

Abstract
Apolipoprotein B (ApoB) is a nonexchangeable apolipoprotein that dictates the synthesis of chylomicrons and very low density lipoproteins. ApoB is the major protein in low density lipoprotein, also known as the “bad cholesterol” that is directly implicated in atherosclerosis. It has been suggested that the N-terminal domain of apoB plays a critical role in the formation of apoB-containing lipoproteins through the initial recruitment of phospholipids in the endoplasmic reticulum. However, very little is known about the mechanism of lipoprotein nucleation by apoB. Here we demonstrate that a strong phospholipid remodeling function is associated with the predicted α-helical and C-sheet domains in the N-terminal 17% of apoB (B17). Using dimyristoylphosphatidylcholine (DMPC) as a model lipid, these domains can convert multilamellar DMPC vesicles into discoidal-shaped particles. The nascent particles reconstituted from different apoB domains are distinctive and compositionally homogenous. This phospholipid remodeling activity is also observed with egg phosphatidylcholine (egg PC) and is therefore not DMPC dependent. Using kinetic analysis of the DMPC clearance assay, we show that the identified phospholipid binding sequences all map to the surface of the lipid binding pocket in the B17 model based on the homologous protein, lipovitellin. Since both B17 and microsomal triglyceride transfer protein (MTP), a critical chaperone during lipoprotein assembly, are homologous to lipovitellin, the identification of these phospholipid remodeling sequences in B17 provides important insights into the potential mechanism that initiates the assembly of apoB-containing lipoproteins.
Apolipoprotein B (apoB) is the only known nonexchangeable apolipoprotein in humans. It is the major protein component of low density lipoprotein (LDL), a major risk factor for atherosclerosis. Two forms of apoB are synthesized in humans, B48 from in small intestine and B100 in the liver (1). The production of B48 is the result of a tissue specific mRNA processing occurring in the small intestine (2, 3). The translation of B48 and B100 in the endoplasmic reticulum (ER) is directly coupled with the assembly of two major classes of lipoproteins: chylomicrons and very low density lipoproteins (VLDL) (4).
Although an apoB structure at atomic resolution is still unavailable, it is generally accepted that the full length apoB has an βα1-β1-α2-β2-α3 pentapartite domain organization, in which α represents predominantly α-helical structures and β corresponds to β-sheets (5). According to this model, apoB is depicted as a belt wrapped around a lipoprotein particle (6). Evidence from electron microscopy indicates that the N-terminus of apoB, largely the βα1 domain, is globularly folded and protrudes away from the LDL particle (7, 8). These findings are consistent with the observation that the N-terminal ∼20% of apoB is homologous to a globular protein, lipovitellin (9, 10). Lipovitellin is a nutrient storage protein in the egg yolk derived from vitellogenesis (11). The crystal structure of lipovitellin indicated that the protein has a funnel-shaped lipid binding cavity lined extensively by hydrophobic β-sheets (12). Despite the low sequence identity, the structural homology between the N-terminal 17% of apoB (B17) and lipovitellin is supported by limited proteolysis and biophysical characterization of the predicted domains (13).
The discovery of microsomal triglyceride transfer protein (MTP) was a key breakthrough in the understanding of lipoprotein assembly in the ER. MTP is a heterodimeric ER resident chaperone. The large subunit of MTP is constantly associated with one copy of protein disulfide isomerase (PDI) (14). In vitro, MTP mobilizes neutral lipids from donor vesicles to acceptor vesicles (15). This lipid transfer activity of MTP has been proposed to be indispensable for the lipidation of apoB (16). Defects in MTP result in abetalipoproteinemia, an autosomal recessive disease characterized by exceedingly low levels of apoB-containing lipoproteins in the plasma (17, 18). Interestingly, the N-terminal ∼60% of MTP is also homologous to lipovitellin and apoB (9, 19). The two MTP binding sites on apoB are in the N-terminal 6% and in the residues between 10−13% (19, 20). The fact that lipovitellin crystallizes as a dimer raises the possibility that there are similar heterodimeric interactions between apoB and MTP (19, 21).
Mounting evidence indicates that the N-terminus of apoB plays a critical role in lipoprotein assembly. This idea first emerged from the observation that the reduction of the disulfide bonds in the N-terminus blocked apoB secretion (22, 23). Similar defects were reported when cysteine pairs were mutated to alanine or serine (24, 25). More recently, two groups reported that in cells with MTP, apoB sequences as short as ∼20% of B100 were sufficient for the assembly of a lipoprotein particle containing neutral lipids (26, 27). Although there were discrepancies regarding the minimal required length of apoB, both studies provided evidence that the N-terminal sequence of apoB, approximate to the lipovitellin homologous region, had all the required elements to form a lipoprotein particle.
We hypothesize that the N-terminal βα1 superdomain of apoB contains phospholipid recruiting sequences that nucleate lipoprotein assembly. In fact, B17 binds both phospholipid vesicles and phospholipid-coated triglyceride emulsions in vitro (28-30). It also has the potential to convert multilamellar DMPC into smaller particles (29). However, the exact phospholipid binding sequences within B17 have not been identified and the structural basis for the protein-phospholipid binding is not understood. Based on the lipovitellin homology model, B17 has three distinct domains, the β-barrel (B5.9), α-helical domain (B6.4−13) and C-sheet (B13−17) (Fig. 1a, b). Here we dissect B17 into individual domains and subdomains in order to identify their potential phospholipid recruiting sequences that may be involved in the early stages of lipoprotein assembly.
Figure 1. Proposed domain organization in B17.

a. The domain comparison between lipovitellin (LV) and apoB. The homology between LV and apoB extends to approximately B20. The five domains in LV are colored by β-barrel, green; α-helical, cyan; C-sheet, red; A-sheet, dark blue (12). Regions missing in the crystal structure of LV are in white. ApoB sequences not homologous to LV are shown in gray. b. The three dimensional model of B17 based on LV. The three domains in B17 are colored in the same scheme.
EXPERIMENTAL PROCEDURES
Cloning, expression and purification of the domain constructs within B17
All apoB domains were cloned from B37 initially described by Carraway et al (31). The following constructs were generated for this study: B5.9 (residues 1−264), B6.4−10 (residues 292−469), B6.4−13 (residues 292−593), B6.4−15 (residues 292−680), B6.4−17 (residues 292−782), B9−13 (residues 430−593), B13−15 (residues 611−680) and B13−17 (residues 611−782). Cys451 was mutated to Ala using Quikchange mutagenesis (Stratagene) in B6.4−10 since its disulfide bond partner was excluded in the construct. All constructs, except for B5.9 were cloned into a pET 24a vector (Novagen) and sequenced at the Boston University Genetics Core facility, prior to the expression in BL21 (DE3) cells (Novagen). B5.9 was cloned in baculovirus using the Gateway System (Invitrogen) and expressed in Sf9 cells. Proteins were purified as previously described (13). All protein concentrations were calculated based on their UV absorbance at 280 nm (32).
Preparation of phospholipid vesicles
To prepare multilamellar vesicles (MLVs) with dimyristoylphosphatidylcholine (DMPC) or egg phosphatidylcholine (egg PC), 10 mg of phospholipids (Avanti Polar Lipids) in chloroform were transferred to a 20 ml glass vial and put under nitrogen to evaporate the solvent. The thin layer of DMPC was placed in a vacuum for at least one hour and then hydrated with TS buffer (10 mM Tris, 150 mM sodium chloride, pH 7.5) at room temperature to form MLVs. Egg PC MLVs were prepared with the same buffer, but at 4 °C. The small unilamellar vesicles (SUVs) were prepared from MLVs by sonication with a Branson sonicator for 30 min on ice. The residual MLVs and metal debris were removed by centrifugation at 14,000 g after sonication.
