | 47±7* |
| BP, mmHg | 116±13 | 121±12 | 119±11 | 126±17 |
| Abdominal girth, cm | 8.5±2 | 15.5±3* | 15.8±2* | 15±4* |
| Total cholesterol, mg/dl | 80±5 | 156±8* | 153±5* | 146±7* |
| HDL cholesterol, mg/dl | 56±3 | 89±2 | 93±5 | 97±4 |
| Triglycerides, mg/dl | 62±8 | 96±13* | 97±10* | 90±8* |
| Fasting glucose, mg/dl | 137±19 | 329±21* | 327±20* | 325±27* |
| Insulin, ng/ml | 0.3±0.1 | 13±4* | 10±3* | 11±2* |
| Blood HbA1c, % | 4.6±0.3 | 7.8±0.6* | 5.2±0.5† | 5.0±0.4† |
Values are means ± SE hemodynamic and metabolic parameters in control mice, diabetic mice, diabetic mice treated with anti-TNF, and dbTFN−/dbTNF− mice (n = 10). BP, blood pressure; HbA1c, hemoglobin A1c.
P < 0.05 vs. control;
P < 0.05 vs. diabetic.
Serum AGE production and mRNA expression of RAGE.
Serum AGE production (Fig. 1A) in diabetic mice was significantly increased compared with control mice, but aminoguanidine (inhibitor of AGE formation) greatly attenuated serum AGE production in diabetic mice without affecting serum AGE production in control mice. Normalization of RAGE transcripts to those of β-actin demonstrated significantly greater (∼9-fold) mRNA expression of RAGE in the coronary arterioles of diabetic mice than in control mice. Moreover, aminoguanidine attenuated RAGE expression in diabetic mice but did not affect that in control mice (Fig. 1B). Serum AGE production and mRNA expression of RAGE are significantly attenuated in dbTNF−/dbTNF− mice compared with diabetic mice (Fig. 1).
Fig. 1.

A: serum advanced glycation end products (AGE) production in control, diabetic, and dbTNF−/dbTNF− mice and control and diabetic mice treated with aminoguanidine (AGD). B: expression of receptor for AGE (RAGE) mRNA in isolated coronary arterioles of control and diabetic mice, control and diabetic mice treated with AGD, and dbTNF−/dbTNF− mice. Data are means ± SD; n = 8. *P < 0.05 vs. control, #P < 0.05 vs. diabetic in each group.
Role of RAGE in ACh-induced NO-mediated vasodilation.
Figure 2A shows responses to ACh in control mice after incubation of the vessels with SNP (0.1 μM) and after incubation of the vessels with S100b in the presence of SNP (0.1 μM). Figure 2B shows responses to ACh in diabetic mice after incubation of the vessels with SNP (0.1 μM) and after incubation of the vessels with S100b in the presence of SNP (0.1 μM). We were expecting a parallel shift in the line due to the constant amount of SNP, but the amplification of dilation was unexpected. Nonetheless, activation of RAGE with S100b eliminated this effect.
Fig. 2.

A: ACh-induced vasodilation in control mice after incubation with sodium nitroprusside (SNP) alone or both SNP and S100b was identical to control (n = 6). B: vasodilation to ACh was potentiated in diabetic mice after incubation with SNP (diabetic vs. diabetic+SNP), but this SNP-potentiated dilation to ACh was abolished after incubation with both SNP and S100b (diabetic+SNP vs. diabetic+SNP+S100b; n = 9). *P < 0.05 vs. diabetic, #P < 0.05 vs. diabetic+SNP+S100b.
Role of RAGE in endothelial dysfunction and NAD(P)H oxidase activity in Type 2 diabetes.