Preparation of DMPC reconstituted particles and kinetic measurements
The reconstituted DMPC particles were prepared by incubating 1 mg/ml protein and DMPC MLVs at specified ratios in TS buffer with 10 mM EDTA, 0.05% sodium azide at 24 °C for 16−24 hours. Since all lipid-binding experiments were performed and compared using weight ratios, the L/P molar ratio varies as the protein molecular weight changes. For instance a 1:1 L/P weight ratio corresponds to molar ratios of 45:1 for B5.9, 31:1 for B6.4−10, 52:1 for B6.4−13, 66:1 for B6.4−15, 83:1 for B6.4−17, 29:1 for B9−13, 13:1 for B13−15 and 29:1 for B13−17.
The kinetic measurements of the DMPC clearance assay were performed on a CARY 300 UV/Vis spectrometer (Varian). Protein samples were prepared at 80 μg/ml in TS buffer and background corrected in quartz cuvettes. DMPC MLVs were added to a final concentration of 80 μg/ml to the protein sample (1:1 L/P ratio) and vigorously mixed just before the kinetic measurement. The clearance of DMPC was monitored at 325 nm as a function of time at 24 °C. Every data point was an average of 0.5 seconds. The measurement for each construct was repeated at least three times.
Size exclusion chromatography
Proteins or protein-DMPC complexes were prepared at 0.5−1 mg/ml protein concentration and specified L/P ratios and then injected onto a Superdex GL-200 column (Amersham Biosciences). The column was eluted with TS buffer at 0.5 ml/min and 4 °C on a FPLC system (Amersham Biosciences) and monitored by UV at 280 nm.
Phospholipid concentration measurement
Phospholipid concentrations were determined by measuring the inorganic phosphorus concentration using the Bartlett assay (33). A series of phosphorus standards (Sigma) from 16 to 130 nmol were prepared together with the DMPC complex samples in glass tubes containing 1 ml distilled water. Each tube was then supplemented with 0.5 ml 10 N sulfuric acid, covered by a marble and incubated at 150−160 °C overnight. On the second day, 0.5 ml 30% (w/w) hydrogen peroxide (Sigma) was added to all samples and incubated at 150−160 °C for 3 hours. After the tubes were cooled, 4.6 ml of 0.22% ammonium molybdate was pipetted to each sample, followed immediately by the addition of 0.2 ml freshly prepared 0.16 mg/ml Fiske SubbaRow reagent (Sigma). The solution was mixed thoroughly and incubated in a 90 °C water bath for 15 min. The colorimetric reading was obtained by measuring the absorbance at 830 nm on a Perkin Elmer UV/Vis spectrometer after the solution cooled. The linear absorbance of the standards was used to determine the sample phosphorus concentrations.
Negative stain electron microscopy
Reconstituted DMPC complex (4 μl) at 0.1−0.5 mg/ml (protein concentration) were loaded on a carbon-coated and glow-discharged copper grid (SPI Supplies) for 1 min, washed with 10 drops of distilled H2O and stained with 1% sodium phosphotungstate, pH 7.5 for 30 s (34). All samples were imaged on a Philips CM12 transmission electron microscope operated at 120 kV with LaB6 filament and recorded on SO-163 EM (Kodak) film at 45,000 × magnifications under minimal electron dose conditions. The film was processed with undiluted Kodak D-19 developer for 12 min and Kodak rapid fixer for 5 min. Electron micrographs were digitized on a Creo IQ Smart2 Scanner (Global Imaging) at 1270 dpi. Particle measurements were performed with Scion Image (Scion Corp.).
Circular dichroism
Circular dichroism (CD) spectra were collected on an AVIV 215 instrument. Protein and reconstituted particle samples at ∼ 2 μM protein were dialyzed against 5 mM potassium phosphate at pH 7.5 prior to the experiment. The exact protein concentration was determined by UV absorbance at 280 nm immediately before each wavelength scan. Each reported wavelength scan was an average of 4 scans taken in a 1 mm cuvette with a 5 second averaging time at every nm at 25 °C. The CD spectra for the buffer and cuvette were acquired immediately before the data collection for background correction. Raw data were converted to mean residue ellipticity and plotted in Origin 7.5 (Microcal Inc.).
Molecular modeling of the B17
The B17 structure was modeled based on its homology to lipovitellin (1LSH.pdb) using MODELLER4 (35), as described previously (13). All molecular images were generated with MOLMOL (36)
RESULTS
The α-helical and C-sheet domains form reconstituted particles with DMPC
To identify the regions in B17 responsible for the DMPC clearance activity, we dissected B17 and individually tested its three domains: the β-barrel (B5.9), α-helical (B6.4−13) and C-sheet (B13−17) (Fig. 1b). The proteins were incubated with the turbid DMPC multilamellar vesicles (MLVs) at 24 °C overnight and then analyzed by size exclusion chromatography (SEC). Chromatograms of the protein after incubation in the absence of DMPC are compared with those mixed 1:1 (wt:wt) with DMPC. The β-barrel domain, B5.9, exists in a monomer/dimer equilibrium in solution. Incubation with DMPC does not change either the amount of free protein in solution or the monomer-dimer equilibrium (Fig. 2a). In solution, B6.4−13 is predominantly a dimer and B13−17 is a mixture of dimers and oligomers. In the presence of DMPC, the B6.4−13 and B13−17 peaks shift to higher molecular weight, indicating their incorporation into larger complexes with DMPC (Fig. 2b, c). Thus, these two domains are responsible for the DMPC clearance activity in B17.
Figure 2. Formation of reconstituted particles with DMPC among the domains in B17.

Chromatograms of three individual domains in B17 incubated with DMPC MLVs: the β-barrel (a), α-helical (b) and C-sheet (c). Proteins were prepared in TS buffer with (black) or without (light gray) DMPC MLVs at 1:1 (wt:wt) ratio and incubated at 24 °C for 16 hours. The β-barrel and α-helical domains were prepared at 1 mg/ml, whereas the C-sheet was prepared at 0.2 mg/ml. The protein/lipid mixtures after incubation were centrifuged and then analyzed by a superdex GL200 column at 4 °C.
The reconstituted particles containing different lengths of the C-sheet are homogeneous in composition and distinctive in size
ApoB domain constructs and DMPC were mixed at increasing ratios in order to survey the range of reconstituted particles that can be formed in vitro. The low solubility of the C-sheet construct B13−17 makes direct examination difficult. Therefore, two constructs, B6.4−15 and B6.4−17, were used, each with an increasing proportion of the C-sheet domain.
Increasing amounts of DMPC were incubated with 1 mg/ml of protein at ratios of 0.5, 1, 2, 4 and 6:1 (wt:wt) to examine the phospholipid binding capacity. The protein/DMPC mixtures after incubation at 24 °C were centrifuged at 14,000 g to remove insoluble MLVs and analyzed by SEC. Chromatograms obtained from these three constructs exhibit a similar pattern (Fig. 3a and Supplement Fig. 1a, 2a). As the L/P ratio increases, the free protein peak decreases, indicating more protein is forming reconstituted particles with DMPC. Moreover, the reconstituted particle peaks shift to lower elution volume as the L/P ratio increases, suggesting the formation of larger particles in the presence of excess lipid. Light scattering becomes more significant for particles formed at higher L/P ratios (4:1 and 6:1) since the total peak area exceeds the peak area of the protein control (Fig. 3a).
Figure 3. Characterization of the B6.4−17/DMPC complexes at different L/P ratios.