Coronary arterioles isolated from control and diabetic mice dilated in a concentration-dependent manner to ACh, although dilation was reduced in diabetic mice (Fig. 3A). However, the impaired vasodilation in diabetic mice was improved by 50% after corruption of AGE/RAGE signaling with sRAGE (Fig. 3A). Figure 3B shows NAD(P)H oxidase activity from isolated coronary arterioles examined in control, diabetic, and dbTNF−/dbTNF− mice and diabetic mice treated with sRAGE or MG132. sRAGE and NF-κB inhibitor MG132 decreased NAD(P)H oxidase activity in diabetic mice.
Fig. 3.

A: treatment with soluble RAGE (sRAGE) to neutralize RAGE signaling partially restored ACh-induced vasodilation in diabetic mice (n = 6) but did not affect the vasodilation in control mice (n = 5). B: NAD(P)H oxidase activity was higher in diabetic mice, but sRAGE or nuclear factor-κB (NF-κB) inhibitor MG132 decreased NAD(P)H oxidase activity in diabetic vs. control mice (n = 6). *P < 0.05 vs. control, #P < 0.05 vs. diabetic.
Protein expression of RAGE in isolated coronary arterioles.
The protein expression of RAGE (Fig. 4) was higher in diabetic mice than in control mice, but RAGE expression in TEMPOL-, anti-TNF-, sRAGE-, or MG132-treated diabetic mice was similar compared with that in control mice. sRAGE or MG132 did not affect the protein expression of RAGE in control mice (Fig. 4B). The protein expression of RAGE was greatly attenuated in dbTNF−/dbTNF− mice compared with diabetic mice (Fig. 4B).
Fig. 4.

Western blotting shows protein expression of RAGE in isolated coronary arterioles of control, diabetic, and dbTNF−/dbTNF− mice and diabetic mice treated with anti-TNF or TEMPOL (A) or sRAGE or MG132 (NF-κB inhibitor) (B). Data are means ± SD; n = 4. *P < 0.05 vs. control, #P < 0.05 vs. diabetic in each group.
Protein expression of TNF-α and NF-κB in isolated coronary arterioles.
TNF-α expression (Fig. 5, A and B) was elevated over twofold and NF-κB expression (Fig. 5C) was elevated about fourfold in diabetic mice. sRAGE or MG132 decreased TNF-α and NF-κB expression in diabetic mice but did not affect expression in control mice. NF-κB expression was similar in dbTNF−/dbTNF− mice and control mice.
Fig. 5.

Western blot analysis for TNF-α protein expression (A and B) and NF-κB protein expression (C). Bars represent the increased expression in diabetic mice as fold change of control mice, e.g., 2 represents a doubling of expression. Data are means ± SD; n = 4. *P < 0.05 vs. control mice, #P < 0.05 vs. diabetic mice.
mRNA and protein expression of NAD(P)H oxidase subunits.
mRNA expression (Fig. 6) for NAD(P)H oxidase (NOX-2, p22phox, and p40phox) was higher in diabetic mice than in control mice; mRNA expression of p47phox, p67phox, NOX-1, and NOX-4 (data not shown) was similar in diabetic and control mice; and mRNA expression of NAD(P)H oxidase subunits was greatly attenuated in dbTNF−/dbTNF− mice compared with diabetic mice. Anti-TNF or sRAGE or MG132 significantly attenuated the expression of NAD(P)H oxidase in diabetic mice. Moreover, the results of protein expression for NAD(P)H oxidase (NOX-2, p22phox, and p40phox) were identical to the mRNA expression in isolated coronary arterioles in control, diabetic, and dbTNF−/dbTNF− mice (data not shown).
Fig. 6.

Real-time PCR analysis for NAD(P)H oxidase subunit [NOX-2 (gp91phox, A), p22phox (B), p40phox (C)] mRNA expression (expressed as % of control) from coronary arterioles in control, diabetic, and dbTNF−/dbTNF− mice and diabetic mice treated with sRAGE or MG132. Data are means ± SD; n = 5. *P < 0.05 vs. control mice, #P < 0.05 vs. diabetic mice.