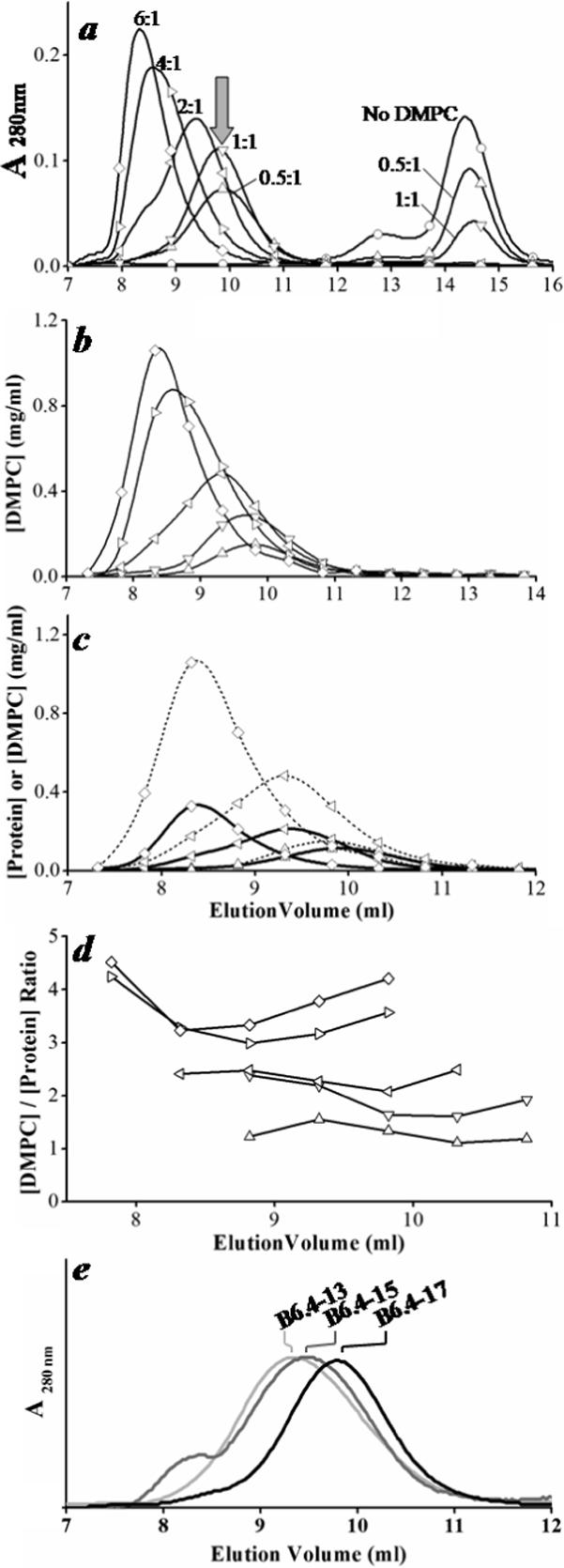
a. SEC analysis of B6.4−17 and its reconstituted DMPC particles prepared at different L/P ratios. B6.4−17 at 1 mg/ml was mixed with DMPC MLVs at 0:1 (○), 0.5:1 (△), 1:1 (▽), 2:1 (◁), 4:1 (▷) and 6:1 (◇) (wt:wt) L/P ratios in 0.6 ml TS buffer with 10 mM EDTA, 0.05% sodium azide. Samples were incubated at 24 °C overnight and analyzed on superdex GL 200 column. Arrow indicates the elution volume of a minimal-sized particle. The labeling scheme in a applies to b-d. b. DMPC concentration in the fractions collected from SEC. DMPC concentration is measured using the Bartlett assay (33). c. The overlay of the lipid and protein concentration. Protein concentration is calculated from UV absorbance at 280 nm. The elution volume of the protein profile has been corrected to match the void volume in the loops from the detector to the fraction collector. d. Actual L/P ratios in each fraction. Only fractions corresponding to the reconstituted particle peak are shown. e. The comparison of the minimal-sized particle prepared by B6.4−13, B6.4−15 and B6.4−17. All three proteins were prepared at 1 mg/ml protein concentration, 1:1 L/P ratios and analyzed by SEC as described previously. UV absorbance was normalized for better comparison.
To determine the elution profile of DMPC, fractions after SEC separation were subjected to phosphorus analysis using the Bartlett assay (33). As the protein concentration is constant in this experiment, an increasing L/P ratio results in an increase in the actual lipid concentration in the product fractions (Fig. 3b, Supplement Fig. 1b, 2b). The coelution of the lipid and protein peaks suggests very few protein-free DMPC vesicles are present (Fig. 3c, Supplement Fig. 1c, 2c). The calculate L/P ratios in the collected fractions reflect the amount of protein and DMPC used initially. Within a single chromatogram, the variation of the measured L/P ratios in different collected fractions is minor (Fig. 3d, Supplement Fig. 1d, 2d). This suggests that in either excess protein or lipid, the reconstituted DMPC complexes are homogeneous.
When protein is in excess, i.e. at L/P ratios of 0.5−1, the elution volume of the reconstituted particle from the SEC column remains unchanged (arrows in Fig. 3a). All lipids are converted to particles with a minimal size, while excess protein is found in the unbound lipid-free fractions. This phenomenon is also observed for B6.4−15 and B6.4−17 (Supplement Fig. 1a, 2a). The presence of a minimal-size particle indicates that the available lipids are not equally distributed to all proteins. Under limited lipid availability, a minimal number of lipids are required to form a reconstituted particle. It is noteworthy that even at 0.5:1 L/P weight ratio of the B6.4−13/DMPC mixture, the molar concentration of DMPC is still 25 times higher than that of the protein. For larger construct B6.4−17, the DMPC molar concentration is 40 times higher than the protein. Since the minimal-size particles are most reproducible and homogeneous, they are the focus of our structural characterization.
A comparison of the minimal-size particles formed with three constructs: B6.4−13, B6.4−15 and B6.4−17, indicates that the size decreases as more C-sheet is included in the particle (Fig. 3e). Since the size of the minimal-size particle is independent from the L/P ratio, the sole determinant of their sizes is the biophysical properties of the protein. For the three tested constructs, the difference is the length of the C-sheet following the α-helical domain.
The reconstituted particles with DMPC are discoidal in shape
The reconstituted particles were further characterized by negative stain electron microscopy (EM). The formation of reconstituted particles from MLVs is observed with B6.4−13 (Fig. 4b, g), B6.4−15 (c, h), B6.4−17 (d, i) and B13−17 (e, j), but not with B5.9 (f). Only large vesicles or very small particles are observed in the turbid B5.9/DMPC samples, which is consistent with the DMPC clearance assay. Those small particles are ∼50 Å in diameter and are likely to be the free B5.9 protein (f, white arrows). The reconstituted particles formed with the α-helical or C-sheet domains (b-e, g-j) look similar to each other and are distinct from DMPC small unilamellar vesicles (SUVs) (a). Three types of morphologies are observed on the electron micrograph: rod-like structures, stacks of rods in rouleaux (b, solid box) and plate-like structures (dotted box). Notably, these observations are very similar to the reconstituted apoA-I/DMPC particles that have been proposed to adopt a discoidal shape (37). The “plates” are the projection of the disks from the top and the “rods” or rouleaux are the disks seen on edge.
Figure 4. Reconstituted DMPC particles imaged by negative stain electron microscopy.

a. Sonicated DMPC SUVs. Black arrows show SUVs with a double bilayer thickness in the rouleaux. b, g. Reconstituted B6.4−13/DMPC particles. The solid square box in b shows the rod-shaped particles forming rouleaux. Dotted box shows a plate-shaped particle. c, h. Reconstituted B6.4−15/DMPC particles. d, i. Reconstituted B6.4−17/DMPC particles. e, j. Reconstituted B13−17/DMPC particles. Samples in b, c, d, e (upper panel) were prepared at 1 mg/ml protein concentration and 1:1 (wt:wt) L/P ratios. Samples in g, h, i, j (lower panel) were prepared at 4:1 L/P ratios. These reconstituted particles were purified by SEC column prior to grid preparation. f. B5.9 incubated with DMPC MLVs. The grid was prepared using turbid protein/DMPC mixtures after incubation. A DMPC MLV is shown in the image. The small particles in the background (white arrows) are probably free proteins. All images were recorded at 45,000× magnification. The magnification bar in j is 500 Å for all images.
Two factors influence the size of the reconstituted particles, the L/P ratio and the protein identity. For all constructs, higher L/P ratios give rise to larger particles. The minimal-size particles prepared with B6.4−13, B6.4−15 and B6.4−17 at 1:1 L/P ratio were measured on electron micrographs and compared with each other. In agreement with the disk model of the reconstituted particles, the rod-shaped particles have a thickness of 46−50 Å (Table 1), very close to the thickness expected for a DMPC bilayer, which is about 42 Å in the gel phase (38). The particle diameter can be derived from two views, the “plate” or the “rod”. Many plate view particles are slightly oblong, giving rise to a longer axis and a shorter axis. The average of the measurements from the “plates” is in close agreement with that from the “rods” for all three constructs. A decrease in particle diameter is observed in the construct with increasing amounts of the C-sheet. Based on the measurements from the “rods”, the average diameter of particles with B6.4−13, B6.4−15 and B6.4−17 are 168±30 Å, 164±22 Å and 152±27 Å, respectively (Table 1). This size difference of the reconstituted particles is also seen in the comparison using SEC methods (Fig. 3e).
Table 1.
Dimensions of the reconstituted protein/DMPC complexes
| B6.4−13 | B6.4−15 | B6.4−17 | |
|---|---|---|---|
| Plate view | |||
| Long Axis (Å) | 178±25 (100)a | 183±27 (100) | 166±21 (100) |
| Short Axis (Å) | 148±22 (100) | 149±23 (100) | 140±16 (100) |
| Average (Å) | 163±28 (200) | 166±30 (200) | 153±23 (200) |
| Rod view | |||
| Thickness (Å) | 48±9.4 (140) | 50±7.6 (103) | 46±9.1 (106) |
| Length (Å) | 168±30 (201)b | 164±22 (228) | 152±27 (207) |
Measurements of particle dimension are performed on the digitized electron micrographs using Scion Image (beta 4.0.2). The average value with standard deviation is shown. Values in parenthesis are the number of particles used for measurement.
Probabilities that differences of the particle length is due to chance, as calculated using student's T test are: B6.4−13 vs. B6.4−15, 8.0%; B6.4−15 vs. B6.4−17, 0.01%; B6.4−17 vs. B6.4−13, 0.01%.
The size of particles formed by B13−17 is not listed here because this construct readily aggregates and a fair comparison cannot be justified without knowing the protein concentration. In addition, B13−17 appears to make particles of a wider range of sizes. For example, at 1:1 L/P ratio B13−17 makes the smallest particles, which are only about 100 Å in diameter (Fig. 4e). Interestingly, B13−17 also makes the largest particles among all constructs at 4:1 L/P ratio (Fig. 4j). The maximum length of the rod-shaped particle reaches ∼400 Å. From the current imaging method, we cannot rule out the possibility that these large particles are collapsed vesicles.
Binding of DMPC does not change the protein secondary structure
The formation of a reconstituted DMPC particle may require conformational changes in the protein, when its aqueous environment changes to a hydrophobic lipid-protein interface. To examine whether the formation of such particles involves changes in the protein secondary structure, we compared the secondary structural content of lipid-free proteins and lipid-bound proteins using circular dichroism (CD). Three constructs, B6.4−13, B6.4−15 and B6.4−17, exhibit helical CD spectra as predicted (Fig. 5a-c, solid triangles). The magnitude of the mean residue ellipticity at 222, 209 and 195 nm deceases from B6.4−13 to B6.4−17 as more of the C-sheet region is included, which is consistent with the homology model where B13−17 consists of predominantly β-sheet structure (13). For all three constructs, the CD spectra of the reconstituted DMPC complexes overlap closely with that of the corresponding lipid-free proteins (Fig. 5a-c). This observation shows that the overall secondary structural content does not change when the protein binds DMPC. It suggests that the formation of secondary structure in the α-helical and the C-sheet domains in apoB is not dependent on the lipid binding.
Figure 5. Comparison of secondary structure upon DMPC binding by CD.
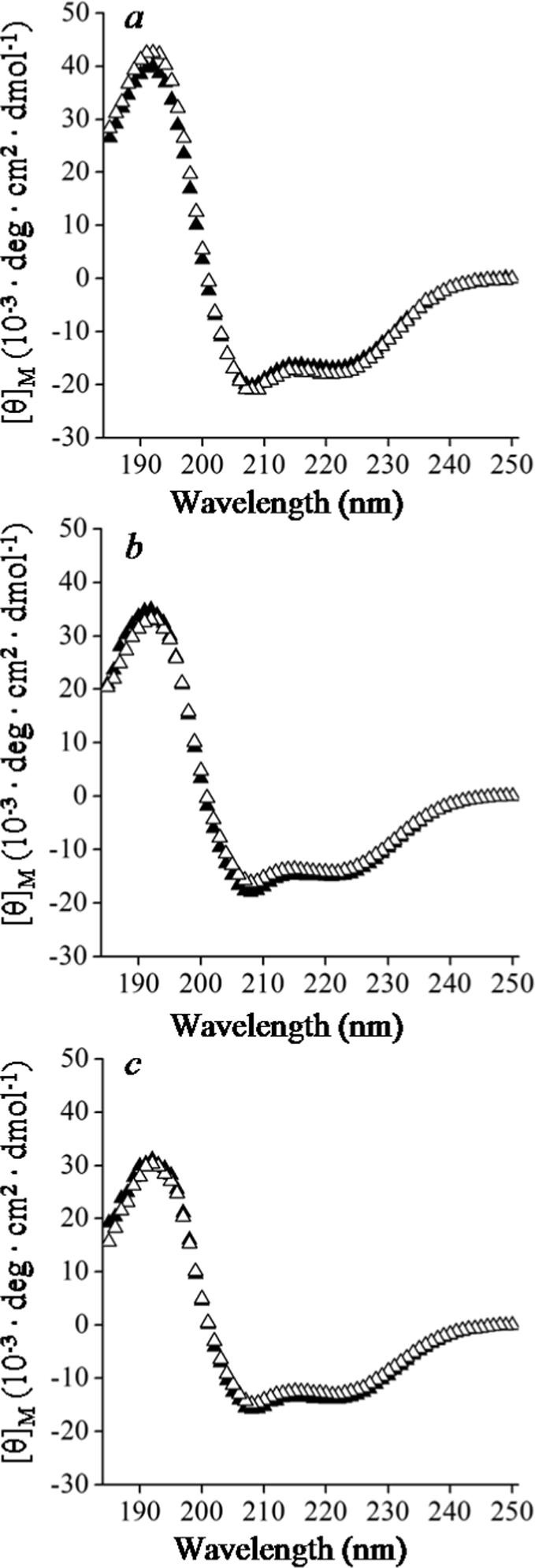
Comparisons of CD wavelength scans of lipid-free proteins (open triangle) and protein-DMPC complexes (solid triangle): a. B6.4−13, b. B6.4−15, and c. B6.4−17. The reconstituted DMPC particles were produced at 1:1 (w:w) P/L ratio and purified by SEC. Each sample contained ∼2 μM protein in 5 mM potassium phosphate at pH 7.5. The exact molar concentration was determined by UV absorbance immediate prior to the CD experiment. Spectra were collected in 1 mm cuvettes at 25 °C. Each spectrum was the average of four scans with an averaging time of 5 s at every 1 nm wavelength and converted to mean residue ellipticity.