DISCUSSION
Our major findings are that overproduction/expression of AGE and RAGE contributes to endothelial dysfunction in Type 2 diabetes. Our findings support the concept that AGE/RAGE and TNF-α signaling is key to the production of ROS; then AGE/RAGE and TNF-α signaling interact to amplify the oxidative stress and induce endothelial dysfunction in diabetic mice.
Role of TNF-α and AGE/RAGE signaling in Type 2 diabetes.
The basis for AGE being involved in the pathophysiological sequelae of vascular dysfunction in Type 2 diabetes stems from hyperglycemia that contributes to the production of AGE. Hyperglycemia and oxidant stress promote nonenzymatic glycoxidation of proteins and lipids (7). AGE-mediated generation of low levels of ROS can result in quenching of the endogenous vasorelaxant NO (7). Activation of RAGE by its various ligands reportedly induces a variety of proinflammatory and procoagulant cellular responses, resulting from the activation of NF-κB, including the expression of vascular cell adhesion molecule-1 (VCAM-1), TNF-α, IL-6, and tissue factor (TF) (24). The interaction of AGE with RAGE induces the production of ROS, which can stimulate the cascade leading to NF-κB-induced transcriptional events. NF-κB will induce expression of TNF-α (5, 23). Accordingly, we determined whether AGE/RAGE signaling leads to TNF-α expression and production in diabetes. Our results showed that protein expression of RAGE, TNF-α, and NF-κB was elevated in Type 2 diabetes. In Type 2 diabetic mice, neutralizing antibody to TNF-α, the free radical scavenger TEMPOL, sRAGE (to corrupt AGE/RAGE signaling), or the NF-κB inhibitor MG132 decreased the protein expression of RAGE, but sRAGE and MG132 also attenuated TNF-α expression. These data provided the evidence for the interaction of AGE/RAGE with TNF-α, which may then stimulate the production of O2•− via NF-κB in Type 2 diabetes.
Moreover, our results showed that vasodilation to ACh was potentiated in diabetic mice after incubation with SNP, but SNP-potentiated dilation to ACh was abolished after incubation with both SNP and the RAGE agonist S100b. These provocative results further support our idea that AGE/RAGE signaling plays a role in endothelial dysfunction in diabetes. We found the fact that the treatment with SNP potentiated the response to ACh in diabetic arterioles puzzling and have only speculation that is unconfirmed. It is possible that administration of SNP, and the subsequent donation of NO, helps the vascular cells scavenge excess superoxide, thus shifting them into a more favorable redox balance. The generation of AGE and augmentation of proinflammatory mechanisms in the vessel, at least in part via accumulation of S100/calgranulins and amphoterin released from activated inflammatory cells, provides a potent feedback loop for sustained oxidant stress, ongoing generation of AGE, and vascular perturbation. It is provocative to note that despite the similarities in glucose, body weight, lipid level, insulin, HbA1c (the index of AGE accumulation), and BP in diabetic animals, endothelial function was better in dbTNF−/dbTNF− and in Leprdb mice treated with anti-TNF or sRAGE. Our results also showed that HbA1c was greater in diabetic vs. control mice, and anti-TNF and dbTNF−/dbTNF− attenuated HbA1c level. Together, our results indicate that the impaired vasodilation may result from increased expression/production of TNF-α and AGE/RAGE signaling in Type 2 diabetes. These pathways stimulate endothelial generation of O2•− radicals in the microvasculature and contribute to the observed endothelial dysfunction.
AGE/RAGE and NO-mediated vasodilation in Type 2 diabetes.