Domains in B17 exhibit distinct kinetics in the remodeling of DMPC MLVs
We measured the kinetics of DMPC clearance as an indicator for the efficiency of phospholipid remodeling. The turbidity of the DMPC MLVs was measured as a function of time in the presence of various apoB domains. Proteins with stronger phospholipid remodeling capability are expected to clear DMPC faster, thus resulting in faster signal decay (Fig. 6).
Figure 6. Kinetics of the DMPC clearance.

The comparison of the DMPC clearance with different apoB constructs. The protein constructs were prepared at 80 μg/ml in TS buffer in quartz cuvettes and background corrected. DMPC MLVs were added to the protein solution at 1:1 (wt:wt) ratio. The mixtures were incubated at 24 °C for 1000 mins and the light scattering of the large MLVs were monitored by UV absorbance at 325 nm. DMPC MLVs, gray curve; B5.9, △; B6.4−10, □; B6.4−13, ▽; B6.4−15, ▷; B6.4−17, ◁; B9−13, ◇; B13−15,  .
.
To make a closer comparison, the clearance curves of the B17 domains were fit to a first order exponential decay y = Ae–x/T1 +B, in which Rc, the reciprocal of the time constant T1, is the rate of clearance (Table 2). Similar to the DMPC MLV control, the proposed N-terminal β-barrel domain (B5.9) exhibits an almost linear curve, which resulted mostly from the precipitation of large MLVs. The N-terminal half of the α-helical domain (B6.4−10) has an Rc value similar to the full length α-helical domain (B6.4−13) (Table 2). In contrast, the C-terminal half of the α-helical domain (B9−13) does not clear DMPC MLVs. Therefore, the phospholipid remodeling elements in the proposed α-helical domain reside primarily in its N-terminal half.
Table 2.
Kinetics of DMPC clearance
|
Constructs |
Rc (min−1)a |
|---|---|
| DMPC | 8.1 × 10−4 |
| B5.9 | 9.5 × 10−4 |
| B6.4−10 | 2.6 × 10−3 |
| B6.4−13 | 2.9 × 10−3 |
| B6.4−15 | 2.7 × 10−2 |
| B6.4−17 | 5.5 × 10−3 |
| B9−13 | 1.1 × 10−3 |
| B13−15b | 1.24 |
Rc is the reciprocal of T1, which is obtained by fitting the curve in Fig. 4 with a first exponential decay y = Ae-x/T1 + B using Origin 7.5.
Only the first 2 minutes in the clearance assay are used in the curve fitting for B13−15.
The N-terminal half of the proposed C-sheet (B13−15) exhibits a unique clearance curve. Its clearance profile indicates three events: a rapid clearance within 2 min, a recovery of signal, and a prolonged signal decay (Fig. 6). This observation suggests that B13−15 very rapidly remodels DMPC MLVs, but it does not stabilize nascent particles, thus these particles aggregate and precipitate out of solution. Notably, the nascent B13−15/DMPC particles settle faster than the DMPC MLVs, suggesting it is compositionally different from DMPC MLVs. When B13−15 is linked to the predicted α-helical domain in B6.4−15 and B6.4−17, its behavior is changed. B6.4−15 and B6.4−17 clear DMPC more rapidly and maintain the stability of the nascent particles. It is likely that the terminal location of B13−15 in B6.4−15 is related to its faster clearance kinetics than that of B6.4−17 (Fig. 6, Table 2). B13−17 was also tested, but the kinetic measurements were not consistent. We suspect that spontaneous protein aggregation is an important factor, resulting in changes in the effective protein concentration in the experiment.
α-helical and C-sheet domains efficiently remodel unilamellar vesicles
The secretion of a well-assembled apoB-containing lipoprotein takes about 30 min (39). Thus, the initial lipid recruitment in the ER should be achieved within a time frame much shorter than 30 min. The DMPC MLV clearance assay provides a measurable approach to compare the speed of phospholipid remodeling by different domains, but it does not reflect the speed of in vivo phospholipid recruitment. Unilamellar vesicles are a closer mimic to the physiological condition in this regard.
When B6.4−17 is added to DMPC SUVs, the morphology of vesicles visualized on the electron micrographs is dramatically changed within the time of grid preparation, which is ∼2 min (Fig. 7a, b). Many rod-shaped particles appeared (white arrowhead), suggesting the formation of reconstituted DMPC complexes. The actual extent of conversion is larger than the ratio between the “rods” and “plates”, since many reconstituted DMPC complexes cannot be visually differentiated from DMPC SUVs in the plate views.
Figure 7. Remodeling of DMPC and Egg PC unilamellar membrane with B6.4−17.

a. Sonicated DMPC SUVs. b. DMPC SUVs with B6.4−17. c. Sonicated Egg PC SUVs. d. Egg PC SUVs with B6.4−17. Protein was 1:1 (wt:wt) mixed with 0.2 mg/ml DMPC and loaded onto EM grid immediately after mixing. White arrowheads show the rod-shaped particles; white arrows show the ring-like structure. The magnification bar is 100 nm.
DMPC is favorable for in vitro lipoprotein reconstitution experiments because its phase transition temperature (24 °C) is close to room temperature. The transition between the liquid and gel phases of DMPC generates extra spaces in the bilayer, thus facilitating remodeling by proteins (40). We found that the surface remodeling activity of B6.4−17 occurs on more heterogeneous bilayers as well, such as egg PC and is not DMPC dependent. Immediately after the protein is added to egg PC SUVs, some rod-shaped particles (Fig. 7d, white arrowheads) are formed, suggesting the conversion of vesicles to reconstituted particles. In addition, we observe many ring-like structures which can be the transitional complex from vesicles to reconstituted particles (white arrows). Clearly, this instantaneously prepared sample contains a mixture of particles, whose structure might reflect the mechanism of phospholipid remodeling.
DISCUSSION
Despite the exciting progress in the study of apolipoprotein B, the formation of an apoB-containing lipoprotein remains an elusive process. Simply mixing the protein and lipid components does not reconstitute the native lipoprotein particle (41). Apparently the ER provides a unique environment for the assembly of apoB-containing lipoproteins by allowing the interaction between proteins, lipids and various chaperones in a well organized manner. Among the many advantages that the ER offers, at least three features can be critical for this assembly process: 1) the location of apoB synthesis is close to the source of lipids, i.e. the ER membrane; 2) efficient apoB-containing lipoprotein assembly requires MTP; 3) the assembly is cotranslational, suggesting that the sequential interaction of the various apoB domains with lipids may be critical.
The N-terminal βα1 superdomain in apoB is the very first region that enters the ER and initiates lipoprotein assembly. It has been proposed that this region has a strong affinity for phospholipids (28, 29, 31). In this study, we use a “divide and conquer” approach to map the phospholipid recruiting elements in the βα1 domain of apoB. Currently our study only encompasses domains within B17, because domain constructs including the proposed A-sheet (B17−20.5) are insoluble in the absence of detergents (unpublished data). According to the lipovitellin based model, the hydrophobic nature of the proposed A-sheet is the likely cause of the protein aggregation when it is refolded without lipids.