TNF-α produces a rapid inhibitory action on NO in the endothelium via activation of a sphingomyelinase/ceramide signaling pathway; this mechanism purportedly mediates the action of TNF-α, thereby contributing to vascular endothelial dysfunction in coronary circulation under different pathological conditions with increased cytokines (12, 15, 29). We found elevations of TNF-α expression in diabetic mice; sRAGE decreased TNF-α expression in diabetic mice but did not affect TNF-α expression in control mice. The exact reason why TNF-α is elevated—obesity, diabetes, hyperlipidemia, or some or all of these conditions—is critical. We examined the role of AGE/RAGE signaling in the expression of TNF-α to better understand signaling mechanisms responsible for TNF-α expression. Our findings indicate that AGE/RAGE signaling plays a pivotal role in regulation of TNF-α expression. Although this does not address any particular risk factor directly, the formation of AGE is known to occur in diabetes, and the oxidative stress induced by AGE/RAGE signaling activates NF-κB.
AGE can also reduce the bioavailability and activity of endothelium-derived NO. Serum AGE in patients with Type 2 diabetes are inversely related to the degree of endothelium-dependent and endothelium-independent vasodilation in the brachial artery (22). Aminoguanidine decreases vascular AGE accumulation and severity of atherosclerotic plaque in diabetic rats (8). In our study, vasodilation to the endothelium-independent vasodilator SNP was identical in control and diabetic mice, and dilation to the endothelium-dependent agonist ACh was reduced in diabetic vs. control mice. These results are consistent with our previous study (10). Blocking the formation of AGE or interaction with RAGE are obvious targets for therapeutics. The studies to address this were conducted with the inhibitor of AGE/RAGE formation, signaling, and interactions, sRAGE, to block the RAGE signaling pathway.
In diabetic ApoE-null mice, in which vascular lesions were already established, treatment with sRAGE induced regression of atherosclerosis and lesion complexity (the percentage of complex lesions decreased as they were converted to fatty streaks) (18). This finding is consistent with ours, in which impaired vasodilation was partially restored by administration of sRAGE, because preservation of endothelial function appears to be anti atherosclerotic. Furthermore, sRAGE decreased TNF-α protein expression in diabetic mice (10). Our results support the concept that the interaction of TNF-α with AGE/RAGE contributes to endothelial dysfunction in diabetic mice. To our knowledge, this is the first study to link the mechanism of coronary arteriolar endothelial dysfunction with AGE/RAGE signaling in Type 2 diabetes.
AGE/RAGE and ROS in Type 2 diabetes.
AGE that bind to RAGE on the endothelial cell surface can lead to a signaling cascade, stimulating NAD(P)H oxidase and increasing the production of ROS (25, 28). The key target of RAGE signaling is NF-κB causing pathological changes in gene expression (3, 25, 28). AGE also may decrease NO availability by decreasing nitric oxide synthase (NOS) activity (4). In our previous study (10), we confirmed the links among TNF-α, NAD(P)H oxidase, ROS, and impaired vasodilation in coronary arterioles in Type 2 diabetes. This study provides further experimental evidence for an interactive signaling pathway of AGE/RAGE and TNF-α. AGE/RAGE appear to sum with TNF-α to induce the endothelial dysfunction in Type 2 diabetes. A previous report showed AGE production increased in diabetic retinal vessels (21) and renal glomeruli (11). High expression of AGE, RAGE, and NF-κB in lacrimal glands of diabetic rats (1) suggests that these factors are involved in signaling and in subsequent inflammatory alterations related to diabetes mellitus. RAGE has been implicated in the pathogenesis of diabetic complications. AGE products and RAGE signaling induces oxidative stress and leads to activation of the transcription factor NF-κB (5, 7). Because TNF-α has four NF-κB sites in its promoter, we postulate that AGE/RAGE signaling increases TNF-α expression.
Our results showed that the free radical scavenger TEMPOL attenuated the protein expression of RAGE in Type 2 diabetes, which suggests a link between RAGE and superoxide. NAD(P)H oxidase inhibitor (apocynin) restored endothelium-dependent dilation in diabetic mice (10). Expression (mRNA and protein) of NAD(P)H oxidase subunits (NOX-2, p22phox, and p40phox) and NAD(P)H oxidase activity were significantly higher in diabetic mice than in control mice. Anti-TNF, sRAGE, or MG132 significantly inhibited the expression of these NAD(P)H subunits in Type 2 diabetes. Moreover, expression of NAD(P)H oxidase subunits was greatly attenuated in dbTNF−/dbTNF− mice compared with diabetic mice, and anti-TNF attenuated the protein expression of RAGE in diabetic mice. These results indicated that the oxidative stress is induced by the production of ROS, which then activates NF-κB. Key to the production of ROS is AGE/RAGE and TNF-α signaling. We have shown interactions among oxidative stress, AGE/RAGE, and TNF-α, because oxidative stress induces NF-κB, and this transcription factor induced both RAGE and TNF-α expression and TNF-α induced RAGE expression (Fig. 7).
Fig. 7.

Schematic figure showing the interactions among TNF-α, AGE/RAGE, and NF-κB signaling. In brief, central to the endothelial dysfunction is oxidative stress. The oxidative stress is induced by the production of reactive oxygen species (ROS), and this induces NF-κB activation. Key to the production of ROS is AGE/RAGE and TNF-α signaling. We have shown interactions among oxidative stress, AGE/RAGE, and TNF-α because oxidative stress induces NF-κB, this transcription factor can induce both RAGE and TNF-α expression, and TNF-α can induce RAGE expression. Thus the oxidative stress of diabetes begets more oxidative stress, eventually inducing endothelial dysfunction, because of decreased bioavailability of nitric oxide (NO) (due to the reaction between NO and O2•−). EC, endothelial cells; VSMCs, vascular smooth muscle cells; TNFR, TNF-α receptor.
In conclusion, our results indicate that the oxidative stress of diabetes begets more oxidative stress, eventually inducing endothelial dysfunction, because of decreased bioavailability of NO. We believe that understanding endothelial dysfunction is critical because the progression of vascular disease may be halted if endothelial dysfunction is rectified. Our study suggests that central to the endothelial dysfunction is oxidative stress; the oxidative stress is induced by the production of ROS, and this induces NF-κB. We envision a scheme in which oxidative stress produced by one of the stimuli begets further oxidative stress by the other, and this becomes the basis for a pathological spiral leading to vascular disease. We believe that TNF-α is further upstream in the hierarchy because TNF-α induces endothelial dysfunction in the prediabetic metabolic syndrome—presumably before AGE/RAGE signaling is pivotal. Our data demonstrated relevance and translation to the cardiovascular disease. Together, these results provide support for our idea that the interaction between TNF-α and RAGE contributes, perhaps even by amplifying one another, toward the evolution of endothelial dysfunction and vascular disease in diabetes. These findings may provide further insight into a novel therapeutic target for cardiovascular diseases associated with TNF-α and AGE/RAGE signaling.
GRANTS
This study was supported by Pfizer Atorvastatin Research Award grants (2004-37), an American Heart Association Scientist Development Grant (110350047A), and National Heart, Lung, and Blood Institute Grants (RO1-HL-077566 and RO1-HL-085119) to C. Zhang.