The formation of discoidal particles by the in vitro incubation of purified B17 with DMPC suspensions was demonstrated by Herscovitz et al. (29) Here we examine the interaction of the individual domains that comprise B17 with DMPC. The N-terminal B5.9 adopts a β-barrel fold and has extensively hydrophilic surfaces according to the lipovitellin based model (Fig. 8c). This domain does not actively remodel phospholipid membranes. The predicted α-helical domain (B6.4−13) can interact with DMPC, break the multilamellar bilayers and form reconstituted DMPC particles. The kinetic analysis indicates this DMPC remodeling property resides primarily in its N-terminal half, i.e. B6.4−10 (Fig. 6). A strong phospholipid perturbation capability is observed in the first half of the C-sheet, yet this region by itself fails to stabilize nascent DMPC complexes. Constructs containing B13−15 (B6.4−15 and B6.4−17) exhibit faster lipid remodeling and are competent to stabilize the nascent DMPC complexes. Although we could not reproducibly measure the kinetics of DMPC clearance by B13−17, this construct clearly stabilizes reconstituted DMPC particles (Fig. 4e, j). Thus, B15−17 is expected to be capable of binding phospholipids as well. The fact that B6.4−17 has slower DMPC clearance kinetics than B6.4−15 suggests that B15−17 does not remodel phospholipids as efficiently as B13−15 (Fig. 5). These clearance assays demonstrate a strong interaction between the N-terminal half of the helical domain and the C-sheet domain of B17 with phospholipids.
Figure 8. Phospholipid remodeling elements in B17.
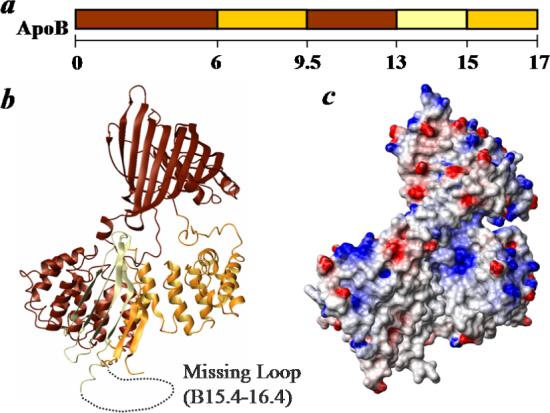
a. Domain map of B17 colored by the phospholipid remodeling capability. The color scheme is based on the kinetic measurements of the DMPC clearance summarized in Figure 6 and Table 2. Lighter color represents more efficient phospholipid remodeling. b. Ribbon model of B17 colored by the phospholipid remodeling capability. Notably, these phospholipid remodeling sequences form an arch that is present on the interaction surface of the lipid binding pocket in the lipovitellin structure. The missing region, B15.4−16.4 is shown by a dotted curve. c. Surface potential representation of the B17 model based on lipovitellin. Negative potential is shown in red and positive in blue. The α-helical and C-sheet domain arch is mainly hydrophobic in the model.
Using negative stain EM we demonstrate that constructs containing the proposed α-helical and C-sheet domains convert DMPC MLVs into relatively homogenous particles. The morphologies of these reconstituted particles are reminiscent of discoidal high density lipoprotein formed with exchangeable apolipoproteins (40). However, the protein sequence in the βα1 superdomain lacks the Y or A type amphipathic α-helices that are the basic building blocks for exchangeable apolipoproteins and for the α2 and α3 superdomains of apoB (5, 42). Moreover, the size of the particles reconstituted by B6.4−17 is larger than that formed with apoA-I. ApoA-I has a molecular weight similar to B6.4−13, but it forms discoidal particles with DMPC as small as 90 Å in diameter (37). In contrast, the minimal-size particle with B6.4−17 is approximately 150 Å in diameter. More interestingly, the overall secondary structure of the constructs containing the α-helical domain and the C-sheet domain remains unchanged when these proteins form reconstituted particles with DMPC. This observation is in contrast to apoA-I, whose α-helical content increases upon phospholipid binding (43). These differences indicate structural inelasticity in apoB, which is likely to be a result of protein tertiary folding. ApoA-I is a molten globule in lipid-free solution and probably forms a double α-helical belt in a phospholipid complex (44, 45). This double belt model may not apply to the B6.4−17/DMPC complex.
Our analysis provides insights into the folding of the protein during phospholipid remodeling. We previously demonstrated that lipid-free B17 adopts a structure very close to the prediction of the lipovitellin-based homology model (13). Our current data further support this hypothesis by mapping the phospholipid binding sequences in B17. When the domains in B17 are colored by their DMPC clearance rates, the phospholipid binding elements generate a continuous surface for lipid binding (Fig. 8a, b). Surface potential calculation indicates this surface is largely hydrophobic (Fig. 8c).
Richardson et al. reported a model of B22.5 with 48 molecules of POPC assembled in an asymmetric bilayer made primarily by comparative modeling based on the crystal structure of lipovitellin (10). Our mapping provides direct experimental support that the lipid binding components in their model within B17 are essentially correct. They hypothesized that sequences in B15.4−16.4 form an amphipathic helix-loop-helix and close the phospholipid pocket in B22.5. This hypothesis is consistent with our previous circular dichroism measurements, which show that B13−17 is about 30% α-helical (13). The proposed α-helices may account for the observed α-helical content as the rest of the domain is composed of β-sheets. This hypothesis is also compatible with the current observation that B13−17 forms stable complexes with DMPC, but B13−15 does not. However, their B22.5 model cannot easily explain how B6.4−17 forms a discoidal complex with DMPC, since the phospholipid binding elements in this model are not aligned in parallel with each other to cover the hydrophobic hydrocarbon chains in DMPC. This discrepancy may be resolved if the subdomains in their model are allowed to realign. We suggest a potential conformational adaptability is a property of the N-terminal domains of apoB when they undergo lipoprotein assembly.
It is clear that the reconstituted DMPC complexes observed in our study do not exist during in vivo lipoprotein maturation. However, the intrinsic phospholipid remodeling properties we identify in the structural domains of the N-terminal region of apoB may shed light on the initiation of lipoprotein assembly. The sequential entry of these domains into the ER lumen may not be random, but rather structurally advantageous for successful lipoprotein assembly. The very N-terminal ∼6% of apoB is proposed to fold into a β-barrel that we demonstrate lacks phospholipid remodeling capability. Yet this domain binds MTP and may serve as an important docking site for MTP during the early stages of lipoprotein assembly (9). Phospholipid binding could begin as early as during the translation of the first half of the proposed α-helical domain, B6.4−10. Another strong phospholipid remodeling sequence resides in the C-sheet, especially B13−15. Overall, although it is stable by itself, B17 has the tendency to bind both neutral lipids and phospholipids (30). It has the capacity to spontaneously break the phospholipid bilayer and stabilize pieces of bilayer, unless this intrinsic activity is inhibited by chaperones in the ER lumen or the inaccessibility of lipids in the ER membrane.
Since MTP is homologous to the N-terminal region of apoB and binds to it, MTP may have similar phospholipid remodeling properties. It is possible that the formation of an initiation complex in vivo is directly assisted by MTP, which is artificially replaced by apoB in our in vitro study. Further apoB translation into the β1 superdomain allows apoB alone to stabilize a lipoprotein and may push off MTP. Recently, it was reported that dMTP, a drosophila MTP homolog that lacks triglyceride transfer activity, promoted apoB-containing lipoprotein assembly in COS cells (46). The authors concluded that the phospholipid transfer activity of dMTP was responsible for this function. Yet, this observation could also be interpreted by the hypothesis that MTP behaves as a structural chaperone and stabilizes the transient particles during lipoprotein assembly.
While we have demonstrated that specific regions of B17 have strong lipid binding and remodeling activity and form discoidal particles in vitro, B17 is secreted from cells in a lipid poor form (29, 47). How do we reconcile our results with the in vivo secretion data? We note that a similar paradox is seen with apoA-I secretion. Significant amounts of apoA-I are secreted in lipid-poor form and lipidation occurs extracelluarly (48). Thus, lipid-interactive proteins can be secreted from cells without lipid. This may be due to a lack of available lipids or binding of chaperones that prevent acquisition of lipid, or both.
In vivo secretion studies have suggested that the translation of B19.5-B22, including most of the regions homologous to the A-sheet in the lipovitellin structure, enables the transition of apoB secretion from a lipid-poor form to a lipoprotein form (26). Although the exact length required for apoB to form a lipoprotein is still under debate (26, 27, 31), these seemingly inconsistent observations might reflect an intrinsic property of apoB during the assembly of apoB-containing lipoproteins. There may be flexibility in the assembly of these lipoproteins in response to the environment, in particular the lipid profile and MTP availability. This flexibility presents a unique challenge that awaits new strategies to describe and characterize.
Supplementary Material
AKNOWLEDGEMENT
We thank Drs. Donald Small, David Atkinson, Haya Herscovitz, Margaretha Carraway and Libo Wang for their insightful discussions, and Cheryl England and Michael Gigliotti for technical assistance.
This work was supported by a grant from the National Institute of Health (Grant HL-26335).
The abbreviations used are
- apoB
apolipoprotein B
- CD
circular dichroism
- DMPC
dimyristoylphosphatidylcholine
- ER
endoplasmic reticulum
- MTP
microsomal triglyceride transfer protein
- LDL
low density lipoprotein
- MLVs
multilamellar vesicles
- SUVs
small unilamellar vesicles
- VLDL
very low density lipoprotein
Footnotes
Supporting Information Available. The supporting information consisting of two figures similar to Figure 3 that show the characterization of B6.4−13 and B6.4−15 complexed to DMPC at different peptide to lipid ratios. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Knott TJ, Rall SC, Jr., Innerarity TL, Jacobson SF, Urdea MS, et al. Human apolipoprotein B: structure of carboxyl-terminal domains, sites of gene expression, and chromosomal localization. Science. 1985;230:37–43. doi: 10.1126/science.2994225. [DOI] [PubMed] [Google Scholar]
- 2.Chen SH, Habib G, Yang CY, Gu ZW, Lee BR, et al. Apolipoprotein B-48 is the product of a messenger RNA with an organ-specific in-frame stop codon. Science. 1987;238:363–6. doi: 10.1126/science.3659919. [DOI] [PubMed] [Google Scholar]
- 3.Powell LM, Wallis SC, Pease RJ, Edwards YH, Knott TJ, et al. A novel form of tissue-specific RNA processing produces apolipoprotein-B48 in intestine. Cell. 1987;50:831–40. doi: 10.1016/0092-8674(87)90510-1. [DOI] [PubMed] [Google Scholar]
- 4.Mahley RW, Innerarity TL, Rall SC, Jr., Weisgraber KH. Plasma lipoproteins: apolipoprotein structure and function. J Lipid Res. 1984;25:1277–94. [PubMed] [Google Scholar]
- 5.Segrest JP, Jones MK, Mishra VK, Anantharamaiah GM, Garber DW. apoB-100 has a pentapartite structure composed of three amphipathic alpha-helical domains alternating with two amphipathic beta-strand domains. Detection by the computer program LOCATE. Arterioscler Thromb. 1994;14:1674–85. doi: 10.1161/01.atv.14.10.1674. [DOI] [PubMed] [Google Scholar]
- 6.Segrest JP, Jones MK, De Loof H, Dashti N. Structure of apolipoprotein B-100 in low density lipoproteins. J Lipid Res. 2001;42:1346–67. [PubMed] [Google Scholar]
- 7.Spin JM, Atkinson D. Cryoelectron microscopy of low density lipoprotein in vitreous ice. Biophys J. 1995;68:2115–23. doi: 10.1016/S0006-3495(95)80392-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orlova EV, Sherman MB, Chiu W, Mowri H, Smith LC, et al. Three-dimensional structure of low density lipoproteins by electron cryomicroscopy. Proc Natl Acad Sci U S A. 1999;96:8420–5. doi: 10.1073/pnas.96.15.8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mann CJ, Anderson TA, Read J, Chester SA, Harrison GB, et al. The structure of vitellogenin provides a molecular model for the assembly and secretion of atherogenic lipoproteins. J Mol Biol. 1999;285:391–408. doi: 10.1006/jmbi.1998.2298. [DOI] [PubMed] [Google Scholar]
- 10.Richardson PE, Manchekar M, Dashti N, Jones MK, Beigneux A, et al. Assembly of lipoprotein particles containing apolipoprotein-B: structural model for the nascent lipoprotein particle. Biophys J. 2005;88:2789–800. doi: 10.1529/biophysj.104.046235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharrock WJ, Rosenwasser TA, Gould J, Knott J, Hussey D, et al. Sequence of lamprey vitellogenin. Implications for the lipovitellin crystal structure. J Mol Biol. 1992;226:903–7. doi: 10.1016/0022-2836(92)90642-w. [DOI] [PubMed] [Google Scholar]
- 12.Anderson TA, Levitt DG, Banaszak LJ. The structural basis of lipid interactions in lipovitellin, a soluble lipoprotein. Structure. 1998;6:895–909. doi: 10.1016/s0969-2126(98)00091-4. [DOI] [PubMed] [Google Scholar]
- 13.Jiang ZG, Carraway M, McKnight CJ. Limited proteolysis and biophysical characterization of the lipovitellin homology region in apolipoprotein B. Biochemistry. 2005;44:1163–73. doi: 10.1021/bi048286y. [DOI] [PubMed] [Google Scholar]
- 14.Wetterau JR, Aggerbeck LP, Laplaud PM, McLean LR. Structural properties of the microsomal triglyceride-transfer protein complex. Biochemistry. 1991;30:4406–12. doi: 10.1021/bi00232a006. [DOI] [PubMed] [Google Scholar]
- 15.Atzel A, Wetterau JR. Mechanism of microsomal triglyceride transfer protein catalyzed lipid transport. Biochemistry. 1993;32:10444–50. doi: 10.1021/bi00090a021. [DOI] [PubMed] [Google Scholar]
- 16.Hussain MM, Shi J, Dreizen P. Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J Lipid Res. 2003;44:22–32. doi: 10.1194/jlr.r200014-jlr200. [DOI] [PubMed] [Google Scholar]
- 17.Wetterau JR, Aggerbeck LP, Bouma ME, Eisenberg C, Munck A, et al. Absence of microsomal triglyceride transfer protein in individuals with abetalipoproteinemia. Science. 1992;258:999–1001. doi: 10.1126/science.1439810. [DOI] [PubMed] [Google Scholar]
- 18.Sharp D, Blinderman L, Combs KA, Kienzle B, Ricci B, et al. Cloning and gene defects in microsomal triglyceride transfer protein associated with abetalipoproteinaemia. Nature. 1993;365:65–9. doi: 10.1038/365065a0. [DOI] [PubMed] [Google Scholar]