Acknowledgments
The design of Fig. 7 was created by Dr. Xiuping Chen from the laboratory of C. Zhang.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Alves M, Calegari VC, Cunha DA, Saad MJ, Velloso LA, Rocha EM. Increased expression of advanced glycation end-products and their receptor, and activation of nuclear factor kappa-B in lacrimal glands of diabetic rats. Diabetologia 48: 2675–2681, 2005. [DOI] [PubMed] [Google Scholar]
- 2.Beckman JA, Creager MA, Libby P. Diabetes and atherosclerosis: epidemiology, pathophysiology, and management. JAMA 287: 2570–2581, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Bierhaus A, Schiekofer S, Schwaninger M, Andrassy M, Humpert PM, Chen J, Hong M, Luther T, Henle T, Kloting I, Morcos M, Hofmann M, Tritschler H, Weigle B, Kasper M, Smith M, Perry G, Schmidt AM, Stern DM, Haring HU, Schleicher E, Nawroth PP. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes 50: 2792–2808, 2001. [DOI] [PubMed] [Google Scholar]
- 4.Bucala R, Tracey KJ, Cerami A. Advanced glycosylation products quench nitric oxide and mediate defective endothelium-dependent vasodilatation in experimental diabetes. J Clin Invest 87: 432–438, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Martin R, Hoeth M, Hofer-Warbinek R, Schmid JA. The transcription factor NF-kappa B and the regulation of vascular cell function. Arterioscler Thromb Vasc Biol 20: E83–E88, 2000. [DOI] [PubMed] [Google Scholar]
- 6.Diamanti-Kandarakis E, Piperi C, Kalofoutis A, Creatsas G. Increased levels of serum advanced glycation end-products in women with polycystic ovary syndrome. Clin Endocrinol (Oxf) 62: 37–43, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Feng L, Matsumoto C, Schwartz A, Schmidt AM, Stern DM, Pile-Spellman J. Chronic vascular inflammation in patients with type 2 diabetes: endothelial biopsy and RT-PCR analysis. Diabetes Care 28: 379–384, 2005. [DOI] [PubMed] [Google Scholar]
- 8.Forbes JM, Yee LT, Thallas V, Lassila M, Candido R, Jandeleit-Dahm KA, Thomas MC, Burns WC, Deemer EK, Thorpe SR, Cooper ME, Allen TJ. Advanced glycation end product interventions reduce diabetes-accelerated atherosclerosis. Diabetes 53: 1813–1823, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288: 373–376, 1980. [DOI] [PubMed] [Google Scholar]
- 10.Gao X, Belmadani S, Picchi A, Xu X, Potter BJ, Tewari-Singh N, Capobianco S, Chilian WM, Zhang C. Tumor necrosis factor-alpha induces endothelial dysfunction in Leprdb mice. Circulation 115: 245–254, 2007. [DOI] [PubMed] [Google Scholar]
- 11.Horie K, Miyata T, Maeda K, Miyata S, Sugiyama S, Sakai H, van Ypersole de Strihou C, Monnier VM, Witztum JL, Kurokawa K. Immunohistochemical colocalization of glycoxidation products and lipid peroxidation products in diabetic renal glomerular lesions. Implication for glycoxidative stress in the pathogenesis of diabetic nephropathy. J Clin Invest 100: 2995–3004, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keller M, Lidington D, Vogel L, Peter BF, Sohn HY, Pagano PJ, Pitson S, Spiegel S, Pohl U, Bolz SS. Sphingosine kinase functionally links elevated transmural pressure and increased reactive oxygen species formation in resistance arteries. FASEB J 20: 702–704, 2006. [DOI] [PubMed] [Google Scholar]
- 13.Kihara M, Schmelzer JD, Poduslo JF, Curran GL, Nickander KK, Low PA. Aminoguanidine effects on nerve blood flow, vascular permeability, electrophysiology, and oxygen free radicals. Proc Natl Acad Sci USA 88: 6107–6111, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuo L, Davis MJ, Chilian WM. Myogenic activity in isolated subepicardial and subendocardial coronary arterioles. Am J Physiol Heart Circ Physiol 255: H1558–H1562, 1988. [DOI] [PubMed] [Google Scholar]