- 19.Segrest JP, Jones MK, Dashti N. N-terminal domain of apolipoprotein B has structural homology to lipovitellin and microsomal triglyceride transfer protein: a “lipid pocket” model for self-assembly of apob-containing lipoprotein particles. J Lipid Res. 1999;40:1401–16. [PubMed] [Google Scholar]
- 20.Hussain MM, Bakillah A, Nayak N, Shelness GS. Amino acids 430−570 in apolipoprotein B are critical for its binding to microsomal triglyceride transfer protein. J Biol Chem. 1998;273:25612–5. doi: 10.1074/jbc.273.40.25612. [DOI] [PubMed] [Google Scholar]
- 21.Dashti N, Gandhi M, Liu X, Lin X, Segrest JP. The N-terminal 1000 residues of apolipoprotein B associate with microsomal triglyceride transfer protein to create a lipid transfer pocket required for lipoprotein assembly. Biochemistry. 2002;41:6978–87. doi: 10.1021/bi011757l. [DOI] [PubMed] [Google Scholar]
- 22.Burch WL, Herscovitz H. Disulfide bonds are required for folding and secretion of apolipoprotein B regardless of its lipidation state. J Biol Chem. 2000;275:16267–74. doi: 10.1074/jbc.M000446200. [DOI] [PubMed] [Google Scholar]
- 23.Ingram MF, Shelness GS. Folding of the amino-terminal domain of apolipoprotein B initiates microsomal triglyceride transfer protein-dependent lipid transfer to nascent very low density lipoprotein. J Biol Chem. 1997;272:10279–86. doi: 10.1074/jbc.272.15.10279. [DOI] [PubMed] [Google Scholar]
- 24.Huang XF, Shelness GS. Identification of cysteine pairs within the amino-terminal 5% of apolipoprotein B essential for hepatic lipoprotein assembly and secretion. J Biol Chem. 1997;272:31872–6. doi: 10.1074/jbc.272.50.31872. [DOI] [PubMed] [Google Scholar]
- 25.Tran K, Boren J, Macri J, Wang Y, McLeod R, et al. Functional analysis of disulfide linkages clustered within the amino terminus of human apolipoprotein B. J Biol Chem. 1998;273:7244–51. doi: 10.1074/jbc.273.13.7244. [DOI] [PubMed] [Google Scholar]
- 26.Shelness GS, Hou L, Ledford AS, Parks JS, Weinberg RB. Identification of the lipoprotein initiating domain of apolipoprotein B. J Biol Chem. 2003;278:44702–7. doi: 10.1074/jbc.M307562200. [DOI] [PubMed] [Google Scholar]
- 27.Manchekar M, Richardson PE, Forte TM, Datta G, Segrest JP, et al. Apolipoprotein B-containing lipoprotein particle assembly: lipid capacity of the nascent lipoprotein particle. J Biol Chem. 2004;279:39757–66. doi: 10.1074/jbc.M406302200. [DOI] [PubMed] [Google Scholar]
- 28.DeLozier JA, Parks JS, Shelness GS. Vesicle-binding properties of wild-type and cysteine mutant forms of alpha(1) domain of apolipoprotein B. J Lipid Res. 2001;42:399–406. [PubMed] [Google Scholar]
- 29.Herscovitz H, Hadzopoulou-Cladaras M, Walsh MT, Cladaras C, Zannis VI, et al. Expression, secretion, and lipid-binding characterization of the N- terminal 17% of apolipoprotein B. Proc Natl Acad Sci U S A. 1991;88:7313–7. doi: 10.1073/pnas.88.16.7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herscovitz H, Derksen A, Walsh MT, McKnight CJ, Gantz DL, et al. The N-terminal 17% of apoB binds tightly and irreversibly to emulsions modeling nascent very low density lipoproteins. J Lipid Res. 2001;42:51–9. [PubMed] [Google Scholar]
- 31.Carraway M, Herscovitz H, Zannis V, Small DM. Specificity of lipid incorporation is determined by sequences in the N- terminal 37 of apoB. Biochemistry. 2000;39:9737–45. doi: 10.1021/bi000791h. [DOI] [PubMed] [Google Scholar]
- 32.Edelhoch H. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 1967;6:1948–54. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- 33.Bartlett GR. Phosphorus assay in column chromatography. J Biol Chem. 1959;234:466–8. [PubMed] [Google Scholar]
- 34.Harris JR, Horne RW. In: Harris JR, editor. IRL Press; Oxford, UK: 1991. pp. 203–228. [Google Scholar]
- 35.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 36.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–5. doi: 10.1016/0263-7855(96)00009-4. 29−32. [DOI] [PubMed] [Google Scholar]
- 37.Tall AR, Small DM, Deckelbaum RJ, Shipley GG. Structure and thermodynamic properties of high density lipoprotein recombinants. J Biol Chem. 1977;252:4701–11. [PubMed] [Google Scholar]
- 38.Hughes AV, Roser SJ, Gerstenberg M, Goldar A, Stidder B, et al. Phase Behavior of DMPC Free Supported Bilayers Studied by Neutron Reflectivity. Langmuir. 2002;18:8161–8171. [Google Scholar]
- 39.Borchardt RA, Davis RA. Intrahepatic assembly of very low density lipoproteins. Rate of transport out of the endoplasmic reticulum determines rate of secretion. J Biol Chem. 1987;262:16394–402. [PubMed] [Google Scholar]
- 40.Atkinson D, Small DM. Recombinant lipoproteins: implications for structure and assembly of native lipoproteins. Annu Rev Biophys Biophys Chem. 1986;15:403–56. doi: 10.1146/annurev.bb.15.060186.002155. [DOI] [PubMed] [Google Scholar]
- 41.Walsh MT, Atkinson D. Reassembly of low-density lipoproteins. Methods Enzymol. 1986;128:582–608. doi: 10.1016/0076-6879(86)28093-3. [DOI] [PubMed] [Google Scholar]
- 42.Segrest JP, Jones MK, De Loof H, Brouillette CG, Venkatachalapathi YV, et al. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J Lipid Res. 1992;33:141–66. [PubMed] [Google Scholar]
- 43.Fang Y, Gursky O, Atkinson D. Lipid-binding studies of human apolipoprotein A-I and its terminally truncated mutants. Biochemistry. 2003;42:13260–8. doi: 10.1021/bi0354031. [DOI] [PubMed] [Google Scholar]
- 44.Segrest JP, Jones MK, Klon AE, Sheldahl CJ, Hellinger M, et al. A detailed molecular belt model for apolipoprotein A-I in discoidal high density lipoprotein. J Biol Chem. 1999;274:31755–8. doi: 10.1074/jbc.274.45.31755. [DOI] [PubMed] [Google Scholar]
- 45.Gorshkova IN, Liu T, Zannis VI, Atkinson D. Lipid-free structure and stability of apolipoprotein A-I: probing the central region by mutation. Biochemistry. 2002;41:10529–39. doi: 10.1021/bi025807d. [DOI] [PubMed] [Google Scholar]
- 46.Rava P, Ojakian GK, Shelness GS, Hussain MM. Phospholipid transfer activity of microsomal triglyceride transfer protein is sufficient for the assembly and secretion of apoB-lipoproteins. J Biol Chem. 2006 doi: 10.1074/jbc.M512823200. [DOI] [PubMed] [Google Scholar]
- 47.Yao ZM, Blackhart BD, Linton MF, Taylor SM, Young SG, et al. Expression of carboxyl-terminally truncated forms of human apolipoprotein B in rat hepatoma cells. Evidence that the length of apolipoprotein B has a major effect on the buoyant density of the secreted lipoproteins. J Biol Chem. 1991;266:3300–8. [PubMed] [Google Scholar]
- 48.Chisholm JW, Burleson ER, Shelness GS, Parks JS. ApoA-I secretion from HepG2 cells: evidence for the secretion of both lipid-poor apoA-I and intracellularly assembled nascent HDL. J Lipid Res. 2002;43:36–44. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.