- 15.Lefer AM, Ma XL. Cytokines and growth factors in endothelial dysfunction. Crit Care Med 21: S9–S14, 1993. [DOI] [PubMed] [Google Scholar]
- 16.Letoha T, Somlai C, Takacs T, Szabolcs A, Rakonczay Z Jr, Jarmay K, Szalontai T, Varga I, Kaszaki J, Boros I, Duda E, Hackler L, Kurucz I, Penke B. The proteasome inhibitor MG132 protects against acute pancreatitis. Free Radic Biol Med 39: 1142–1151, 2005. [DOI] [PubMed] [Google Scholar]
-
17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2
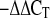 method. Methods 25: 402–408, 2001. [DOI] [PubMed] [Google Scholar]
method. Methods 25: 402–408, 2001. [DOI] [PubMed] [Google Scholar] - 18.Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ Jr, Chow WS, Stern D, Schmidt AM. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med 4: 1025–1031, 1998. [DOI] [PubMed] [Google Scholar]
- 19.Picchi A, Gao X, Belmadani S, Potter BJ, Focardi M, Chilian WM, Zhang C. Tumor necrosis factor-alpha induces endothelial dysfunction in the prediabetic metabolic syndrome. Circ Res 99: 69–77, 2006. [DOI] [PubMed] [Google Scholar]
- 20.Rask-Madsen C, Dominguez H, Ihlemann N, Hermann T, Kober L, Torp-Pedersen C. Tumor necrosis factor-alpha inhibits insulin's stimulating effect on glucose uptake and endothelium-dependent vasodilation in humans. Circulation 108: 1815–1821, 2003. [DOI] [PubMed] [Google Scholar]
- 21.Stitt AW, Li YM, Gardiner TA, Bucala R, Archer DB, Vlassara H. Advanced glycation end products (AGEs) co-localize with AGE receptors in the retinal vasculature of diabetic and of AGE-infused rats. Am J Pathol 150: 523–531, 1997. [PMC free article] [PubMed] [Google Scholar]
- 22.Tan KC, Chow WS, Ai VH, Metz C, Bucala R, Lam KS. Advanced glycation end products and endothelial dysfunction in Type 2 diabetes. Diabetes Care 25: 1055–1059, 2002. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka N, Yonekura H, Yamagishi S, Fujimori H, Yamamoto Y, Yamamoto H. The receptor for advanced glycation end products is induced by the glycation products themselves and tumor necrosis factor-alpha through nuclear factor-kappa B, and by 17beta-estradiol through Sp-1 in human vascular endothelial cells. J Biol Chem 275: 25781–25790, 2000. [DOI] [PubMed] [Google Scholar]
- 24.Valencia JV, Mone M, Zhang J, Weetall M, Buxton FP, Hughes TE. Divergent pathways of gene expression are activated by the RAGE ligands S100b and AGE-BSA. Diabetes 53: 743–751, 2004. [DOI] [PubMed] [Google Scholar]
- 25.Wautier MP, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier JL. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab 280: E685–E694, 2001. [DOI] [PubMed] [Google Scholar]
- 26.Wendt T, Harja E, Bucciarelli L, Qu W, Lu Y, Rong LL, Jenkins DG, Stein G, Schmidt AM, Yan SF. RAGE modulates vascular inflammation and atherosclerosis in a murine model of Type 2 diabetes. Atherosclerosis 185: 70–77, 2006. [DOI] [PubMed] [Google Scholar]
- 27.Yagihashi S, Kamijo M, Baba M, Yagihashi N, Nagai K. Effect of aminoguanidine on functional and structural abnormalities in peripheral nerve of STZ-induced diabetic rats. Diabetes 41: 47–52, 1992. [DOI] [PubMed] [Google Scholar]
- 28.Yan SD, Schmidt AM, Anderson GM, Zhang J, Brett J, Zou YS, Pinsky D, Stern D. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem 269: 9889–9897, 1994. [PubMed] [Google Scholar]
- 29.Zhang C, Xu X, Potter BJ, Wang W, Kuo L, Michael L, Bagby GJ, Chilian WM. TNF-alpha contributes to endothelial dysfunction in ischemia/reperfusion injury. Arterioscler Thromb Vasc Biol 26: 475–480, 2006. [DOI] [PubMed] [Google Scholar]